
Assay Development and Identification of the First Plasmodium falciparum 7,8-dihydro-6-hydroxymethylpterin-pyrophosphokinase Inhibitors

 , ,
, ,
Abstract
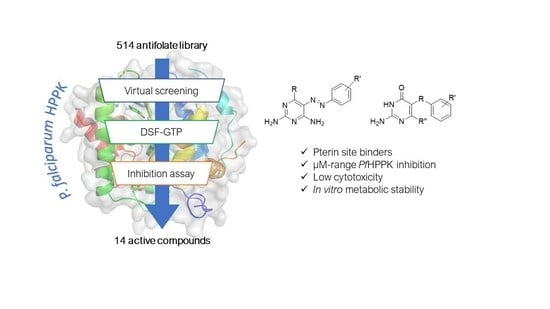
:
1. Introduction
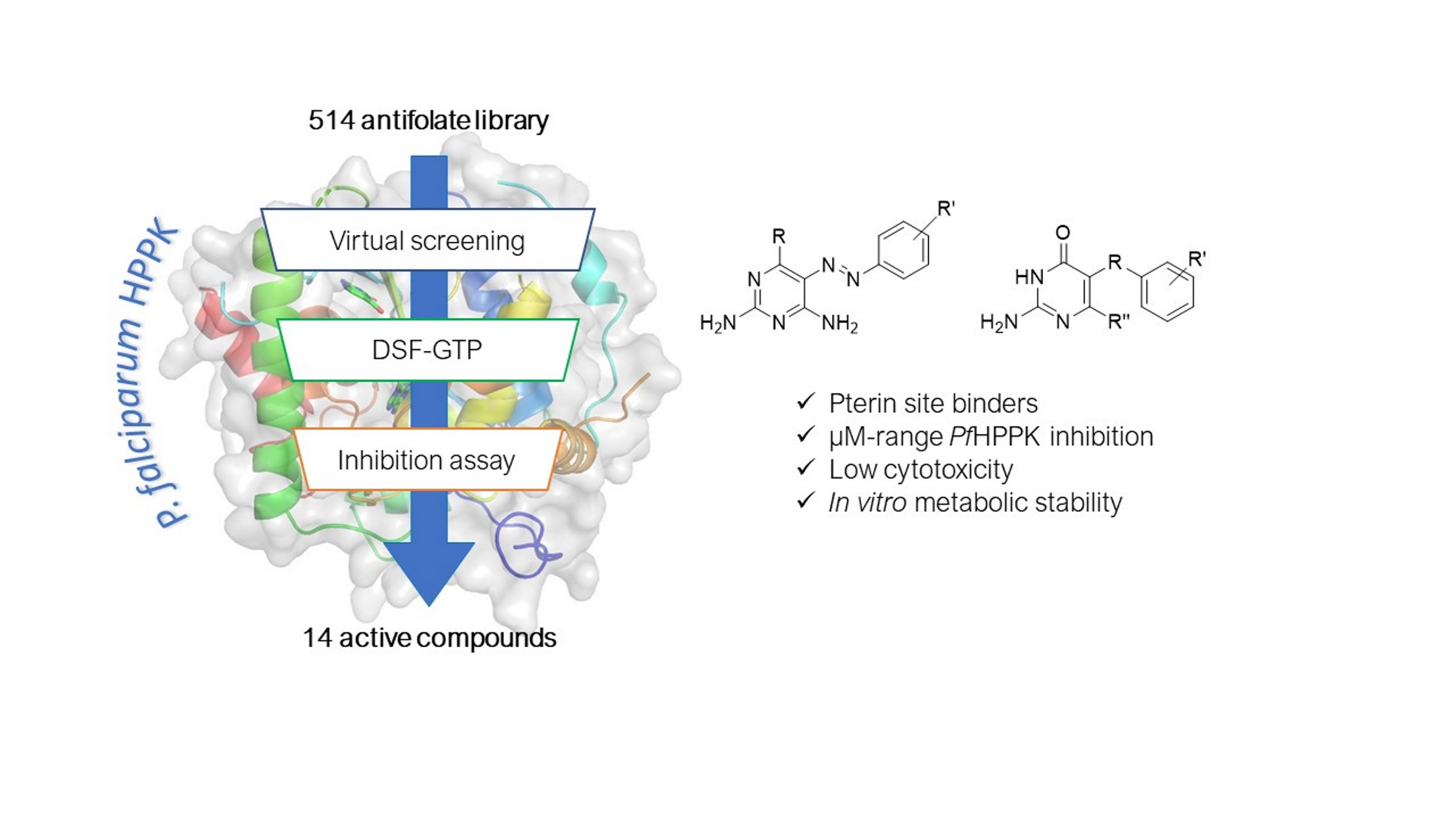
2. Results
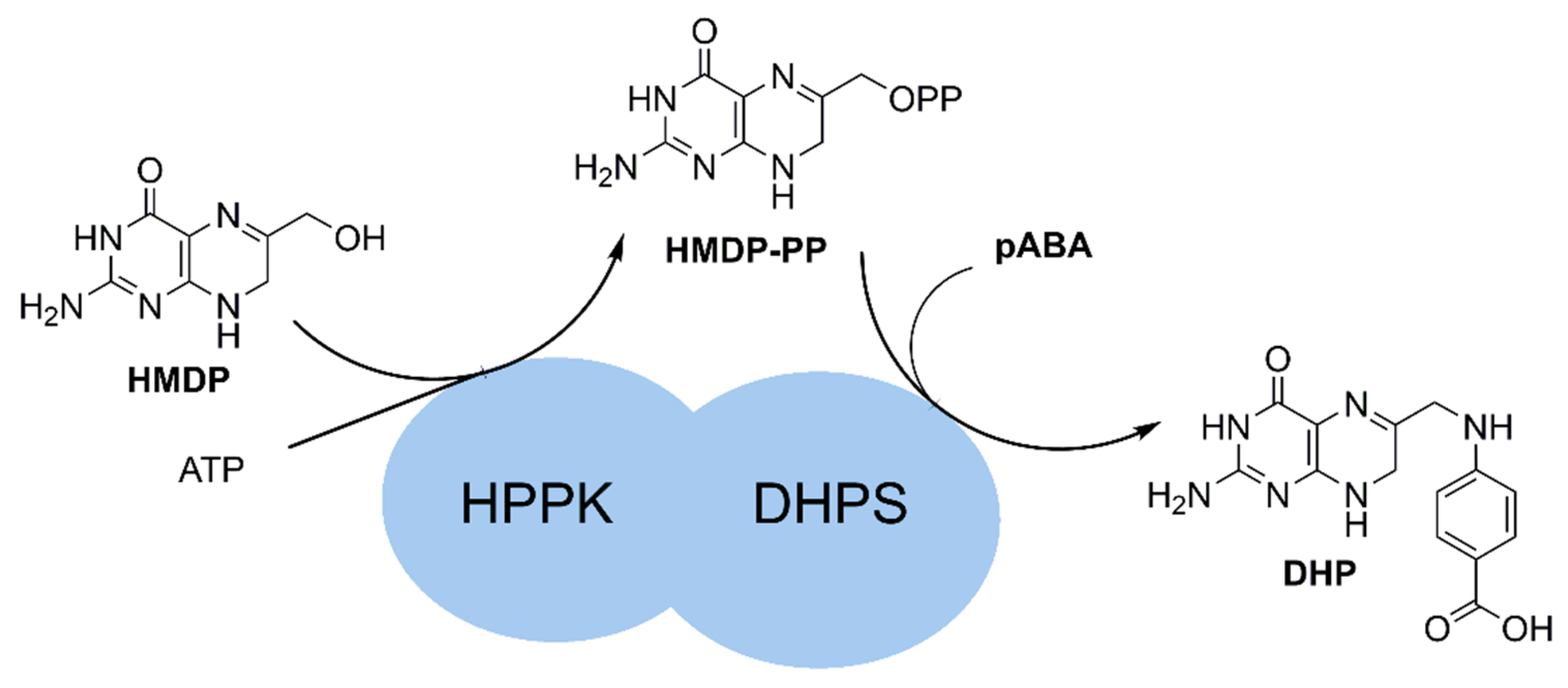
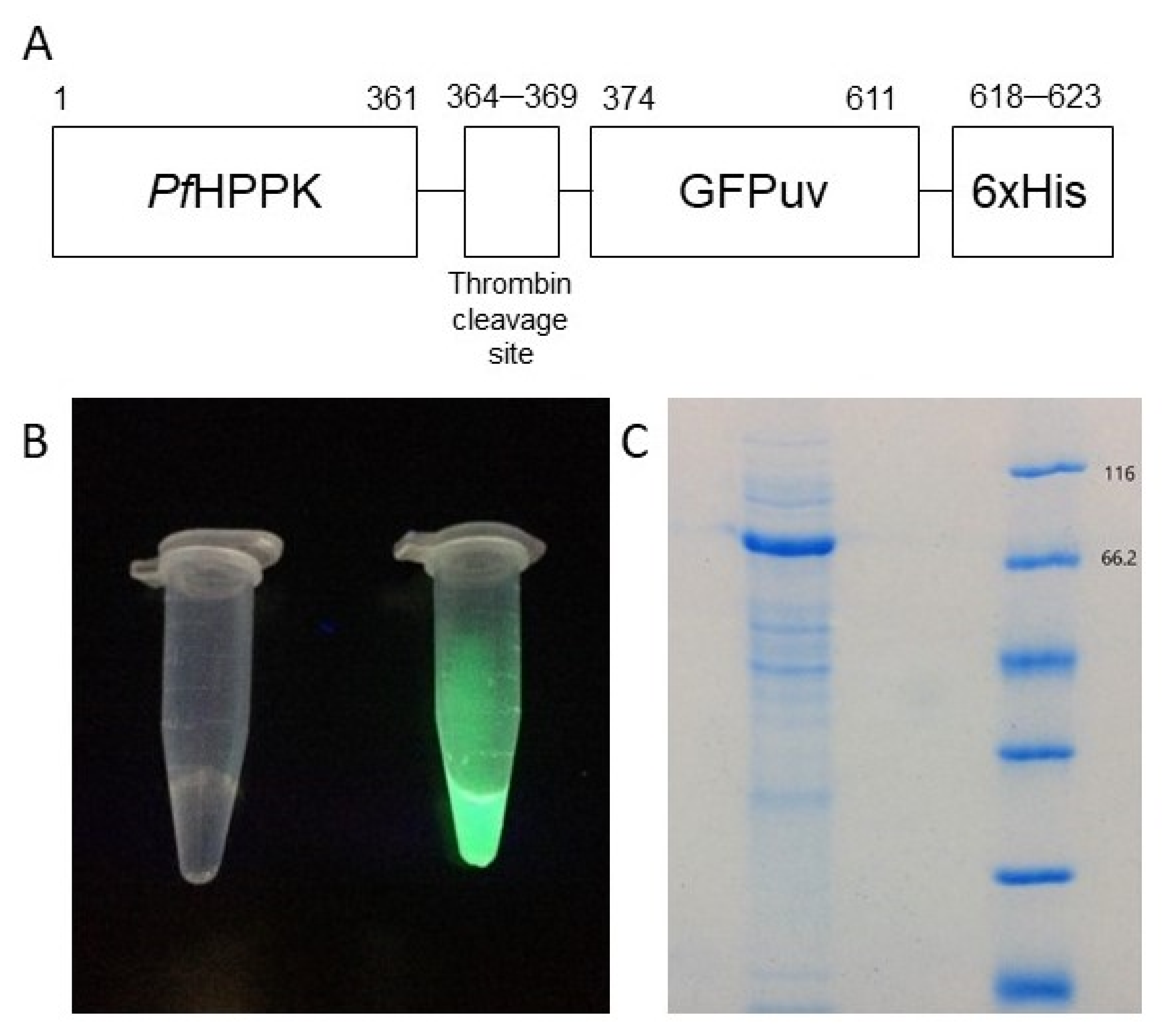
2.1. Design of the Protein Construct
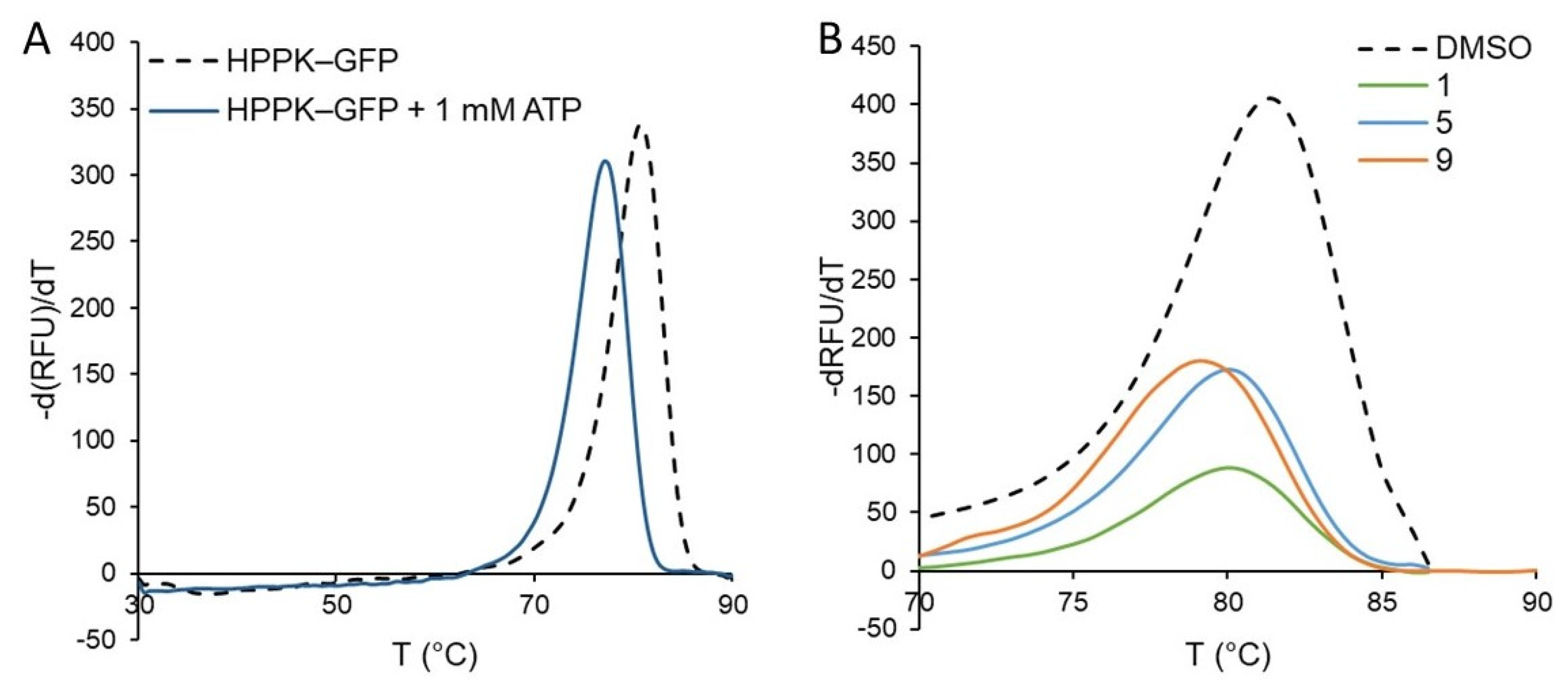
2.2. Assay Development
2.3. Screening of the Antifolate Library
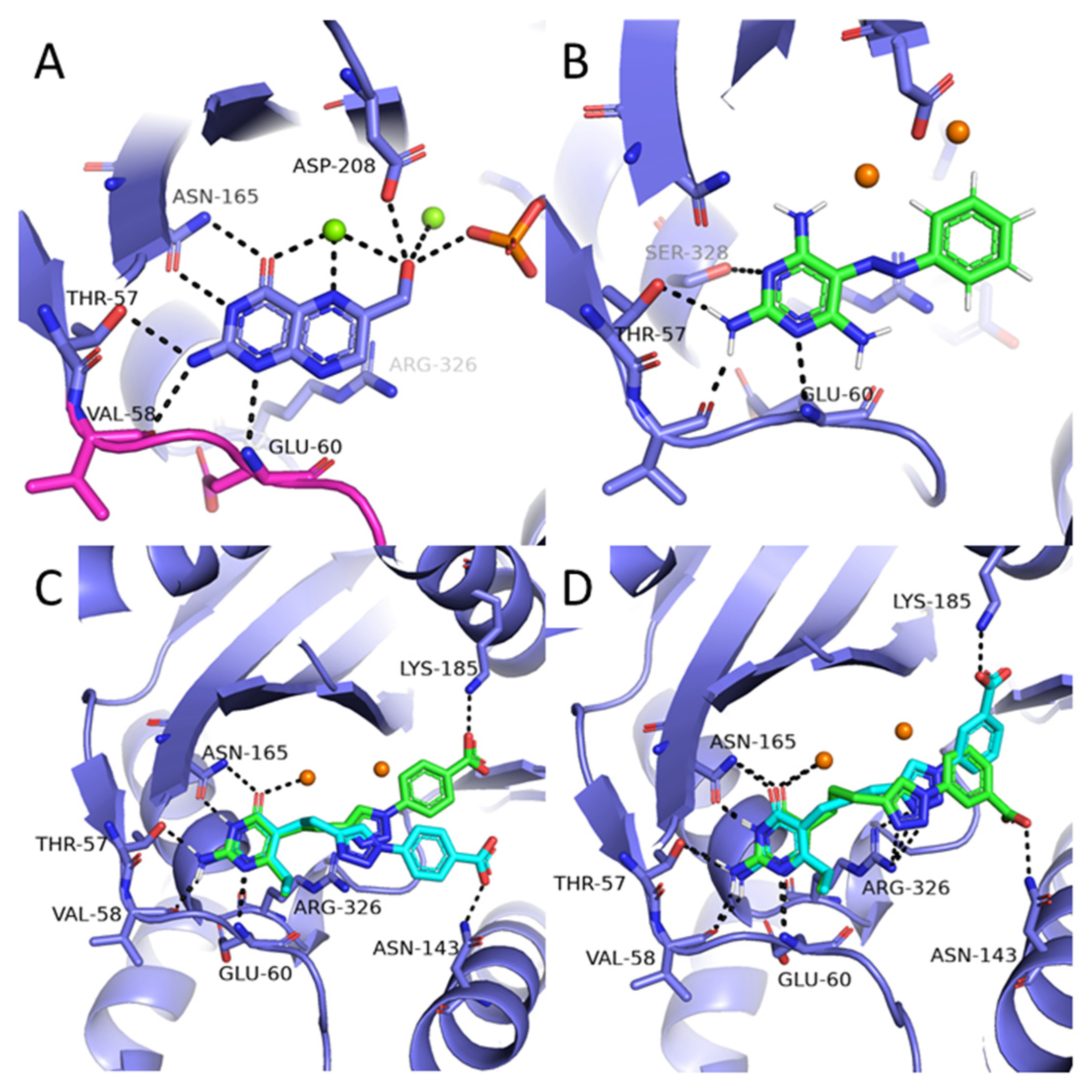
2.4. Molecular Docking
2.5. In Vitro ADME, Cell Toxicity and Antiparasitic Activity
3. Discussion
4. Experimental Methods
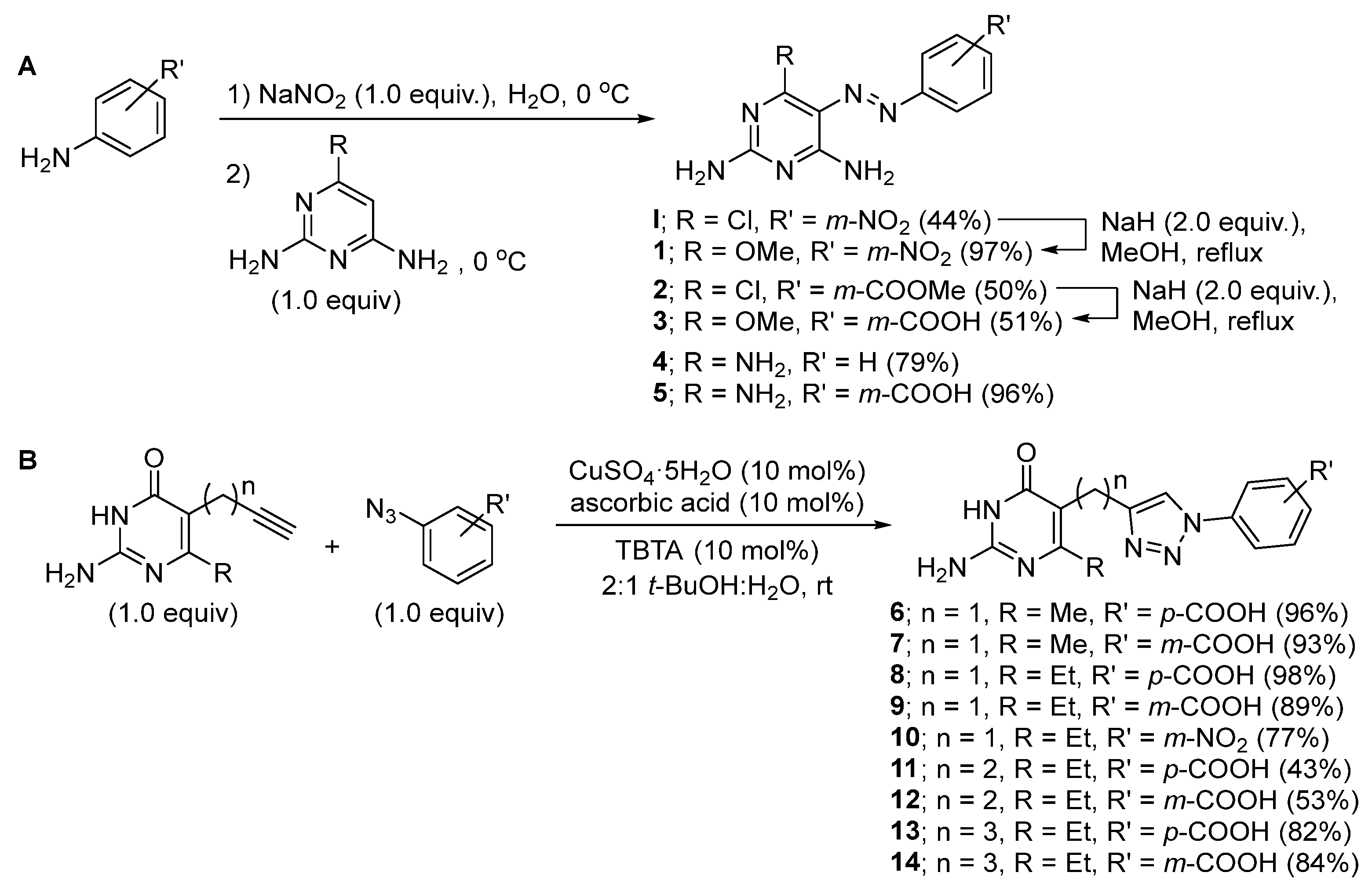
4.1. Synthesis
4.2. Cloning
4.3. Protein Expression and Purification
4.4. DSF-GTP
4.5. Enzymatic Assay
4.6. Molecular Docking
4.7. ADME Properties
4.8. Cytotoxicity Testing
4.9. Parasite Testing
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Nzila, A. Inhibitors of de Novo Folate Enzymes in Plasmodium Falciparum. Drug Discov. Today 2006, 11, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Nzila, A.; Ward, S.A.; Marsh, K.; Sims, P.F.G.; Hyde, J.E. Comparative Folate Metabolism in Humans and Malaria Parasites (Part II): Activities as yet Untargeted or Specific to Plasmodium. Trends Parasitol. 2005, 21, 334–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, I.B.; Hyde, J.E. Folate Metabolism in Human Malaria Parasites—75 Years On. Mol. Biochem. Parasitol. 2013, 188, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Müller, I.B.; Hyde, J.E.; Wrenger, C. Vitamin B Metabolism in Plasmodium Falciparum as a Source of Drug Targets. Trends Parasitol. 2010, 26, 35–43. [Google Scholar] [CrossRef]
