
Recent Developments in the Photochemical Synthesis of Functionalized Imidazopyridines


{kind=link}
{kind=link}
{kind=link}
{kind=link}
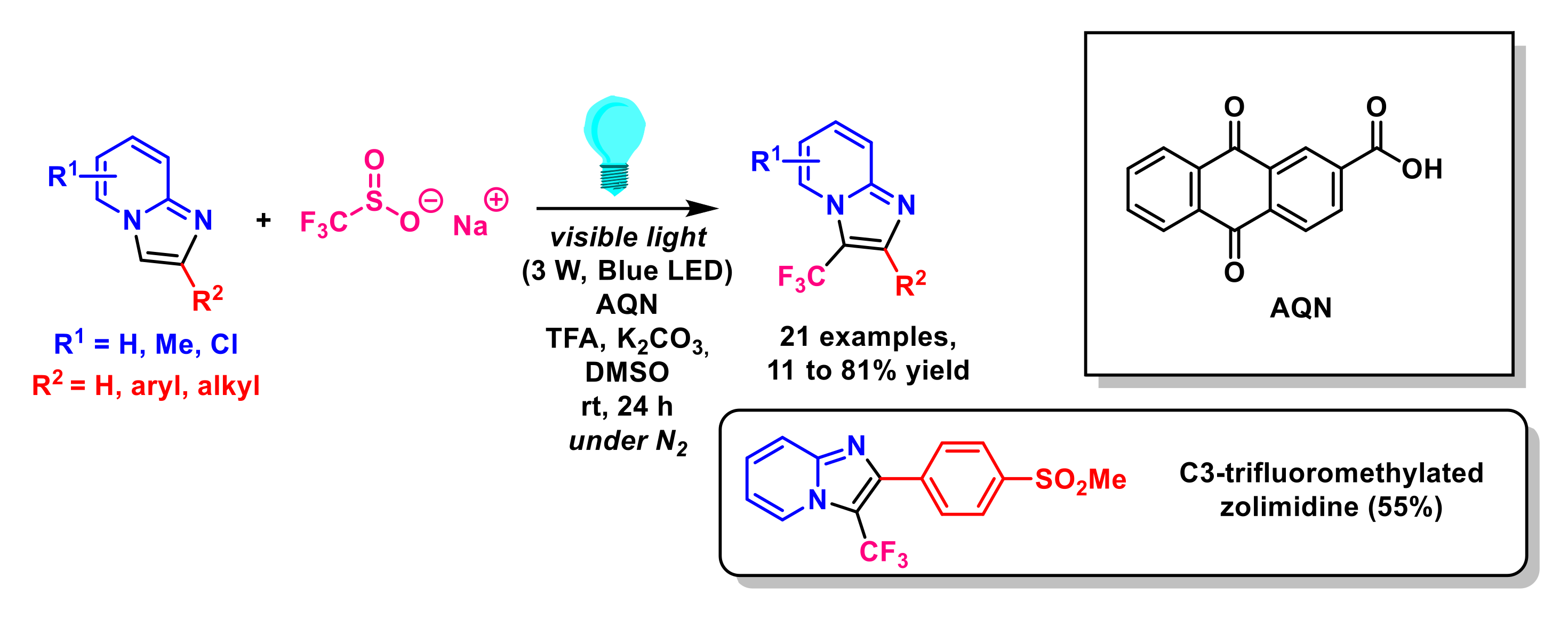{kind=link}
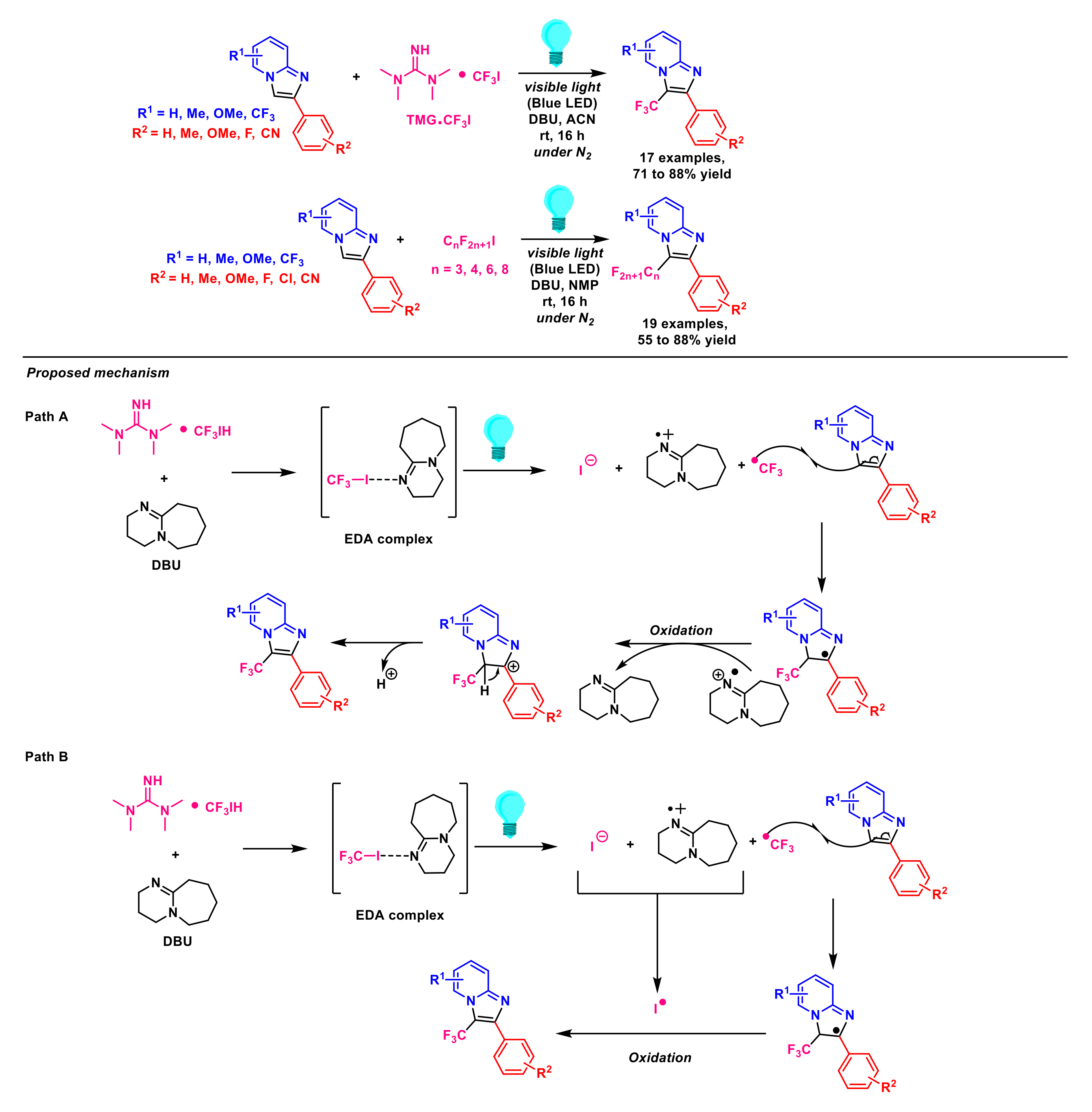{kind=link}
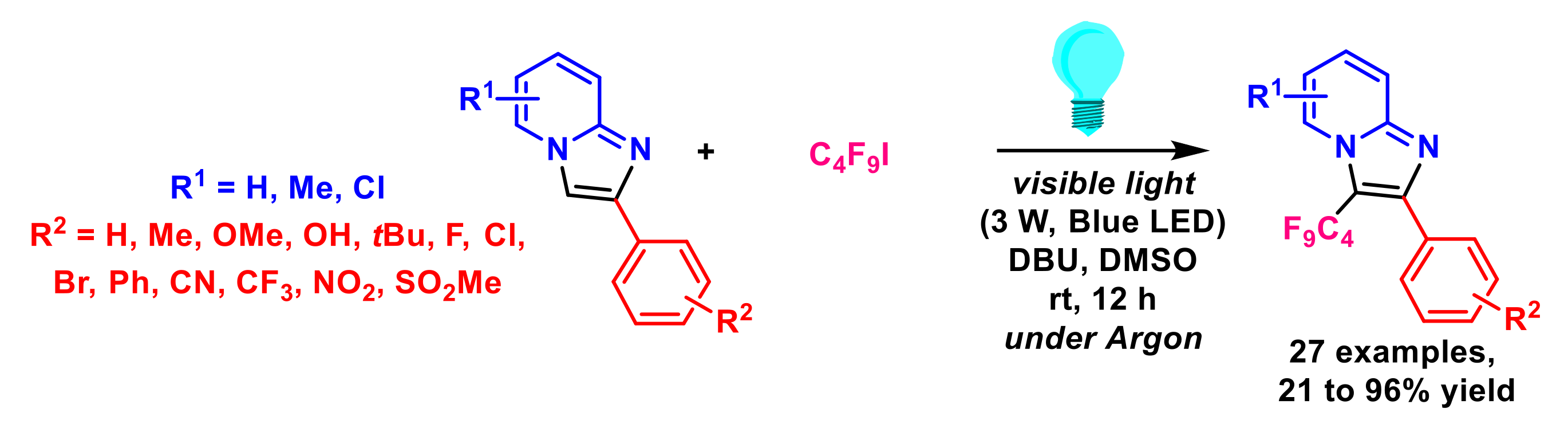