
Methylenedioxy Piperamide-Derived Compound D5 Regulates Inflammatory Cytokine Secretion in a Culture of Human Glial Cells


Abstract
:1. Highlights
- -
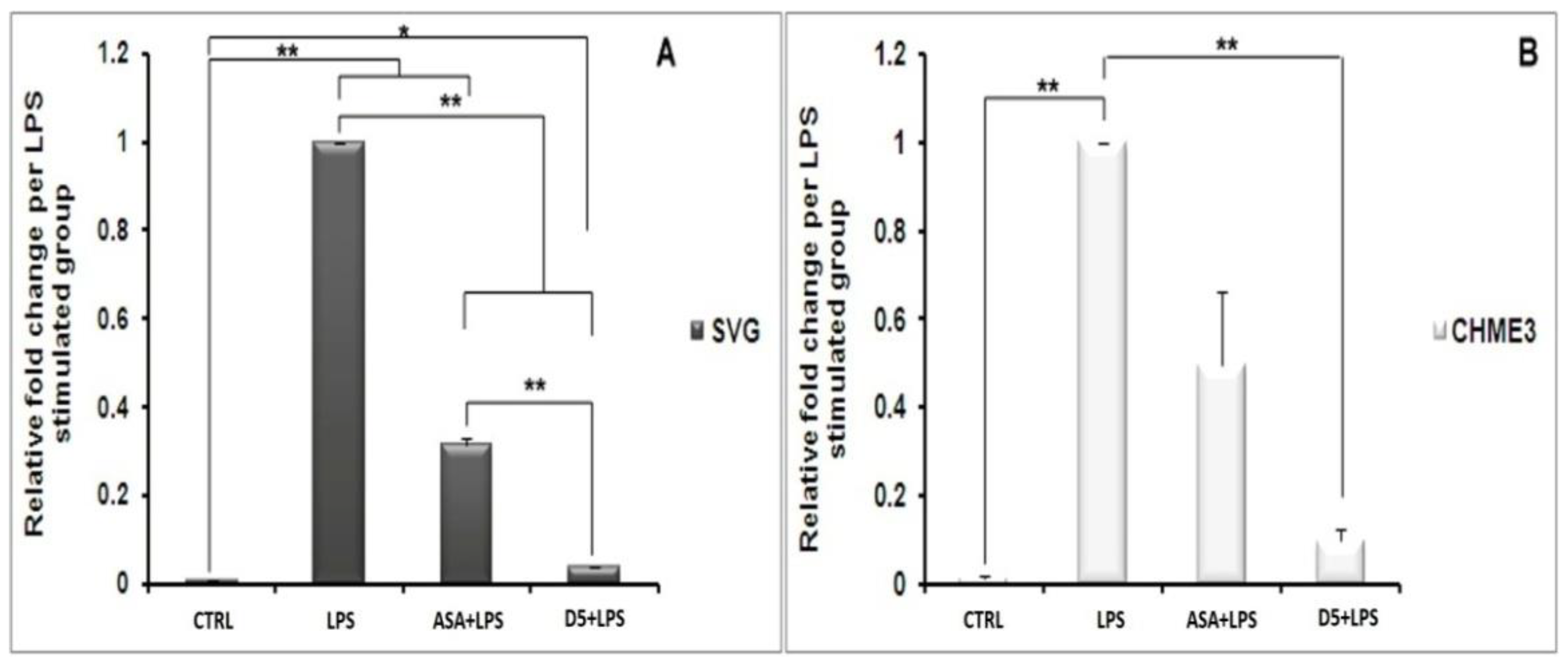
- D5 reduced the level of pro-inflammatory cytokines, through up-regulating PPAR-γ expression, suppressing IKK-β, iNOS, NO production, and NF-κB activation in inflamed astrocytes (SVG) and microglia (CHME3) after 24 h of incubation.
- -
- D5 showed higher efficacy than ASA on the inhibition of the NF-κB pathway leading to neuroinflammation.
- -
- D5 acts as a potent anti-neuroinflammatory agent.
2. Introduction
3. Materials and Methods
3.1. In Vitro Study
3.1.1. Cell Culture