- Swarbrick, J.; Iliades, P.; Simpson, J.; Macreadie, I. Folate Biosynthesis- Reappraisal of Old and Novel Targets in the Search for New Antimicrobials. Open Enzym. Inhib. J. 2008, 1, 12–33. [Google Scholar] [CrossRef] [Green Version]
- Kirkland, L.O.; McInnes, C. Non-ATP Competitive Protein Kinase Inhibitors as Anti-Tumor Therapeutics. Biochem. Pharmacol. 2009, 77, 1561–1571. [Google Scholar] [CrossRef]
- Hallyburton, I.; Grimaldi, R.; Woodland, A.; Baragaña, B.; Luksch, T.; Spinks, D.; James, D.; Leroy, D.; Waterson, D.; Fairlamb, A.H.; et al. Screening a Protein Kinase Inhibitor Library against Plasmodium Falciparum. Malar. J. 2017, 16, 446. [Google Scholar] [CrossRef] [Green Version]
- Lucet, I.S.; Tobin, A.; Drewry, D.; Wilks, A.F.; Doerig, C. Plasmodium Kinases as Targets for New-Generation Antimalarials. Future Med. Chem. 2012, 4, 2295–2310. [Google Scholar] [CrossRef]
- Paquet, T.; Manach, C.L.; Cabrera, D.G.; Younis, Y.; Henrich, P.P.; Abraham, T.S.; Lee, M.C.S.; Basak, R.; Ghidelli-Disse, S.; Lafuente-Monasterio, M.J.; et al. Antimalarial Efficacy of MMV390048, an Inhibitor of Plasmodium Phosphatidylinositol 4-Kinase. Sci. Transl. Med. 2017, 9, eaad9735. [Google Scholar] [CrossRef] [Green Version]
- Jönsson, M.; Swedberg, G. Hydroxymethyldihydropterin Pyrophosphokinase from Plasmodium Falciparum Complements a FolK-Knockout Mutant in E. Coli When Expressed as a Separate Polypeptide Detached from Dihydropteroate Synthase. Mol. Biochem. Parasitol. 2005, 140, 123–125. [Google Scholar] [CrossRef]