{kind=link}
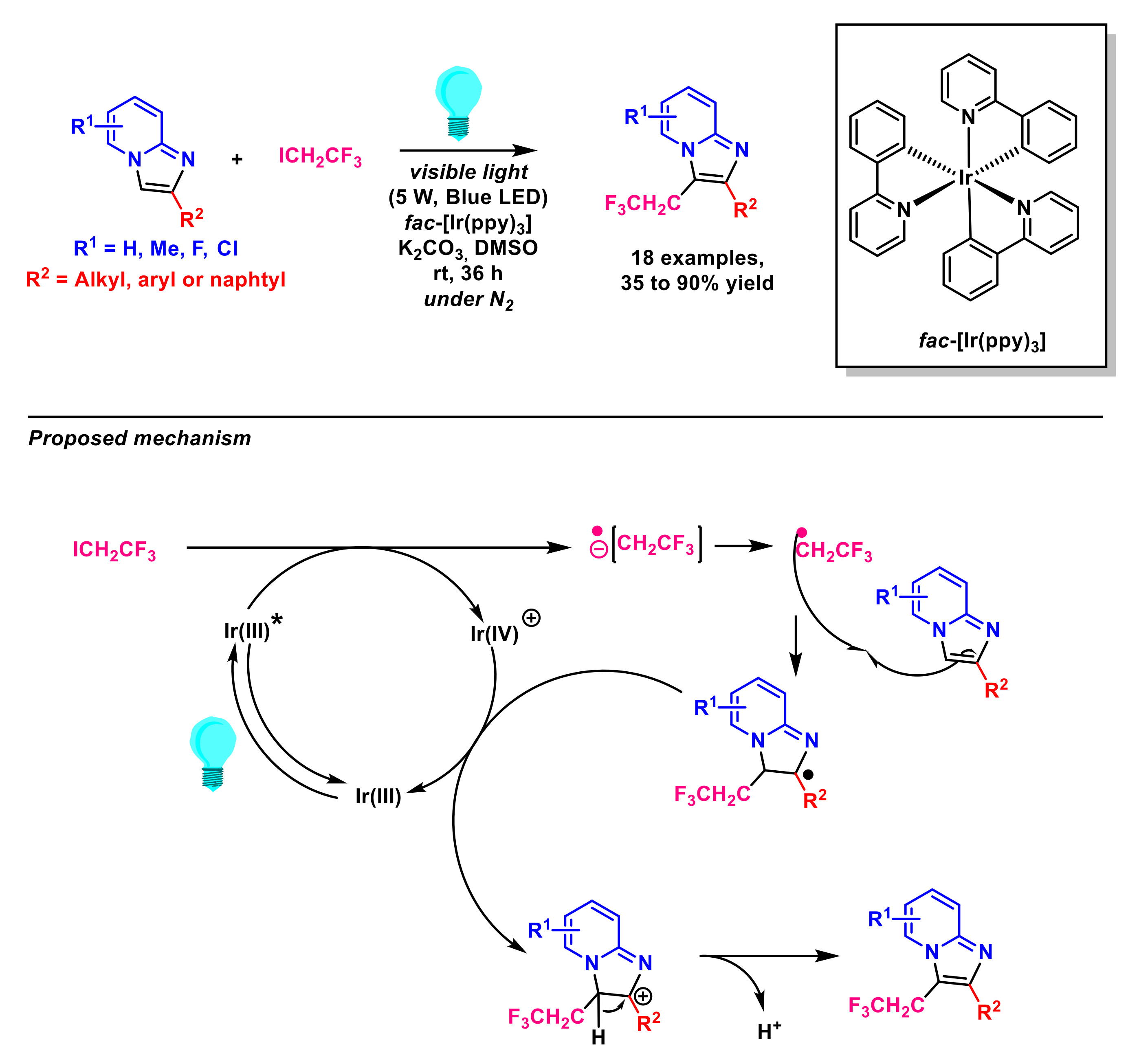{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. C–H Functionalization