3.1.2. Cell Treatment
3.1.3. Cytoprotective Effect of D5 in the Selected Concentration
3.1.4. NO Quantification
3.1.5. Isolation of Total Proteins
3.1.6. Western Blot Analysis
3.1.7. Enzyme-Linked Immunosorbent Assay (ELISA)
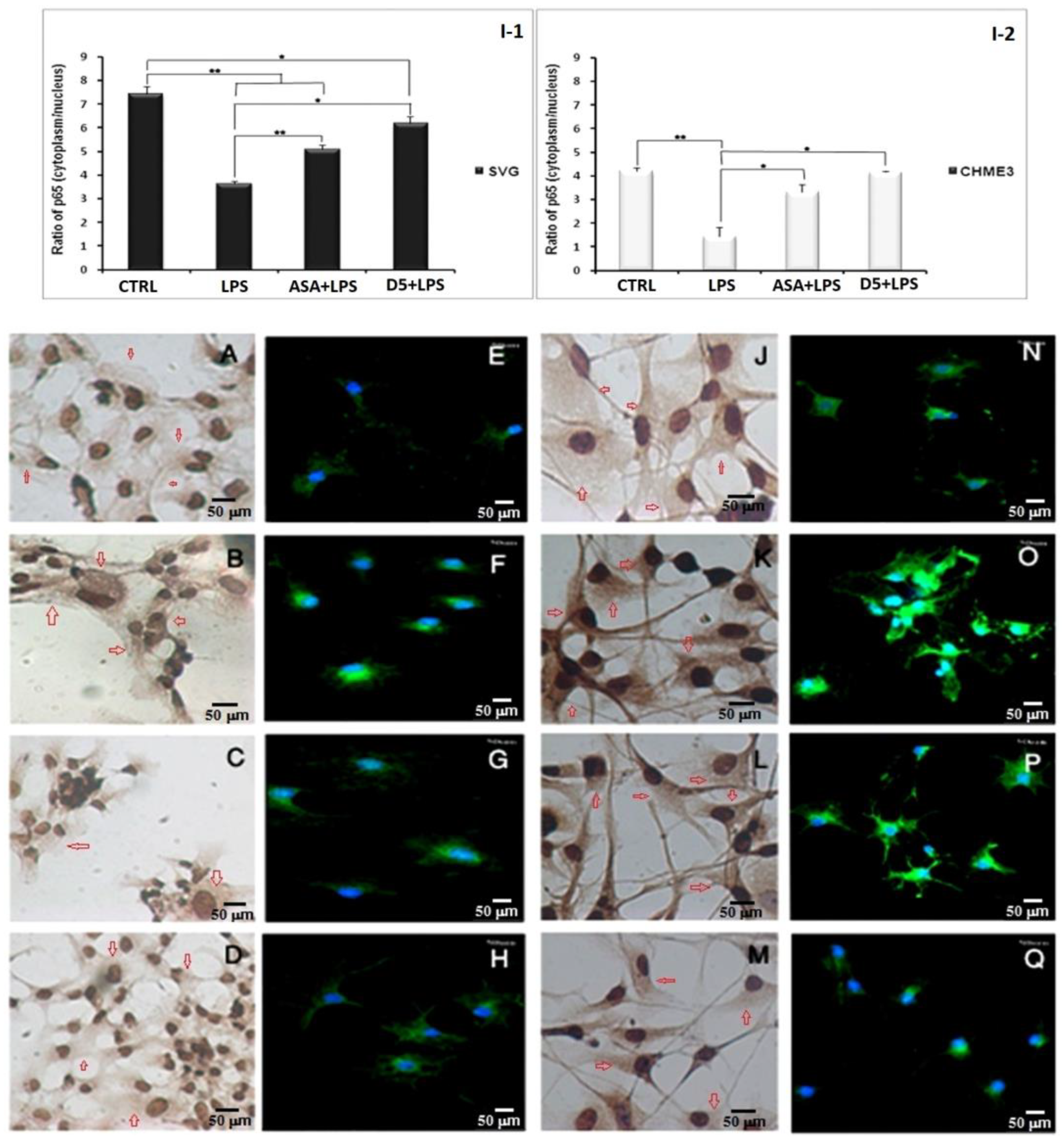
3.1.8. Immunocytochemistry
3.1.9. Total RNA Extraction and Quantitative Real-Time PCR (qPCR)
3.2. The Pharmacological Properties of Methylenedioxy (D5) vs. Hydroxymethyl (D4) Derivatives of Piperamide
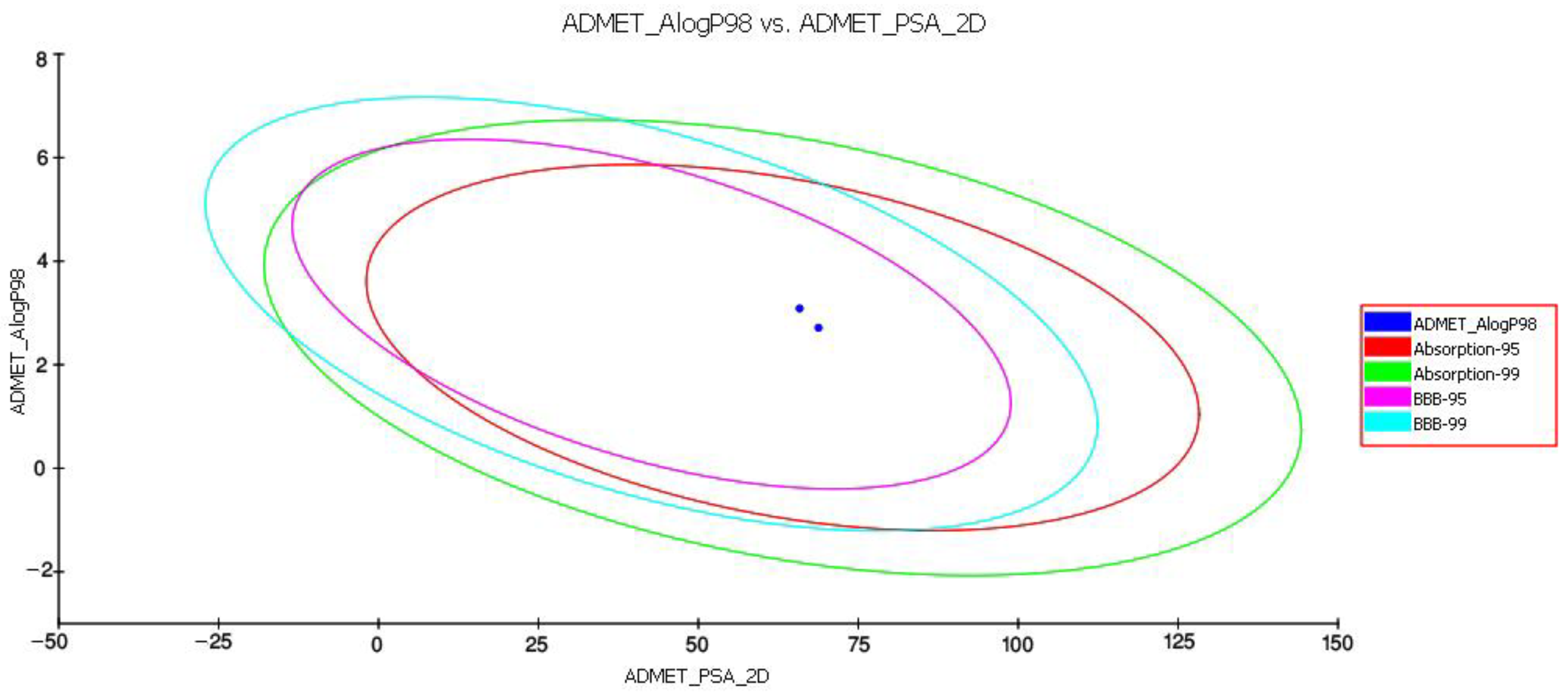
3.2.1. In Silico Evaluation of Pharmacological Properties and Potential Inhibitory Nature
- Comparison of the physicochemical and toxicological properties
- b.