- Rattanachuen, W.; Jönsson, M.; Swedberg, G.; Sirawaraporn, W. Probing the Roles of Non-Homologous Insertions in the N-Terminal Domain of Plasmodium Falciparum Hydroxymethylpterin Pyrophosphokinase–Dihydropteroate Synthase. Mol. Biochem. Parasitol. 2009, 168, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Chitnumsub, P.; Jaruwat, A.; Talawanich, Y.; Noytanom, K.; Liwnaree, B.; Poen, S.; Yuthavong, Y. The Structure of Plasmodium Falciparum Hydroxymethyldihydropterin Pyrophosphokinase-Dihydropteroate Synthase Reveals the Basis of Sulfa Resistance. FEBS J. 2020, 287, 3273–3297. [Google Scholar] [CrossRef] [PubMed]
- Moreau, M.J.J.; Morin, I.; Askin, S.P.; Cooper, A.; Moreland, N.J.; Vasudevan, S.G.; Schaeffer, P.M. Rapid Determination of Protein Stability and Ligand Binding by Differential Scanning Fluorimetry of GFP-Tagged Proteins. RSC Adv. 2012, 2, 11892–11900. [Google Scholar] [CrossRef]
- Schaeffer, P. Protein Stability Assay Using a Fluorescent Reporter of Protein Folding. U.S. Patent Application No. 12/958,581, 2 June 2011. [Google Scholar]
- Sorenson, A.E.; Schaeffer, P.M. High-Throughput Differential Scanning Fluorimetry of GFP-Tagged Proteins. In Targeting Enzymes for Pharmaceutical Development: Methods and Protocols; Methods in Molecular Biology; Labrou, N.E., Ed.; Springer: New York, NY, USA, 2020; pp. 69–85. ISBN 978-1-07-160163-1. [Google Scholar]