2.1. Formation of C–C Bonds
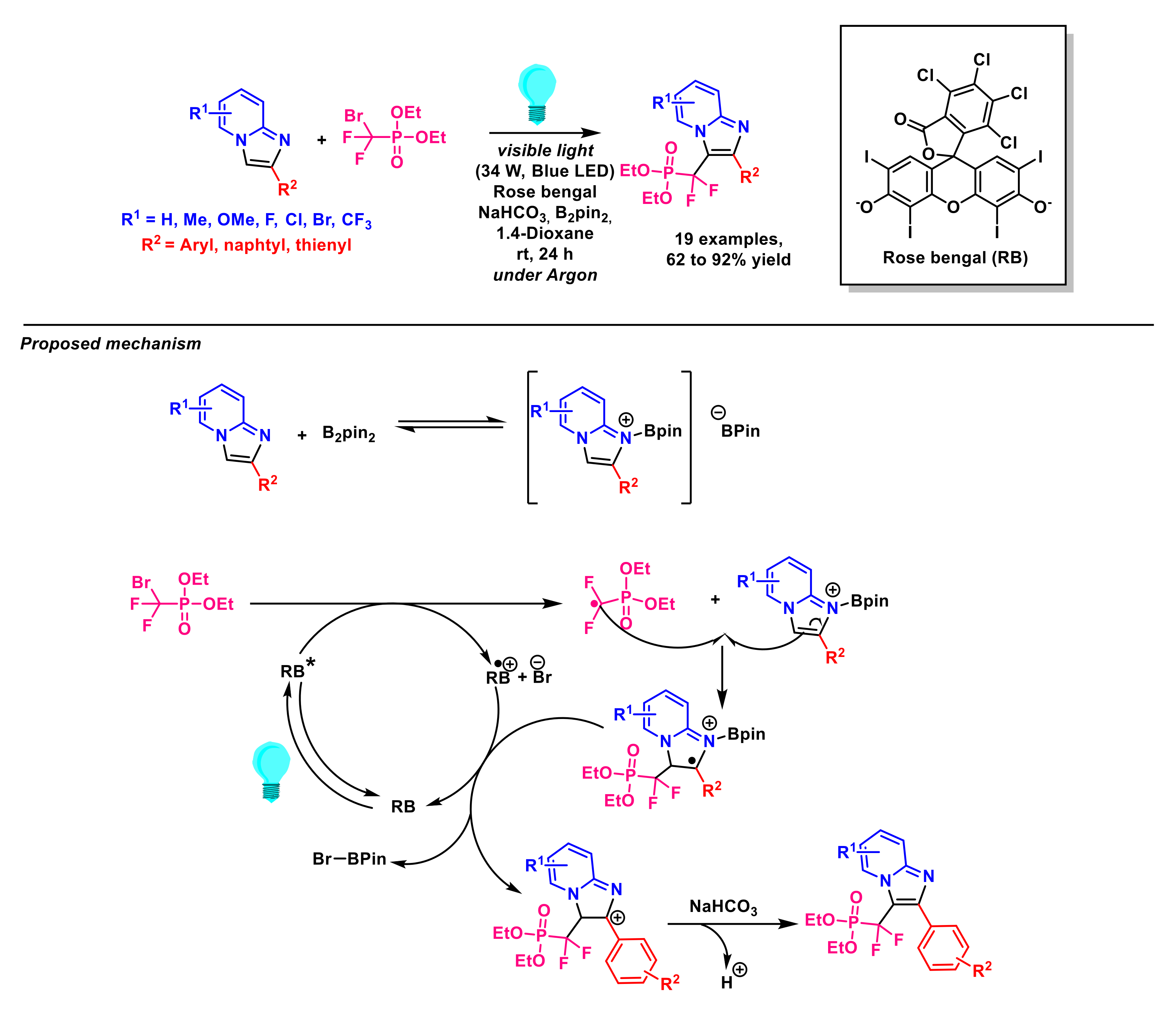
2.1.1. Fluoroalkylation of Imidazopyridines