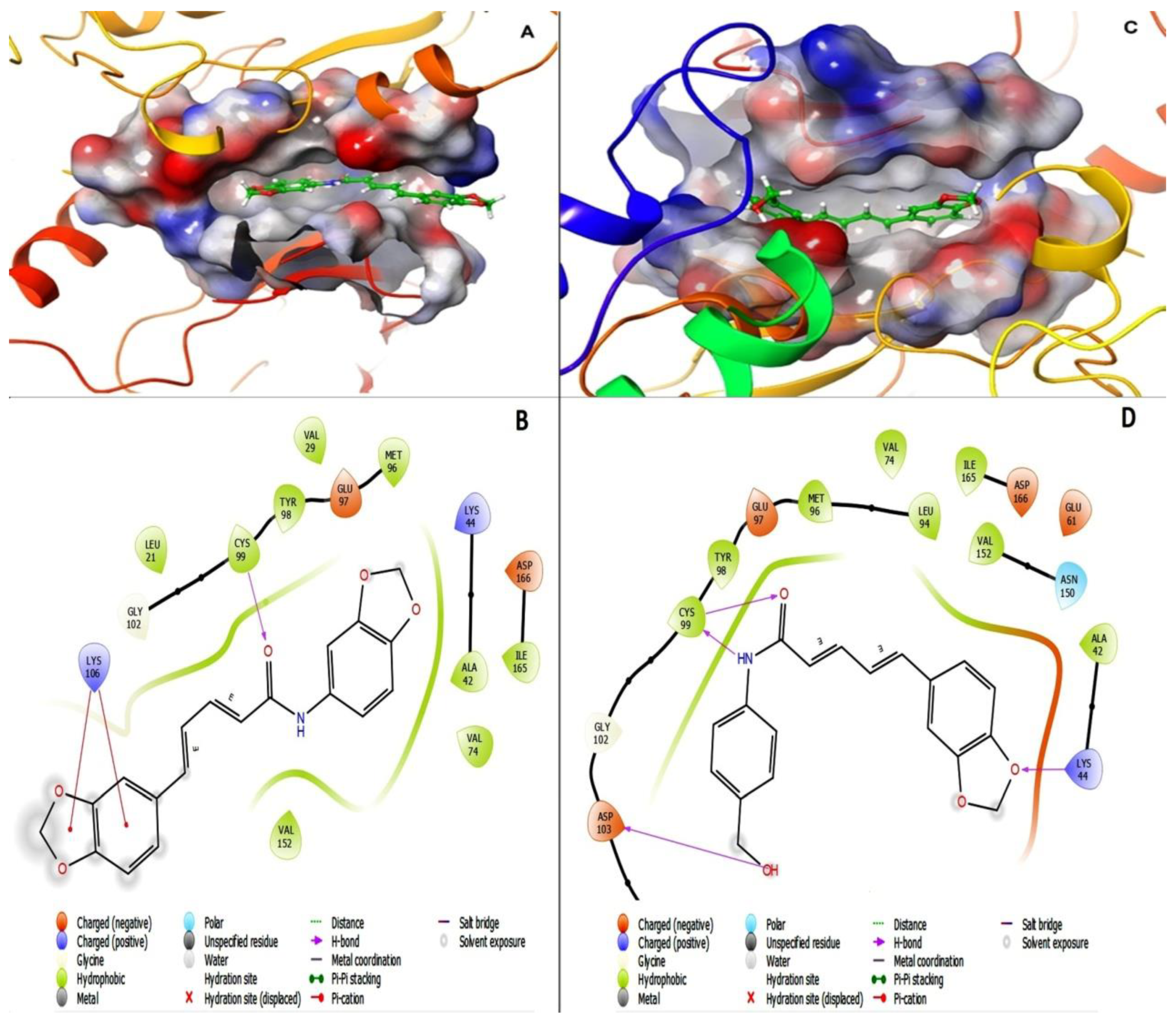
- Comparison of pharmacodynamic properties against IKK-β enzyme
3.2.2. In Vitro Druggability Evaluation of D5 vs. D4 on Neuroinflammatory Progress
3.3. Data Presentation and Statistical Analysis
4. Results
4.1. In Vitro Pharmacological Property Evaluation of D5 against LPS-Induced Inflammation in the Glial Cells
4.1.1. Determination of the Drug’s Optimal Concentrations
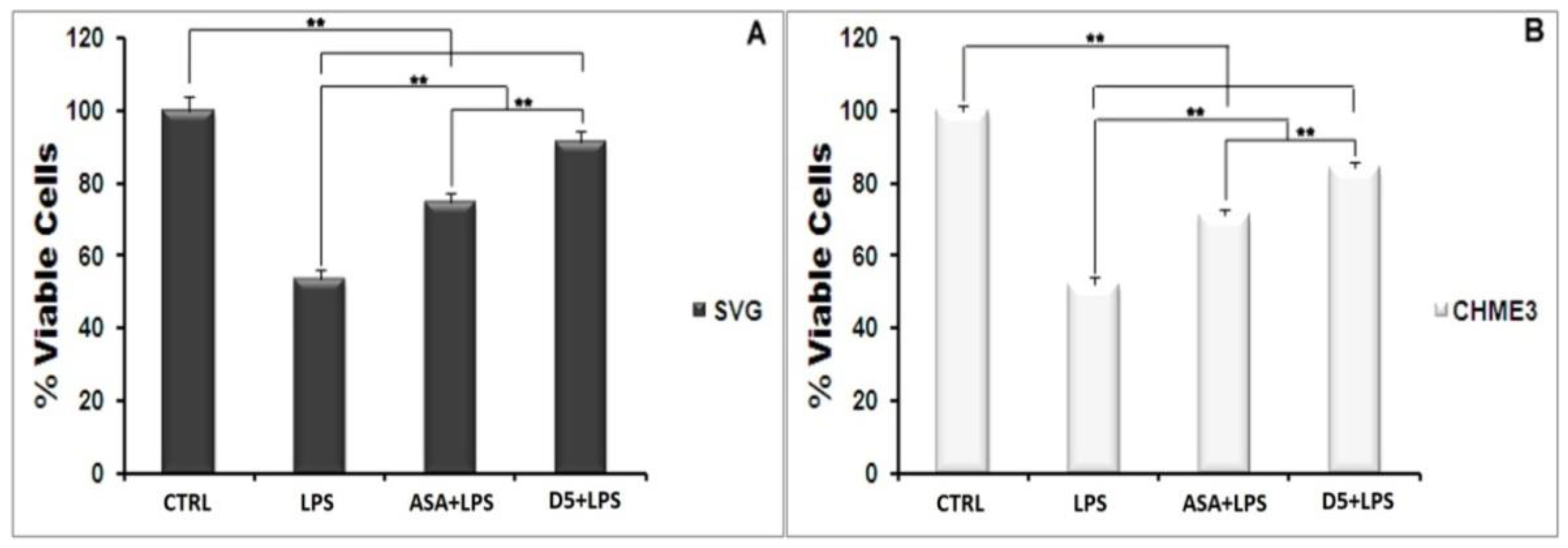
4.1.2. Drug Safety
4.1.3. Inhibitory Effect of D5 on the iNOS Expression Level and Nitric Oxide Secretion in LPS-Stimulated SVG and CHME3 Cell Lines
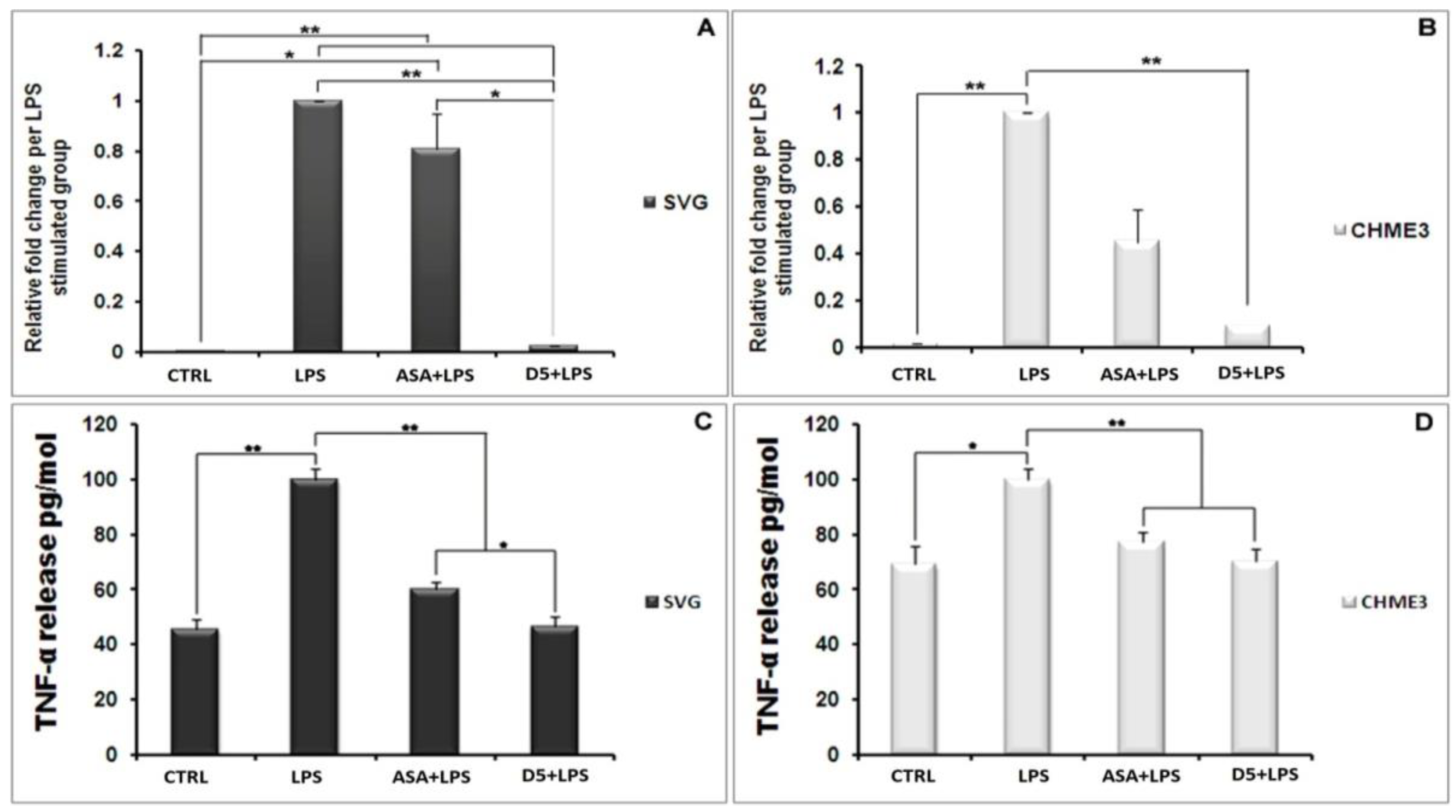
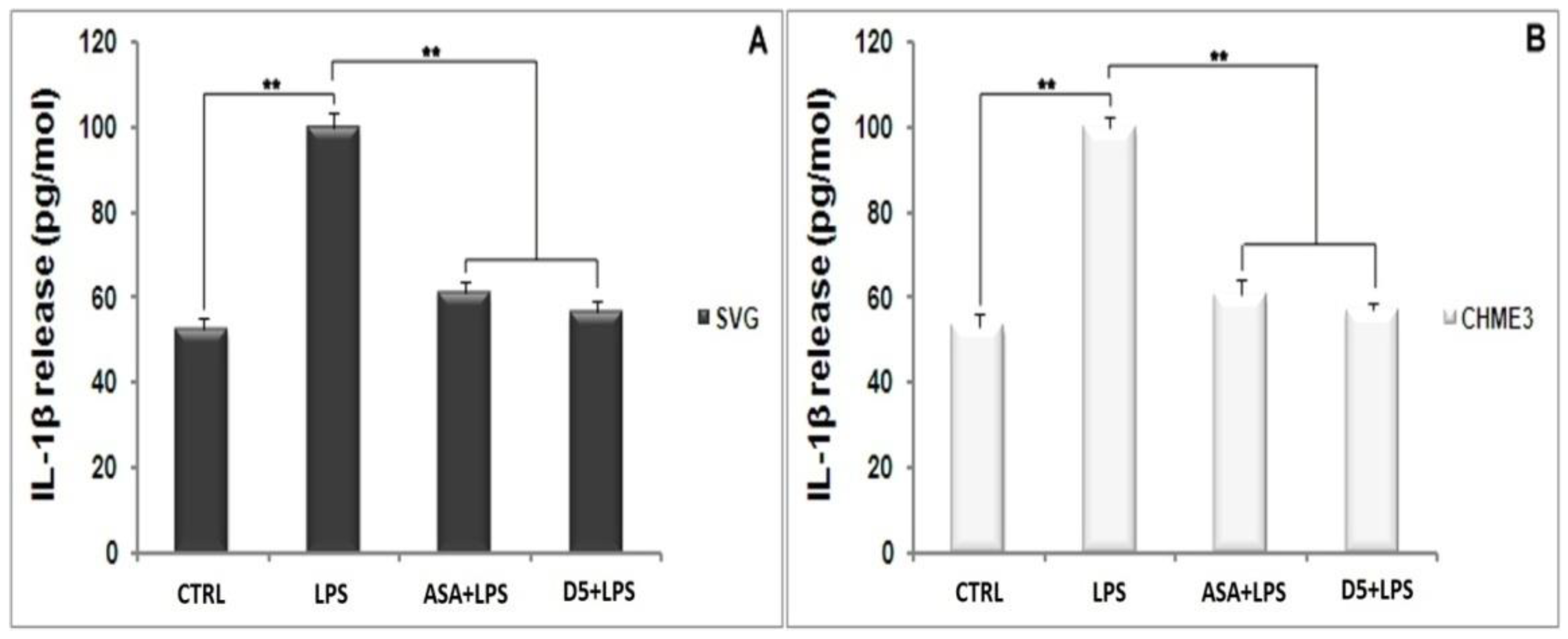
4.1.4. Inhibitory Effects of D5 on TNF-α and IL-1β Expression in LPS-Stimulated SVG and CHME3 Cell Lines
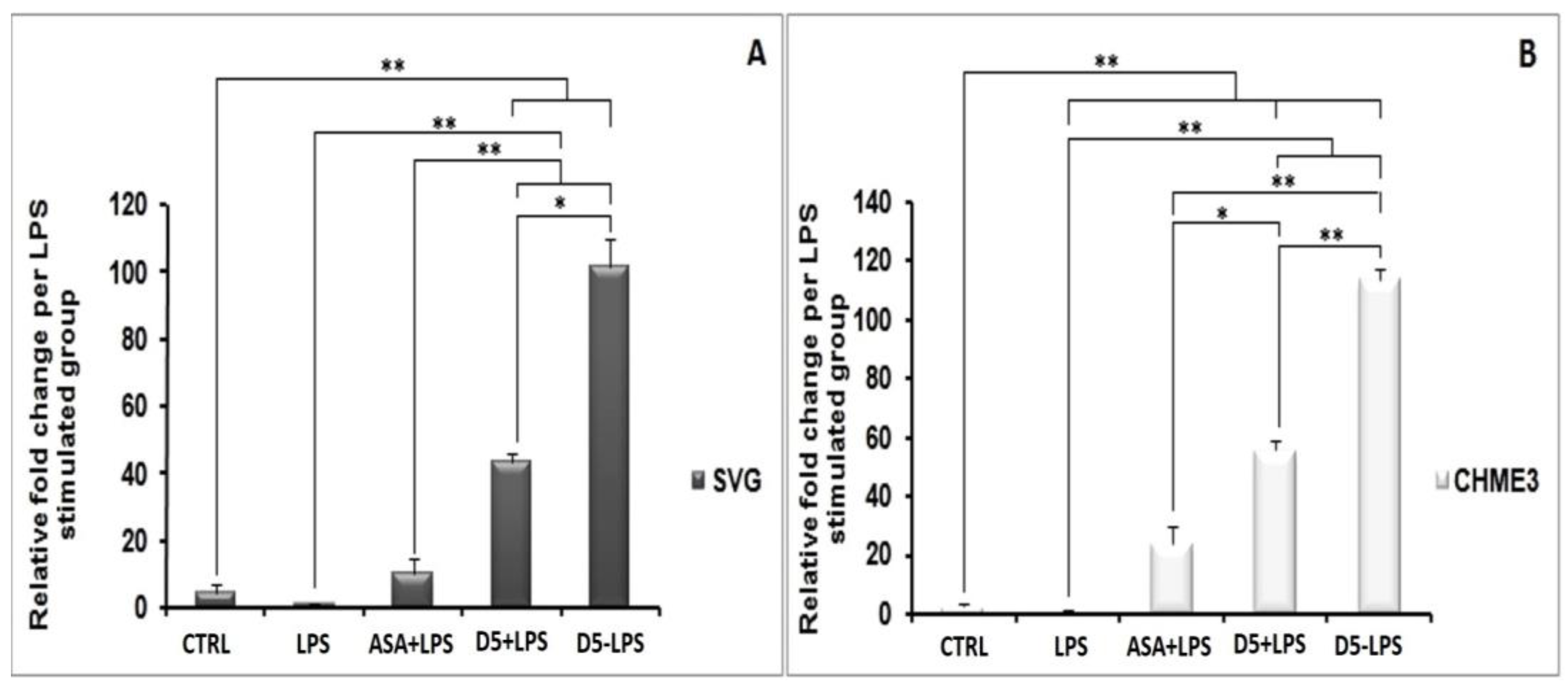
4.1.5. Up-Regulation of PPAR-γ Receptor Expression by D5 in LPS-Stimulated SVG and CHME3 Cell Lines
4.1.6. Inhibitory Effects of D5 on NF-κBSignaling in LPS-Stimulated SVG and CHME3 Cell Lines
4.2. Comparison of the Anti-Neuroinflammatory Potential of Methylenedioxy (D5) and Hydroxymethyl (D4) Piperamide Derivatives
4.2.1. In Silico Comparison between Pharmacological Properties of D5 and D4
- Comparison of the physicochemical and toxicological properties of both compounds
- b.
- Comparison of the pharmacodynamic properties of D4 and D5 against IKK-β enzyme
4.2.2. In Vitro Comparison of the Druggability Potential of Methylenedioxy and Hydroxymethyl Piperamide Derivatives on LPS-Induced inflammation in the Glial Cells
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goshen, I.; Yirmiya, R. The role of pro-inflammatory cytokines in memory processes and neural plasticity. Psychoneuroimmunology 2007, 4, 337–378. [Google Scholar]
- Hwang, J.; Zheng, L.T.; Ock, J.; Lee, M.G.; Kim, S.-H.; Lee, H.-W.; Lee, W.-H.; Park, H.-C.; Suk, K. Inhibition of glial inflammatory activation and neurotoxicity by tricyclic antidepressants. Neuropharmacology 2008, 55, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Zipp, F.; Aktas, O. The brain as a target of inflammation: Common pathways link inflammatory and neurodegenerative diseases. Trends Neurosci. 2006, 29, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Hong, J.-S. Role of microglia in inflammation-mediated neurodegenerative diseases: Mechanisms and strategies for therapeutic intervention. J. Pharmacol. Exp. Ther. 2003, 304, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.H.; Bigbee, M.J.; Boynton, G.E.; Wakeham, C.M.; Rosenheim, H.M.; Staral, C.J.; Morrissey, J.L.; Hund, A.K. Non-steroidal anti-inflammatory drugs in Alzheimer’s disease and Parkinson’s disease: Reconsidering the role of neuroinflammation. Pharmaceuticals 2010, 3, 1812–1841. [Google Scholar] [CrossRef]
- Kolhe, S.R.; Borole, P.; Patel, U. Extraction and evaluation of piperine from Piper nigrum Linn. Int. J. Appl. Biol. Pharm. Technol. 2011, 2, 144–149. [Google Scholar]