- Bond, T.E.H.; Sorenson, A.E.; Schaeffer, P.M. A Green Fluorescent Protein-Based Assay for High-Throughput Ligand-Binding Studies of a Mycobacterial Biotin Protein Ligase. Microbiol. Res. 2017, 205, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Japrung, D.; Chusacultanachai, S.; Yuvaniyama, J.; Wilairat, P.; Yuthavong, Y. A Simple Dual Selection for Functionally Active Mutants of Plasmodium Falciparum Dihydrofolate Reductase with Improved Solubility. Protein Eng. Des. Sel. 2005, 18, 457–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamchonwongpaisan, S.; Charoensetakul, N.; Srisuwannaket, C.; Taweechai, S.; Ratanachak, R.; Vanichtanankul, J.; Vitsupakorn, D.; Arwon, U.; Thongpanchang, C.; Tarnchompoo, B.; et al. Flexible Diaminodihydrotriazine Inhibitors of Plasmodium Falciparum Dihydrofolate Reductase: Binding Strengths, Modes of Binding and Their Antimalarial Activities. Eur. J. Med. Chem. 2020, 195, 112263. [Google Scholar] [CrossRef] [PubMed]
- Tarnchompoo, B.; Chitnumsub, P.; Jaruwat, A.; Shaw, P.J.; Vanichtanankul, J.; Poen, S.; Rattanajak, R.; Wongsombat, C.; Tonsomboon, A.; Decharuangsilp, S.; et al. Hybrid Inhibitors of Malarial Dihydrofolate Reductase with Dual Binding Modes That Can Forestall Resistance. ACS Med. Chem. Lett. 2018, 9, 1235–1240. [Google Scholar] [CrossRef] [Green Version]
- Tarnchompoo, B.; Sirichaiwat, C.; Phupong, W.; Intaraudom, C.; Sirawaraporn, W.; Kamchonwongpaisan, S.; Vanichtanankul, J.; Thebtaranonth, Y.; Yuthavong, Y. Development of 2,4-Diaminopyrimidines as Antimalarials Based on Inhibition of the S108N and C59R+S108N Mutants of Dihydrofolate Reductase from Pyrimethamine-Resistant Plasmodium Falciparum. J. Med. Chem. 2002, 45, 1244–1252. [Google Scholar] [CrossRef]