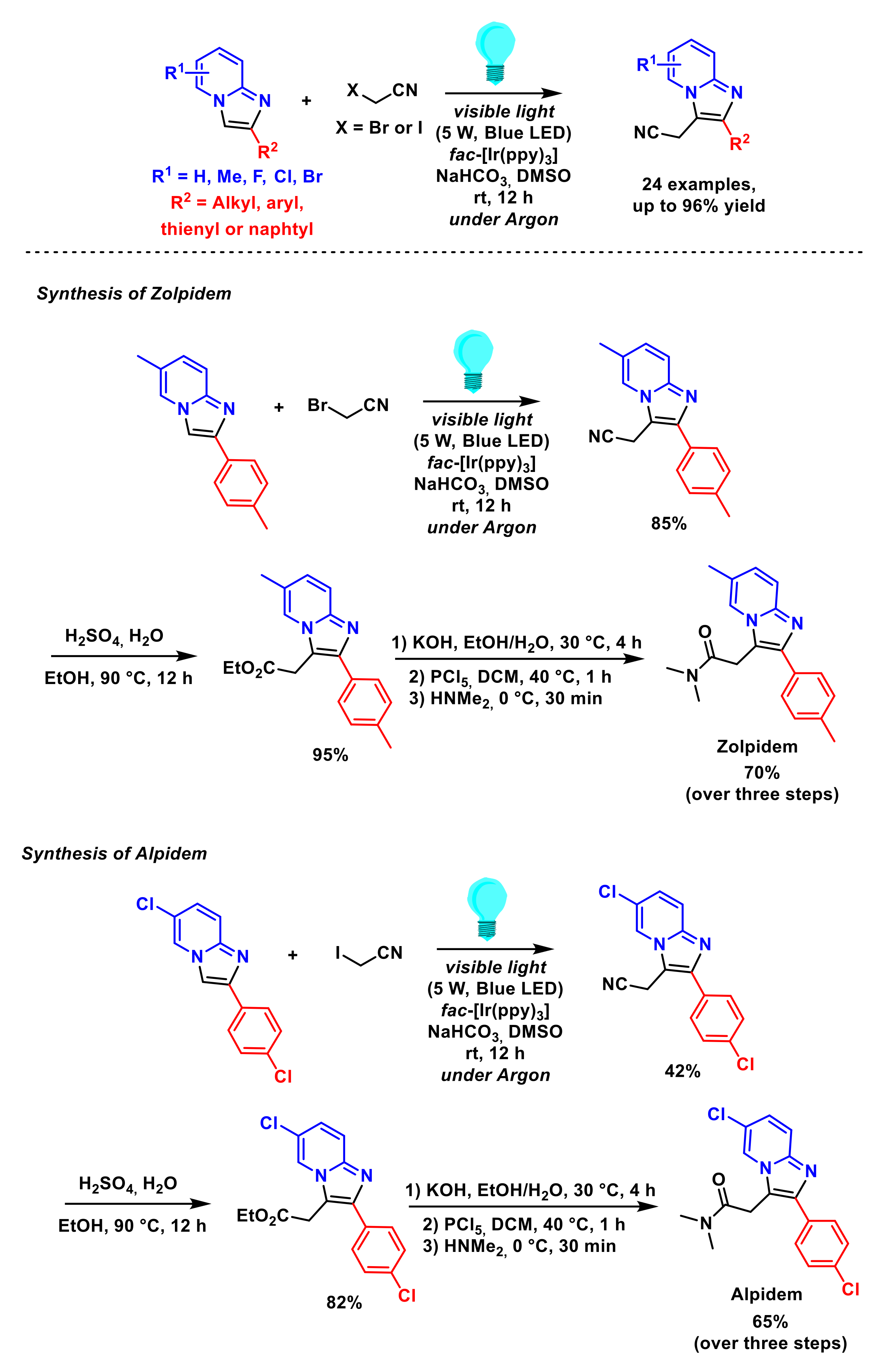
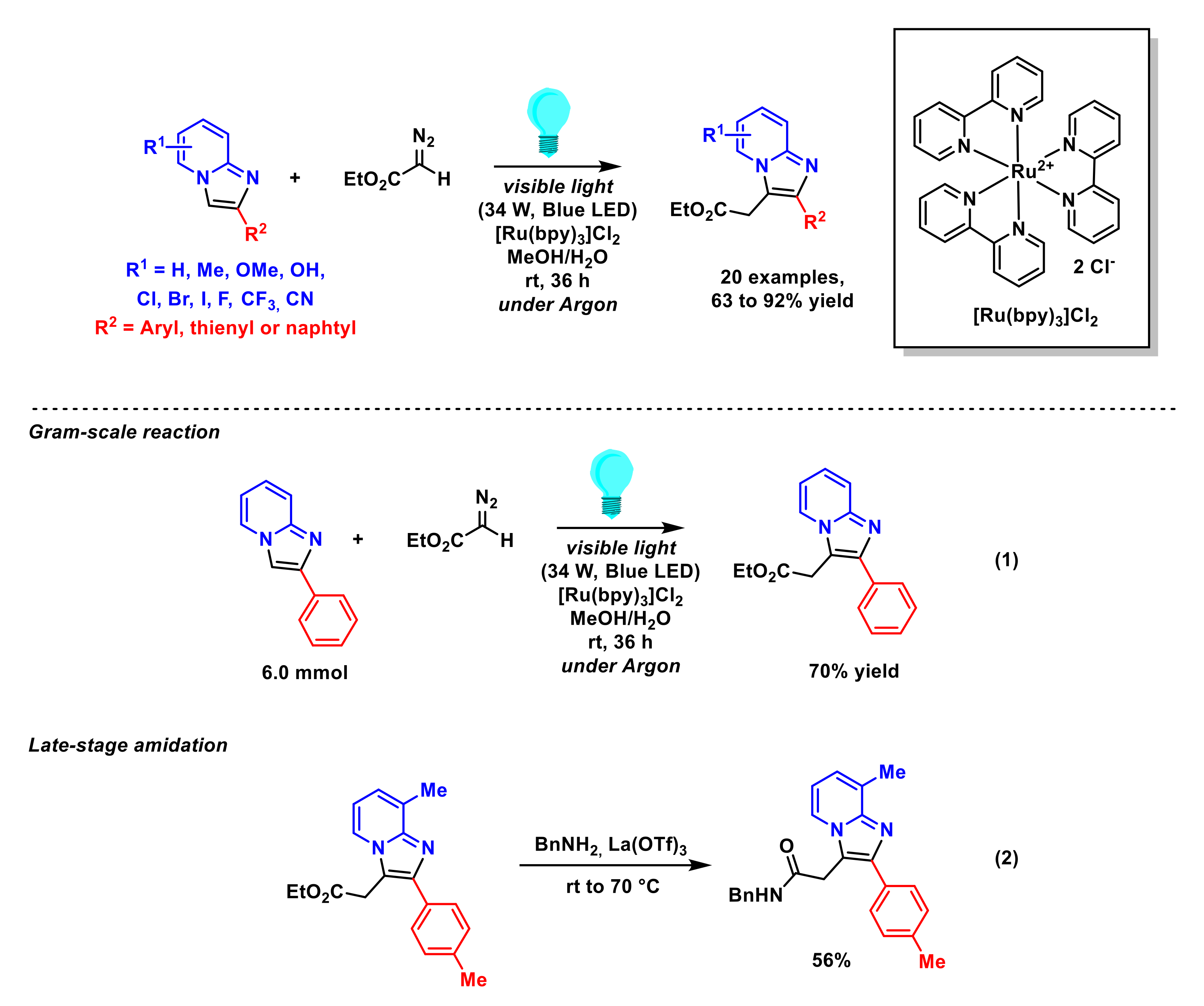
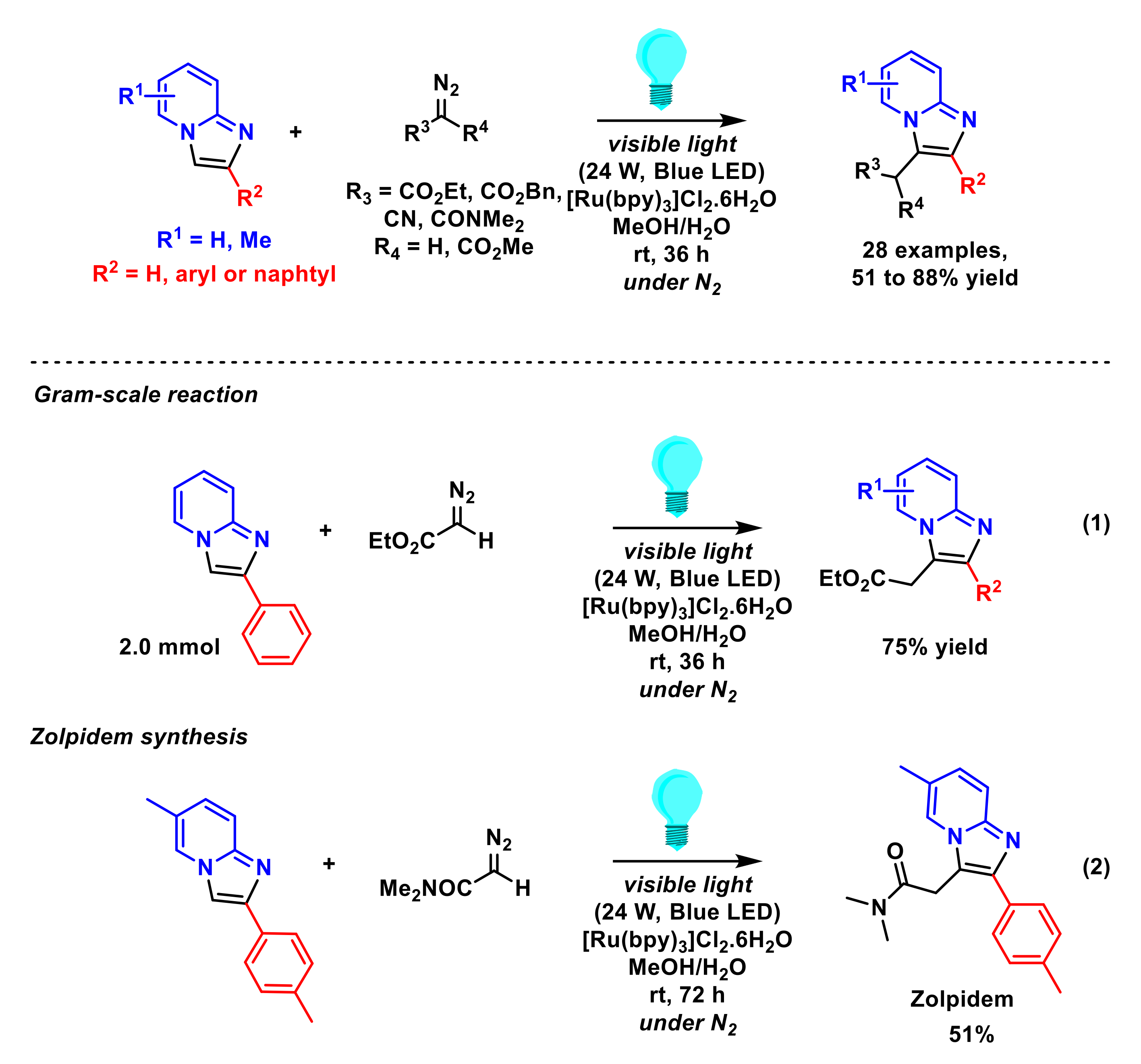
2.1.2. Alkylation of Imidazopyridines