- Chan, G.C.-F.; Cheung, K.-W.; Sze, D.M.-Y. The immunomodulatory and anticancer properties of propolis. Clin. Rev. Allergy Immunol. 2013, 44, 262–273. [Google Scholar] [CrossRef]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef]
- Choi, S.-Y.; Ko, H.-C.; Ko, S.-Y.; Hwang, J.-H.; Park, J.-G.; Kang, S.-H.; Han, S.-H.; Yun, S.-H.; Kim, S.-J. Correlation between flavonoid content and the NO production inhibitory activity of peel extracts from various citrus fruits. Biol. Pharm. Bull. 2007, 30, 772–778. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, M.; Verpoorte, R.; Korthout, H.A.; Mustafa, N.R. Phytochemicals as a potential source for TNF-α inhibitors. Phytochem. Rev. 2013, 12, 65–93. [Google Scholar] [CrossRef]
- Gutiérrez, R.M.P.; Gonzalez, A.M.N.; Hoyo-Vadillo, C. Alkaloids from piper: A review of its phytochemistry and pharmacology. Mini Rev. Med. Chem. 2013, 13, 163–193. [Google Scholar] [PubMed]
- Shinta, D.N.; Choodej, S.; Pudhom, K. Synthesis of Piperine Amide Derivatives and Evaluation of Their Anti-inflammatory Activity. In Proceedings of the 6th RSU International Research Conference, Muang, Thailand, 30 April 2021. [Google Scholar]
- Smilkov, K.; Ackova, D.G.; Cvetkovski, A.; Ruskovska, T.; Vidovic, B.; Atalay, M. Piperine: Old spice and new nutraceutical? Curr. Pharm. Des. 2019, 25, 1729–1739. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.P.; Jain, S.K.; Kaur, A.; Singh, S.; Kumar, R.; Garg, P.; Sharma, S.S.; Arora, S.K. Synthesis and antileishmanial activity of piperoyl-amino acid conjugates. Eur. J. Med. Chem. 2010, 45, 3439–3445. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, K. Black pepper and its pungent principle-piperine: A review of diverse physiological effects. Crit. Rev. Food Sci. Nutr. 2007, 47, 735–748. [Google Scholar] [CrossRef]
- Wang, Y.; Yao, Y.; Liu, J.; Wu, L.; Liu, T.; Cui, J.; Lee, D.Y.-W. Synthesis and biological activity of piperine derivatives as potential PPARγ agonists. Drug Des. Dev. Ther. 2020, 14, 2069. [Google Scholar] [CrossRef] [PubMed]
- Hazra, B.; Chakraborty, S.; Bhaskar, M.; Mukherjee, S.; Mahadevan, A.; Basu, A. miR-301a regulates inflammatory response to Japanese encephalitis virus infection via suppression of NKRF activity. J. Immunol. 2019, 203, 2222–2238. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Mishra, M.K.; Ghosh, J.; Basu, A. Japanese encephalitis virus infection induces IL-18 and IL-1β in microglia and astrocytes: Correlation with in vitro cytokine responsiveness of glial cells and subsequent neuronal death. J. Neuroimmunol. 2008, 195, 60–72. [Google Scholar] [CrossRef]
- Wang, J.-S.; Wu, D.; Huang, D.-Y.; Lin, W.-W. TAK1 inhibition-induced RIP1-dependent apoptosis in murine macrophages relies on constitutive TNF-α signaling and ROS production. J. Biomed. Sci. 2015, 22, 76. [Google Scholar] [CrossRef] [Green Version]
- Rival, Y.; Benéteau, N.; Taillandier, T.; Pezet, M.; Dupont-Passelaigue, E.; Patoiseau, J.-F.; Junquéro, D.; Colpaert, F.C.; Delhon, A. PPARα and PPARδ activators inhibit cytokine-induced nuclear translocation of NF-κB and expression of VCAM-1 in EAhy926 endothelial cells. Eur. J. Pharmacol. 2002, 435, 143–151. [Google Scholar] [CrossRef]