- Kamchonwongpaisan, S.; Quarrell, R.; Charoensetakul, N.; Ponsinet, R.; Vilaivan, T.; Vanichtanankul, J.; Tarnchompoo, B.; Sirawaraporn, W.; Lowe, G.; Yuthavong, Y. Inhibitors of Multiple Mutants of Plasmodium Falciparum Dihydrofolate Reductase and Their Antimalarial Activities. J. Med. Chem. 2004, 47, 673–680. [Google Scholar] [CrossRef]
- Kasekarn, W.; Sirawaraporn, R.; Chahomchuen, T.; Cowman, A.F.; Sirawaraporn, W. Molecular Characterization of Bifunctional Hydroxymethyldihydropterin Pyrophosphokinase-Dihydropteroate Synthase from Plasmodium Falciparum. Mol. Biochem. Parasitol. 2004, 137, 43–53. [Google Scholar] [CrossRef]
- Acker, M.; Auld, D. Considerations for the Design and Reporting of Enzyme Assays in High-Throughput Screening Applications. Perspect. Sci. 2014, 1, 56–73. [Google Scholar] [CrossRef] [Green Version]
- Yuthavong, Y.; Tarnchompoo, B.; Vilaivan, T.; Chitnumsub, P.; Kamchonwongpaisan, S.; Charman, S.A.; McLennan, D.N.; White, K.L.; Vivas, L.; Bongard, E.; et al. Malarial Dihydrofolate Reductase as a Paradigm for Drug Development against a Resistance-Compromised Target. Proc. Natl. Acad. Sci. USA 2012, 109, 16823–16828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chughlay, M.F.; Rossignol, E.; Donini, C.; Gaaloul, M.E.; Lorch, U.; Coates, S.; Langdon, G.; Hammond, T.; Möhrle, J.; Chalon, S. First-in-Human Clinical Trial to Assess the Safety, Tolerability and Pharmacokinetics of P218, a Novel Candidate for Malaria Chemoprotection. Br. J. Clin. Pharmacol. 