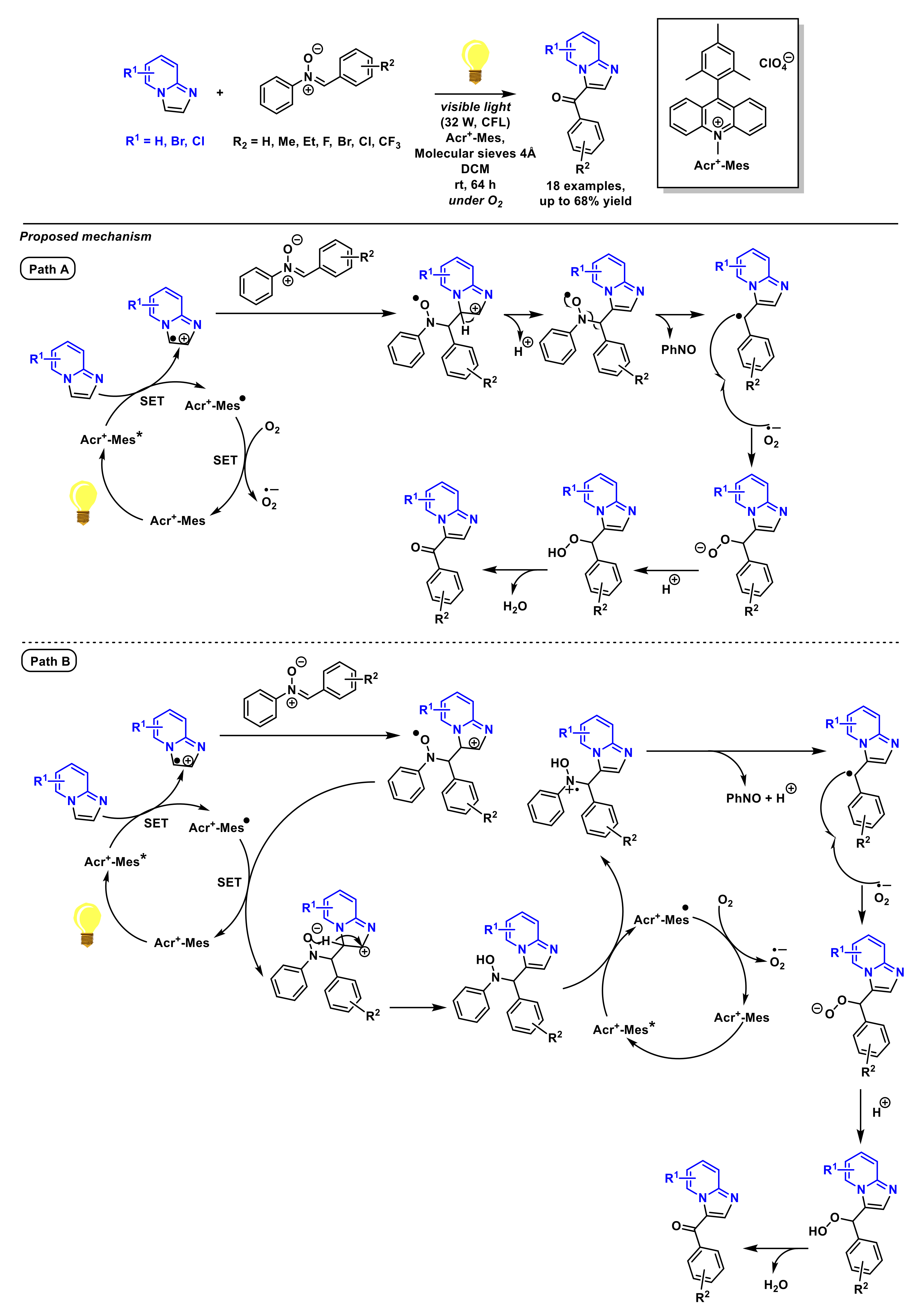
2.1.3. Carbonylalkylation and Carbonylation of Imidazopyridines
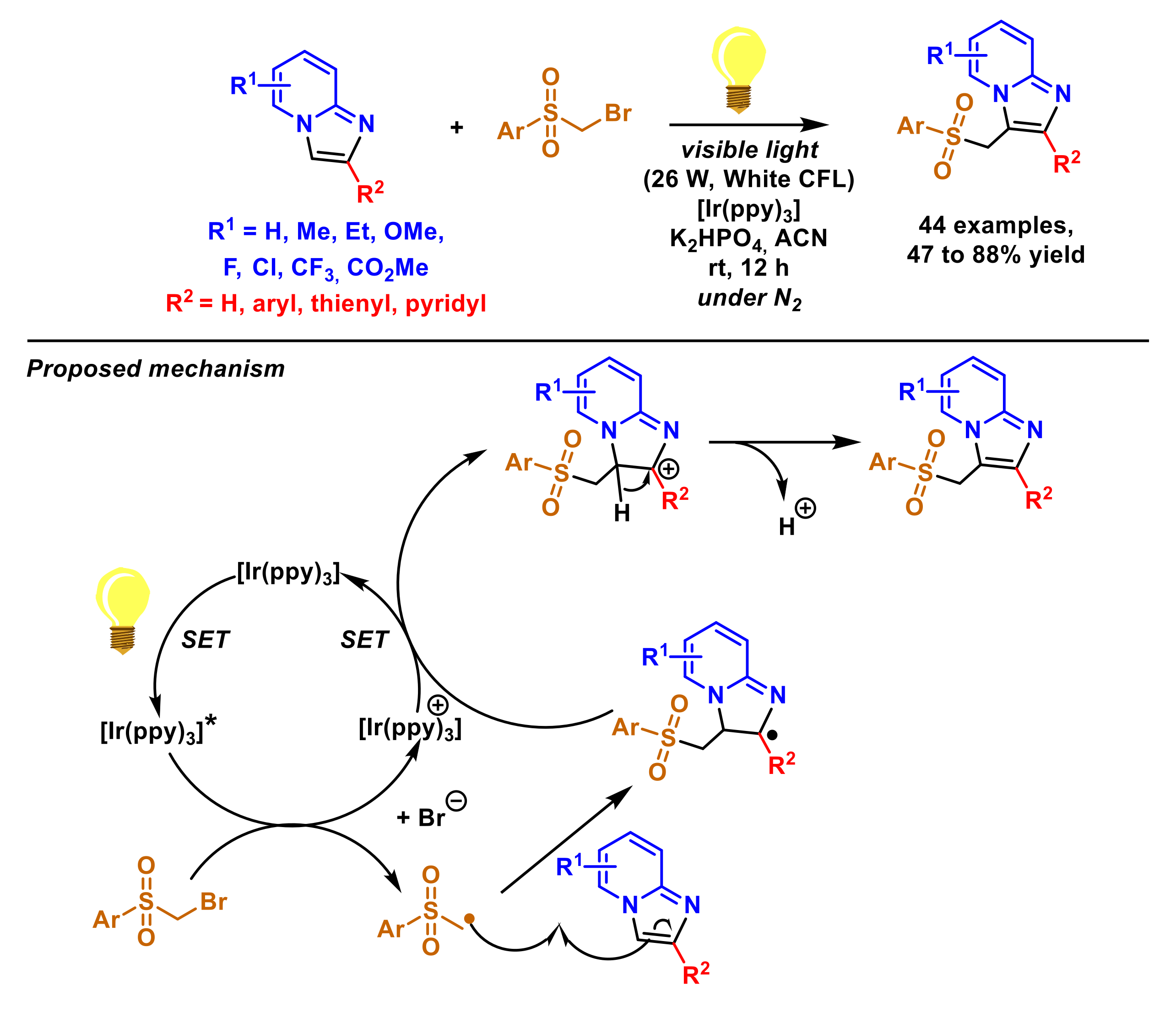