- Giustarini, D.; Rossi, R.; Milzani, A.; Dalle-Donne, I. Nitrite and nitrate measurement by Griess reagent in human plasma: Evaluation of interferences and standardization. Methods Enzymol. 2008, 440, 361–380. [Google Scholar]
- Wei, Z.; Kale, S.; Rabinovsky, R.; El Fatimy, R.; Krichevsky, A.M. Co-cultures of glioma stem cells and primary neurons, astrocytes, microglia, and endothelial cells for investigation of intercellular communication in the brain. Front. Neurosci. 2019, 13, 361. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grellner, W.; Georg, T.; Wilske, J. Quantitative analysis of proinflammatory cytokines (IL-1β, IL-6, TNF-α) in human skin wounds. Forensic Sci. Int. 2000, 113, 251–264. [Google Scholar] [CrossRef]
- Lamprecht, M.R.; Sabatini, D.M.; Carpenter, A.E. CellProfiler™: Free, versatile software for automated biological image analysis. BioTechniques 2007, 42, 71–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eswaran, N.; Sundaram, V.A.; Rao, K.A.; Balasundaram, S.T. Simple isolation and characterization of seminal plasma extracellular vesicle and its total RNA in an academic lab. 3 Biotech 2018, 8, 139. [Google Scholar] [CrossRef]
- Jung, M.; Xu, C.; Ohl, F.; Mager, A.-K.; Jung, K.; Jung, M. Transcriptor First Strand cDNA Synthesis Kit: Efficient and Fast—A Comparison to Other Kits. Gene 2005, 50, 16–17. [Google Scholar]
- Kit, S. iTaq Universal SYBR Green Supermix (Bio-Rad). In Real-Time Primers Targeting SOD2 and GAPDH Were Ob; Bio-Rad: Hercules, CA, USA, 2021. [Google Scholar]
- Karolchik, D.; Hinrichs, A.S.; Kent, W.J. The UCSC genome browser. Curr. Protoc. Bioinform. 2012, 40, 1.4.1–1.4.33. [Google Scholar] [CrossRef]
- Thornton, B.; Basu, C. Real-time PCR (qPCR) primer design using free online software. Biochem. Mol. Biol. Educ. 2011, 39, 145–154. [Google Scholar] [CrossRef]
- Kibbe, W.A. OligoCalc: An online oligonucleotide properties calculator. Nucleic Acids Res. 2007, 35, W43–W46. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, S.; Manogar, P.; Prabhu, S.; Singh, R.A.S. Novel ligand-based docking; molecular dynamic simulations; and absorption, distribution, metabolism, and excretion approach to analyzing potential acetylcholinesterase inhibitors for Alzheimer’s disease. J. Pharm. Anal. 2018, 8, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Adane, L.; Bharatam, P.V. Computer-aided molecular design of 1H-imidazole-2, 4-diamine derivatives as potential inhibitors of Plasmodium falciparum DHFR enzyme. J. Mol. Model. 2011, 17, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H. In Vitro and In Vivo Anti-Inflammatory Effects of Rosmanol and Carnosol Isolated from Rosemary. Ph.D. Thesis, Rutgers University-Graduate School, New Brunswick, NJ, USA, 2010. [Google Scholar]