2020, 86, 1113–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beale, G.H.; Thaithong, S.; Siripool, N. Isolation of Clones of Plasmodium Falciparum by Micromanipulation. Trans. R. Soc. Beale, G.H.; Thaithong, S.; Siripool, N. Isolation of Clones of Plasmodium Falciparum by Micromanipulation. Trans. R. Soc. Trop. Med. Hyg. 1991, 85, 37. [Google Scholar] [CrossRef]
- Peterson, D.S.; Milhous, W.K.; Wellems, T.E. Molecular Basis of Differential Resistance to Cycloguanil and Pyrimethamine in Plasmodium Falciparum Malaria. Proc. Natl. Acad. Sci. USA 1990, 87, 3018–3022. [Google Scholar] [CrossRef] [Green Version]
- Shi, G.; Shaw, G.X.; Zhu, F.; Tarasov, S.G.; Ji, X. Bisubstrate Inhibitors of 6-Hydroxymethyl-7,8-Dihydropterin Pyrophosphokinase: Transition State Analogs for High Affinity Binding. Bioorg. Med. Chem. 2021, 29, 115847. [Google Scholar] [CrossRef]
- Dennis, M.L.; Lee, M.D.; Harjani, J.R.; Ahmed, M.; DeBono, A.J.; Pitcher, N.P.; Wang, Z.-C.; Chhabra, S.; Barlow, N.; Rahmani, R.; et al. 8-Mercaptoguanine Derivatives as Inhibitors of Dihydropteroate Synthase. Chem. Eur. J. 2018, 24, 1922–1930. [Google Scholar] [CrossRef]
- Hevener, K.E.; Yun, M.-K.; Qi, J.; Kerr, I.D.; Babaoglu, K.; Hurdle, J.G.; Balakrishna, K.; White, S.W.; Lee, R.E. Structural Studies of Pterin-Based Inhibitors of Dihydropteroate Synthase. J. Med. Chem. 2010, 53, 166–177. [Google Scholar] [CrossRef]