2.1.4. Sulfonylmethylation of Imidazopyridines
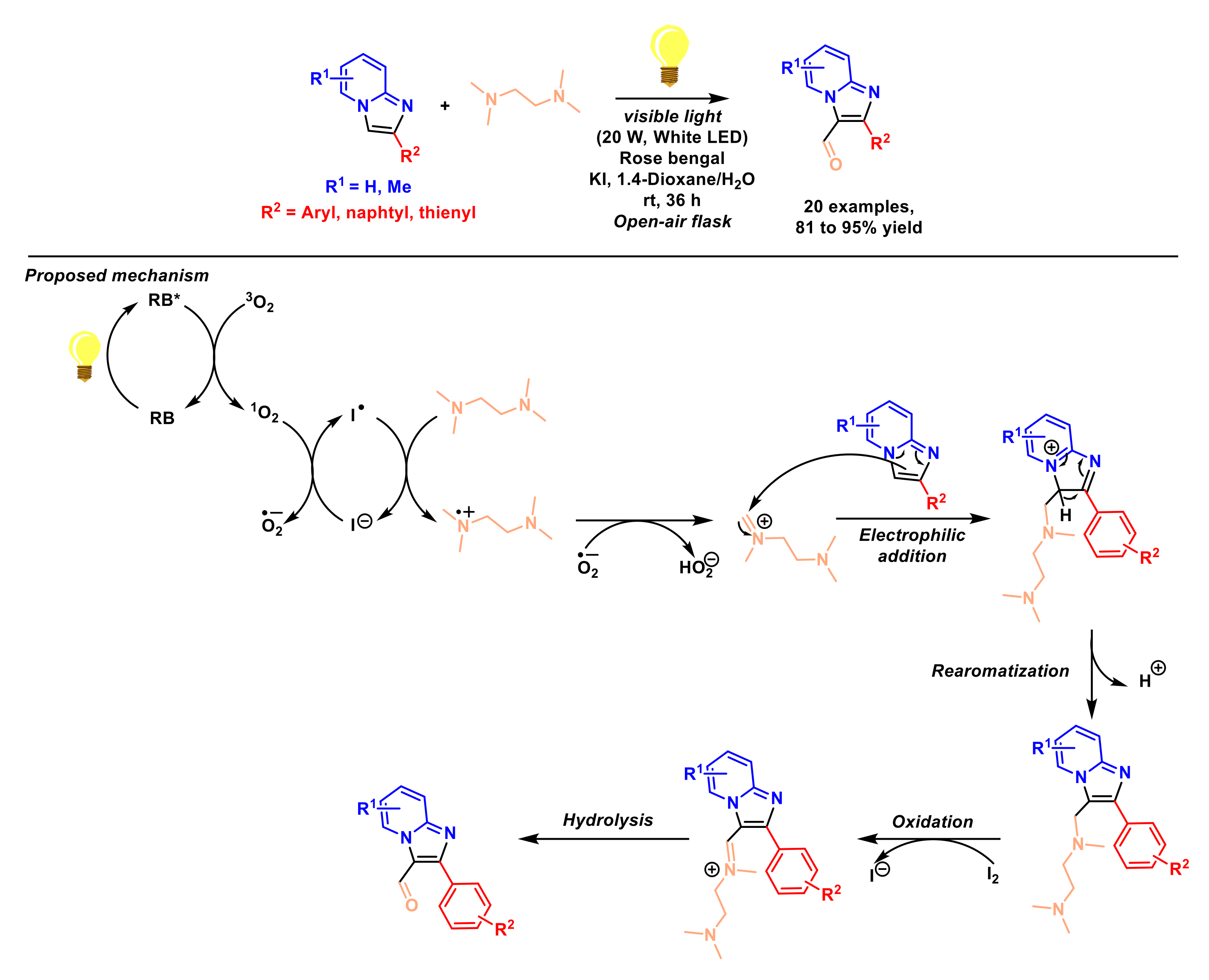
2.1.5. Formylation of Imidazopyridines