- Singh, S.; Singh, T.G. Role of nuclear factor kappa B (NF-κB) signalling in neurodegenerative diseases: An mechanistic approach. Current Neuropharmacology 2020, 18, 918–935. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Hua, F.; Wang, X.; Huang, J.-L. Therapeutic potential of IKK-β inhibitors from natural phenolics for inflammation in cardiovascular diseases. Inflammopharmacology 2020, 28, 19–37. [Google Scholar] [CrossRef]
- Liu, S.; Misquitta, Y.R.; Olland, A.; Johnson, M.A.; Kelleher, K.S.; Kriz, R.; Lin, L.L.; Stahl, M.; Mosyak, L. Crystal Structure of a Human IκB Kinase β Asymmetric Dimer. J. Biol. Chem. 2013, 288, 22758–22767. [Google Scholar] [CrossRef] [Green Version]
- Lyne, P.D.; Kenny, P.W.; Cosgrove, D.A.; Deng, C.; Zabludoff, S.; Wendoloski, J.J.; Ashwell, S. Identification of compounds with nanomolar binding affinity for checkpoint kinase-1 using knowledge-based virtual screening. J. Med. Chem. 2004, 47, 1962–1968. [Google Scholar] [CrossRef]
- Mongkolsapaya, J.; Grimes, J.M.; Chen, N.; Xu, X.-N.; Stuart, D.I.; Jones, E.Y.; Screaton, G.R. Structure of the TRAIL–DR5 complex reveals mechanisms conferring specificity in apoptotic initiation. Nat. Struct. Biol. 1999, 6, 1048–1053. [Google Scholar]
- Peran, D.; Sykora, R.; Vidunova, J.; Krsova, I.; Pekara, J.; Renza, M.; Brizgalova, N.; Cmorej, P.C. Non-technical skills in pre-hospital care in the Czech Republic: A prospective multicentric observational study (NTS study). BMC Emerg. Med. 2022, 22, 83. [Google Scholar] [CrossRef]
- Csermely, P.; Korcsmáros, T.; Kiss, H.J.; London, G.; Nussinov, R. Structure and dynamics of molecular networks: A novel paradigm of drug discovery: A comprehensive review. Pharmacol. Ther. 2013, 138, 333–408. [Google Scholar] [CrossRef] [Green Version]
- Tarakçıoğlu, M.; Erbağci, A.B.; Usalan, C.; Deveci, R.; Kocabaş, R. Acute effect of hemodialysis on serum levels of the proinflammatory cytokines. Mediat. Inflamm. 2003, 12, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, A.; Minghetti, L. PPAR-γ agonists as regulators of microglial activation and brain inflammation. Curr. Pharm. Des. 2006, 12, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Carta, A.R. PPAR-γ: Therapeutic prospects in Parkinson’s disease. Curr. Drug Targets 2013, 14, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Osthoff, K.; Ferrari, D.; Riehemann, K.; Wesselborg, S. Regulation of NF-κB activation by MAP kinase cascades. Immunobiology 1997, 198, 35–49. [Google Scholar] [CrossRef]
- Gveric, D.; Kaltschmidt, C.; Cuzner, M.L.; Newcombe, J. Transcription Factor NF-ΚB and Inhibitor lκBα are Localized in Macrophages in Active Multiple Sclerosis Lesions. J. Neuropathol. Exp. Neurol. 1998, 57, 168–178. [Google Scholar] [CrossRef] [Green Version]
- Puleston, J.M. Molecular Targeting in Inflammatory Bowel Disease an Investigation to Identify Novel Chemokine Targets for the Potential Development of New Therapeutic Agents for Inflammatory Bowel Disease. Ph.D. Thesis, University of London, London, UK, 2005. [Google Scholar]
- Małek, R.; Borowicz, K.K.; Jargiełło, M.; Czuczwar, S.J. Role of nuclear factor kB in the central nervous system. Pharmacol. Rep. 2007, 59, 25–33. [Google Scholar]