- Yun, M.-K.; Wu, Y.; Li, Z.; Zhao, Y.; Waddell, M.B.; Ferreira, A.M.; Lee, R.E.; Bashford, D.; White, S.W. Catalysis and Sulfa Drug Resistance in Dihydropteroate Synthase. Science 2012, 335, 1110–1114. [Google Scholar] [CrossRef] [Green Version]
- Griffith, E.C.; Wallace, M.J.; Wu, Y.; Kumar, G.; Gajewski, S.; Jackson, P.; Phelps, G.A.; Zheng, Z.; Rock, C.O.; Lee, R.E.; et al. The Structural and Functional Basis for Recurring Sulfa Drug Resistance Mutations in Staphylococcus Aureus Dihydropteroate Synthase. Front. Microbiol. 2018, 9, 1369. [Google Scholar] [CrossRef]
- Ferreira, L.L.G.; Andricopulo, A.D. ADMET Modeling Approaches in Drug Discovery. Drug Discov. Today 2019, 24, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Tuntland, T.; Ethell, B.; Kosaka, T.; Blasco, F.; Zang, R.X.; Jain, M.; Gould, T.; Hoffmaster, K. Implementation of Pharmacokinetic and Pharmacodynamic Strategies in Early Research Phases of Drug Discovery and Development at Novartis Institute of Biomedical Research. Front. Pharmacol. 2014, 5, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maestro, Schrödinger LLC: New York, NY, USA, 2020.
- Glide, Schrödinger LLC: New York, NY, USA, 2020.
- The PyMOL Molecular Graphics System, Schrödinger LLC: New York, NY, USA, 2010.
- Abay, E.T.; van der Westuizen, J.H.; Swart, K.J.; Gibhard, L.; Lawrence, N.; Dambuza, N.; Wilhelm, A.; Pravin, K.; Wiesner, L. Efficacy and Pharmacokinetic Evaluation of a Novel Anti-Malarial Compound (NP046) in a Mouse Model. Malar. J. 2015, 14, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marvin, ChemAxon: Budapest, Hungary, 2022.