- Tripathi, P.; Aggarwal, A. NF-kB transcription factor: A key player in the generation of immune response. Curr. Sci. 2006, 90, 519. [Google Scholar]
- Mendonca, J.; Kumar, R.; Owoyemi, O.; Boyapati, K.; Thomas, N.; Kanacharoen, S.; Coffey, M.; Topiwala, D.; Gomes, C.; Ozbek, B. Supraphysiological testosterone induces ferroptosis and activates NF-kappaB mediated immune pathways in prostate cancer through nucleophagy. bioRxiv 2020. [Google Scholar] [CrossRef]
- Rafati, D.S. Roles of NF-kB in Spinal Cord Injury Pathophysiology. Ph.D. Thesis, Fort Worth, University of Texas Medical Branch, Galveston, TX, USA, 2005. [Google Scholar]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [Green Version]
- Mandrekar-Colucci, S.; Karlo, J.C.; Landreth, G.E. Mechanisms underlying the rapid peroxisome proliferator-activated receptor-γ-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer’s disease. J. Neurosci. 2012, 32, 10117–10128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, G.J.; Drego, R.; Hakimian, E.; Masliah, E. Mechanisms of cell signaling and inflammation in Alzheimer’s disease. Curr. Drug Targets Inflamm. Allergy 2005, 4, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Okwute, S.K.; Egharevba, H.O. Piperine-type amides: Review of the chemical and biological characteristics. Int. J. Chem. 2013, 5, 99. [Google Scholar] [CrossRef]
- Kumar, B.; Mantha, A.K.; Kumar, V. Recent developments on the structure–activity relationship studies of MAO inhibitors and their role in different neurological disorders. RSC Adv. 2016, 6, 42660–42683. [Google Scholar] [CrossRef]
- Pedersen, M.E.; Metzler, B.; Stafford, G.I.; Van Staden, J.; Jäger, A.K.; Rasmussen, H.B. Amides from Piper capense with CNS activity—A preliminary SAR analysis. Molecules 2009, 14, 3833–3843. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Amounts | DOSE * | IL-6/24 h | INF-γ/24 h | ||
|---|---|---|---|---|---|---|
| OD | QUANTIFY (pg/mL) | OD | QUANTIFY (pg/mL) | |||
| D4 | ½ EC50 | 140 | 0.32 | 282.11 ± 5.24 | 0.30 | 219.82 ± 10.13 |
| EC50 | 280 | 0.22 | 188.07 ± 6.17 | 0.23 | 163.04 ± 4.52 | |
| 2x EC50 | 560 | 0.17 | 141.04 ± 8.03 | 0.21 | 146.82 ± 5.85 | |
| D5 | ½ EC50 | 120 | 0.28 | 259.08 ± 9.22 | 0.31 | 291.37 ± 5.93 |
| EC50 | 240 | 0.21 | 194.93 ± 5.92 | 0.20 | 186.97 ± 5.28 | |
| 2x EC50 | 480 | 0.19 | 170.98 ± 5.40 | 0.16 | 149.33 ± 7.12 | |
| ASP | ½ EC50 | 4600 | 0.31 | 283.64 ± 5.13 | 0.33 | 309.09 ± 5.23 |
| EC50 | 9100 | 0.27 | 244.04 ± 10.86 | 0.20 | 192.98 ± 6.18 | |
| 2x EC50 | 18,200 | 0.22 | 206.90 ± 8.05 | 0.14 | 131.92 ± 7.77 | |
| CELL + LPS | 0.44 | 402.44 ± 8.29 | 0.43 | 412.54 ± 4.49 | ||
| CELL | 0.16 | 144.28 ± 5.99 | 0.12 | 111.68 ± 7.90 | ||
| Physicochemical Properties | Computed Range | Acceptable Range | |
|---|---|---|---|
| D5 | D4 | ||
| SASA | 565.2 | 611.314 | 300–1000 |
| FOSA | 196.15 | 179.518 | 0–50 |
| FISA | 52.860 | 101.941 | 7–330 |
| Volume | 993.086 | 1044.573 | 500–2000 |
| donorHB | 1 | 2 | 0–6 |
| accptHB | 5.5 | 5.7 | 2–20 |
| CNS | 0 | −1 | −2–2 |
| QPlogPC16 | 10.272 | 11.365 | 4–18 |
| QPlogPoct‡ | 14.632 | 17.079 | 8–35 |
| QPlogPw | 9.148 | 10.906 | 4–45 |
| QPlogPo/w | 3.232 | 3 | −2–6.5 |
| QPlogHERG | −5.44 | −6.108 | <−5 |
| QPlogBB | −0.284 | −0.943 | −3–1.2 |
| QPPMDCK | 1694.260 | 531.993 | <25 poor, >500 great |
| QPlogKp | −0.804 | −1.468 | −8–−1 |
| QPlogKhsa | 0.012 | −0.012 | −1.5–1.5 |
| HOA | 3 | 3 | 1–3 |
| %HOA | 100 | 100 | <25% poor, >80% high |
| ROF | 0 | 0 | Max 5 |
| ROT | 0 | 0 | Max 3 |
| QPlogS | −3.642 | −4.03 | >−5.7 * |
| QPPCaco | 3210.48 | 1069.535 | >22 nm/s * |
| # Primary Metabolites | 0 | 1 | <7 * |
| WOE | Non-carcinogen | Non-carcinogen | |
| Rodent Carcinogenicity | Non-carcinogen | Non-carcinogen | |
| Rodent Mutagenicity | Non-mutagen | Non-mutagen | |
| LD50 | 2.152542 | 4.905009 | g/kg_body_weight |
| LC50 (inhalation) | 8.922757 | 8.173971 | mg/m3/h |
| TD50 | 0.167370 | 0.196023 | g/kg_body_weight |
| EC50 | 0.241032 | 0.560252 | mg/L |
| Name | D4 | D5 | Pvalue | Tcrit | Tstat | df | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SVG | CHME3 | SVG | CHME3 | SVG | CHME3 | SVG | CHME3 | SVG | CHME3 | SVG | CHME3 | |
| * No secretion (normalized by LPS group) | 69.9 ± 4.7 | 87.4 ± 1.0 | 55.6 ± 5.0 | 54.3 ± 4.4 | 0.05 | 0.01 | 2.8 | 4.3 | 3.6 | 12.7 | 4 | 2 |
| % iNOS secretion (normalized by LPS group) | 59.2 ± 3.9 | 79.2 ± 4.8 | 47.2 ± 2.6 | 36.3 ± 3.2 | 0.05 | 0.01 | 3.2 | 2.8 | 4.5 | 12.9 | 3 | 4 |
| % Phospho-IkB-α (normalized by LPS group) | 22.0 ± 1.8 | 32.1 ± 4.7 | 38.8 ± 2.8 | 25.8 ± 3.8 | 0.01 | 0.05 | 3.2 | 2.8 | −8.8 | 3.4 | 3 | 4 |
| % TNF-α secretion (normalized by LPS group) | 127.7 ± 5.4 | 100 ± 9.2 | 101.7 ± 7.7 | 101.6 ± 6.0 | 0.01 | -- | 2.8 | -- | 4.8 | -- | 4 | -- |
| # INF-γ secretion (normalized by LPS group) | 73.2 ± 0.5 | -- | 46.7 ± 1.3 | -- | 0.01 | -- | 3.2 | -- | 32.4 | -- | 3 | -- |
| @ IKK-β gene expression (normalized by LPS group) | 0.062 ± 0.010 | 0.105 ± 0.024 | 0.036 ± 0.003 | 0.098 ± 0.028 | 0.05 | -- | 4.3 | -- | 4.5 | -- | 2 | -- |
| @ TNF-α gene expression (normalized by LPS group) | 0.05 ± 0.008 | 0.054 ± 0.012 | 0.025 ± 0.001 | 0.096 ± 0.027 | 0.05 | -- | 4.3 | -- | 5.7 | -- | 2 | -- |
| @ PPAR-γ gene expression (normalized by LPS group) | 37.365 ± 1.347 | 25.909 ± 1.792 | 43.133 ± 2.836 | 55.624 ± 2.88 | -- | 0.01 | -- | 3.2 | -- | 15.1 | -- | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahbazi, S.; Zakerali, T. Methylenedioxy Piperamide-Derived Compound D5 Regulates Inflammatory Cytokine Secretion in a Culture of Human Glial Cells. Molecules 2022, 27, 3527. https://doi.org/10.3390/molecules27113527
Shahbazi S, Zakerali T. Methylenedioxy Piperamide-Derived Compound D5 Regulates Inflammatory Cytokine Secretion in a Culture of Human Glial Cells. Molecules. 2022; 27(11):3527. https://doi.org/10.3390/molecules27113527
Chicago/Turabian StyleShahbazi, Sajad, and Tara Zakerali. 2022. "Methylenedioxy Piperamide-Derived Compound D5 Regulates Inflammatory Cytokine Secretion in a Culture of Human Glial Cells" Molecules 27, no. 11: 3527. https://doi.org/10.3390/molecules27113527