- ACD/Labs, Advanced Chemistry Development, Inc.: Toronto, ON, Canada, 2022.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Code | R | R’ | ΔTm (°C) 1 | Inhib. at 1 mM (%) 2 |
| 1 | OMe | m-NO2 | −1 ± 0 | 2.5 ± 1.4 |
| 2 | Cl | m-COOMe | −1 ± 0 | 11.0 ± 4.8 |
| 3 | OMe | m-COOH | −1 ± 0 | 23.3 ± 4.8 |
| 4 | NH2 | H | −1 ± 0 | 46.0 ± 7.5 |
| 5 | NH2 | m-COOH | −1 ± 0 | 14.6 ± 3.2 |
 | |||||
|---|---|---|---|---|---|
| Code | n | R | R’ | ΔTm (°C) 1 | Inhib. at 1 mM (%) 2 |
| 6 | 1 | Me | p-COOH | −1.0 ± 0 | 1.8 ± 0.6 |
| 7 | 1 | Me | m-COOH | −1.0 ± 0 | 9.2 ± 1.5 |
| 8 | 1 | Et | p-COOH | −1.2 ± 0.3 | 6.9 ± 2.0 |
| 9 | 1 | Et | m-COOH | −1.0 ± 0 | 16.4 ± 2.9 |
| 10 | 1 | Et | m-NO2 | −1.2 ± 0.3 | 12.1 ± 4.0 |
| 11 | 2 | Et | p-COOH | −1.3 ± 0.3 | 34.9 ± 5.5 |
| 12 | 2 | Et | m-COOH | −1.3 ± 0.3 | 24.5 ± 2.3 |
| 13 | 3 | Et | p-COOH | −1.5 ± 0 | 24.9 ± 3.1 |
| 14 | 3 | Et | m-COOH | −1.2 ± 0.3 | 42.8 ± 5.5 |
| Compound | cLog S7.4 1 | cLog D7.4 1 | cLog P 2 | PSA 1 | CLint (HLM) (µL/min/mg) | CLint (RLM) (µL/min/mg) |
|---|---|---|---|---|---|---|
| P218 | −3.7 | −0.20 | 2.79 | 133.58 | <3 | <3 |
| 11 | −0.24 | −1.23 | 2.11 | 135.49 | <3 | 6.91 |
| 13 | −0.50 | −1.23 | 2.53 | 135.49 | <3 | 7.38 |
| 14 | 0.00 | −1.23 | 2.53 | 135.49 | 4.88 | 7.05 |
| 3 | −1.50 | −0.59 | 2.67 | 149.07 | <3 | <3 |
| 4 | −2.95 | 1.83 | 0.83 | 128.56 | 362.63 | 82.39 |
| IC50 (μM) | ||||
|---|---|---|---|---|
| Compound | TM4/8.2 | V1/S | VERO | KB |
| 1 | >100 | 21.0 | >100 | >100 |
| 2 | >100 | 89.2 | >100 | >100 |
| 3 | >50 | >50 | >100 | >100 |
| 4 | 75.4 | >50 | 29.6 | 62.4 |
| 5 | >10 | >10 | n.d. | n.d. |
| 6 | >25 | >25 | >25 | >25 |
| 7 | >25 | >25 | >25 | >25 |
| 8 | >50 | >50 | >50 | >50 |
| 9 | >50 | >50 | >50 | >50 |
| 10 | >50 | >50 | >50 | >50 |
| 11 | >50 | >50 | >50 | >50 |
| 12 | >50 | >50 | >50 | >50 |
| 13 | >50 | >50 | >50 | >50 |
| 14 | >50 | >50 | >50 | >50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoarau, M.; Suwanakitti, N.; Varatthan, T.; Thiabma, R.; Rattanajak, R.; Charoensetakul, N.; Redman, E.K.; Khotavivattana, T.; Vilaivan, T.; Yuthavong, Y.; et al. Assay Development and Identification of the First Plasmodium falciparum 7,8-dihydro-6-hydroxymethylpterin-pyrophosphokinase Inhibitors. Molecules 2022, 27, 3515. https://doi.org/10.3390/molecules27113515
Hoarau M, Suwanakitti N, Varatthan T, Thiabma R, Rattanajak R, Charoensetakul N, Redman EK, Khotavivattana T, Vilaivan T, Yuthavong Y, et al. Assay Development and Identification of the First Plasmodium falciparum 7,8-dihydro-6-hydroxymethylpterin-pyrophosphokinase Inhibitors. Molecules. 2022; 27(11):3515. https://doi.org/10.3390/molecules27113515
Chicago/Turabian StyleHoarau, Marie, Nattida Suwanakitti, Thaveechai Varatthan, Ratthiya Thiabma, Roonglawan Rattanajak, Netnapa Charoensetakul, Emily K. Redman, Tanatorn Khotavivattana, Tirayut Vilaivan, Yongyuth Yuthavong, and et al. 2022. "Assay Development and Identification of the First Plasmodium falciparum 7,8-dihydro-6-hydroxymethylpterin-pyrophosphokinase Inhibitors" Molecules 27, no. 11: 3515. https://doi.org/10.3390/molecules27113515