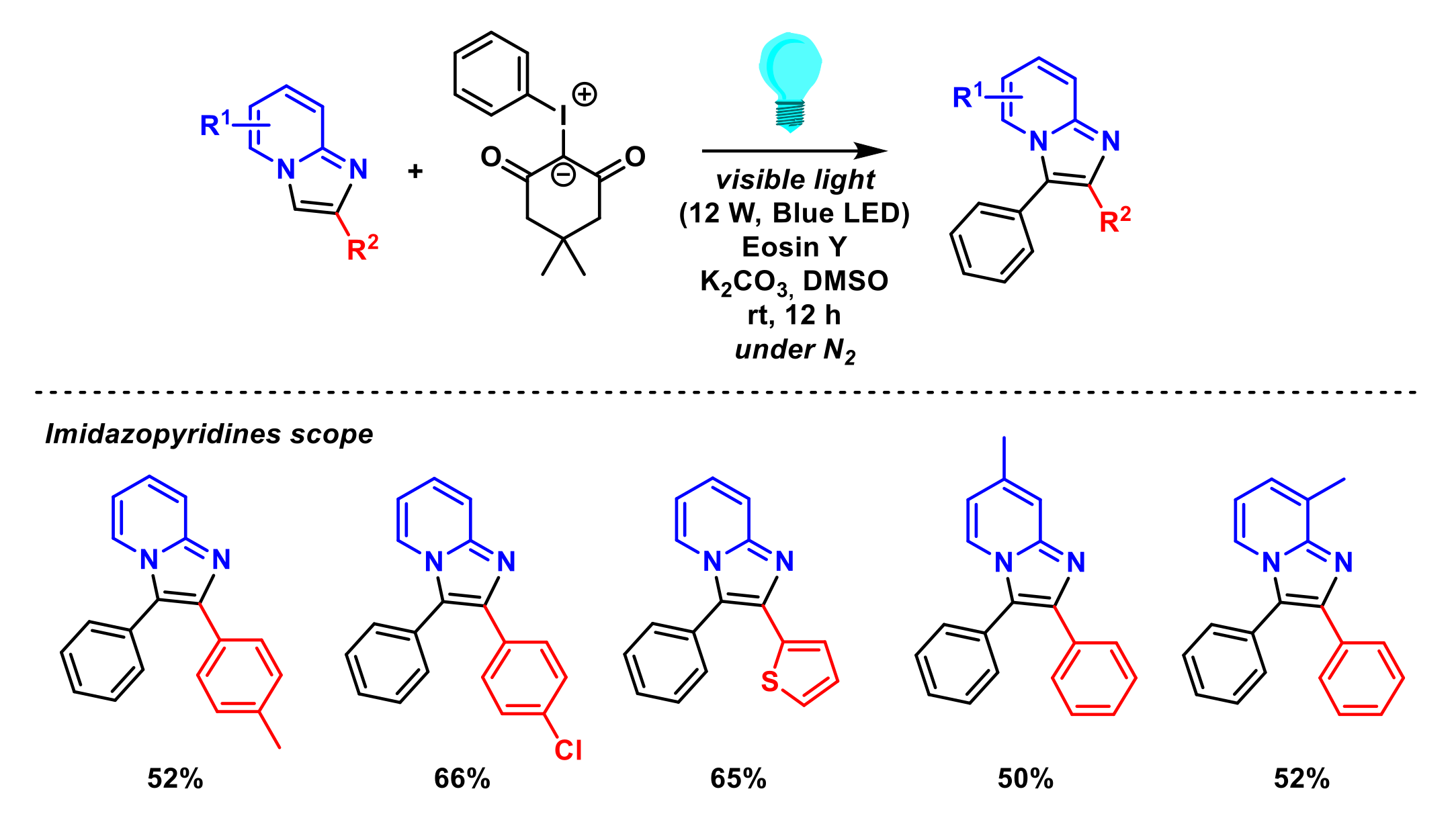
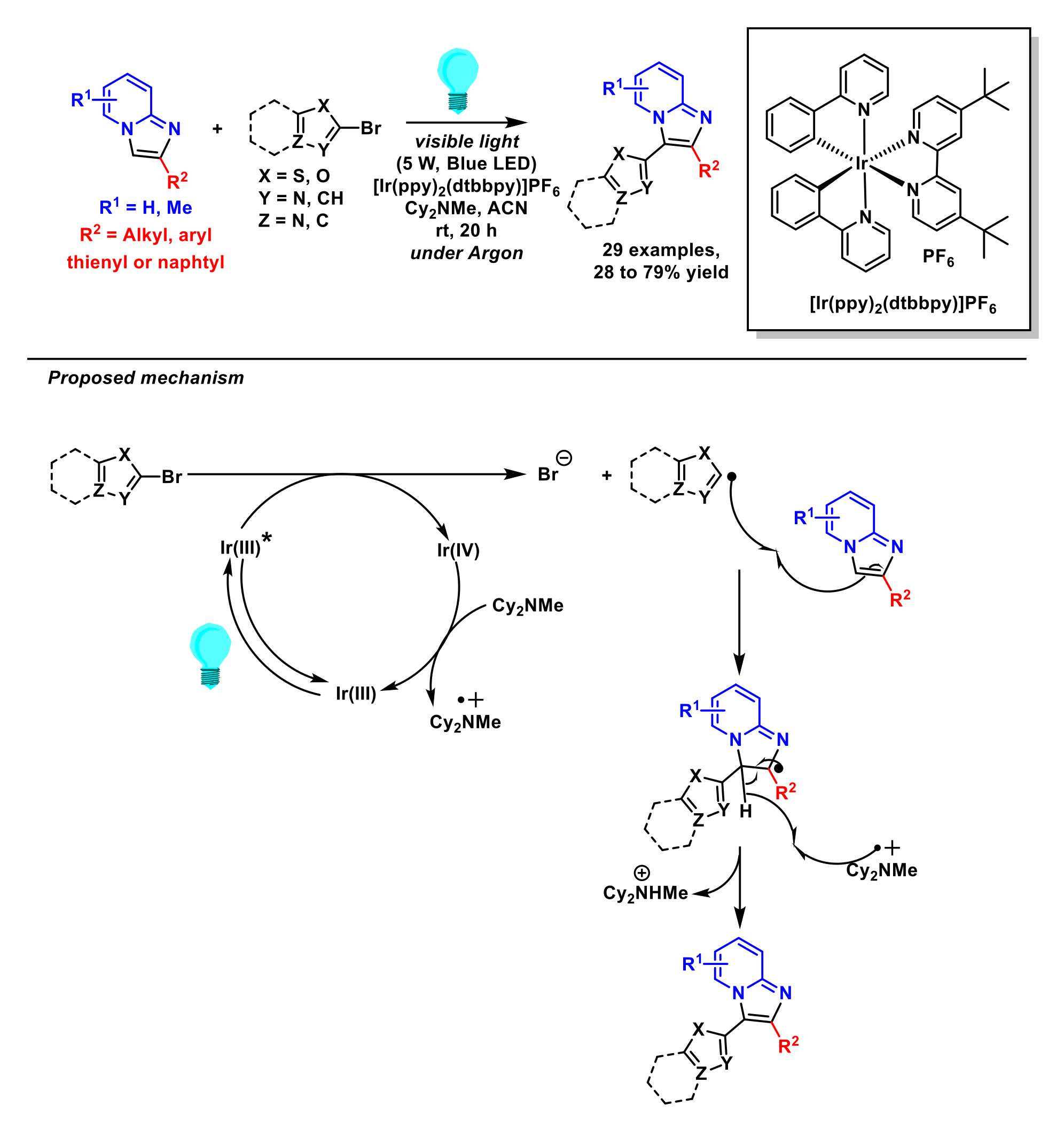
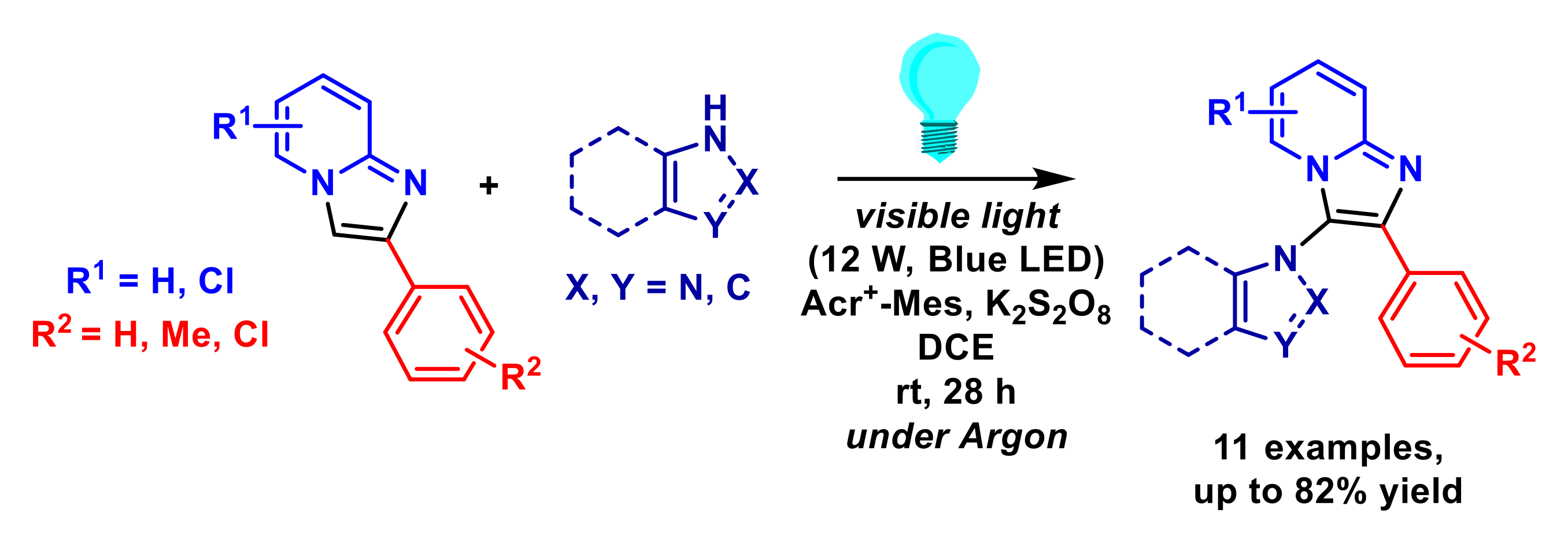
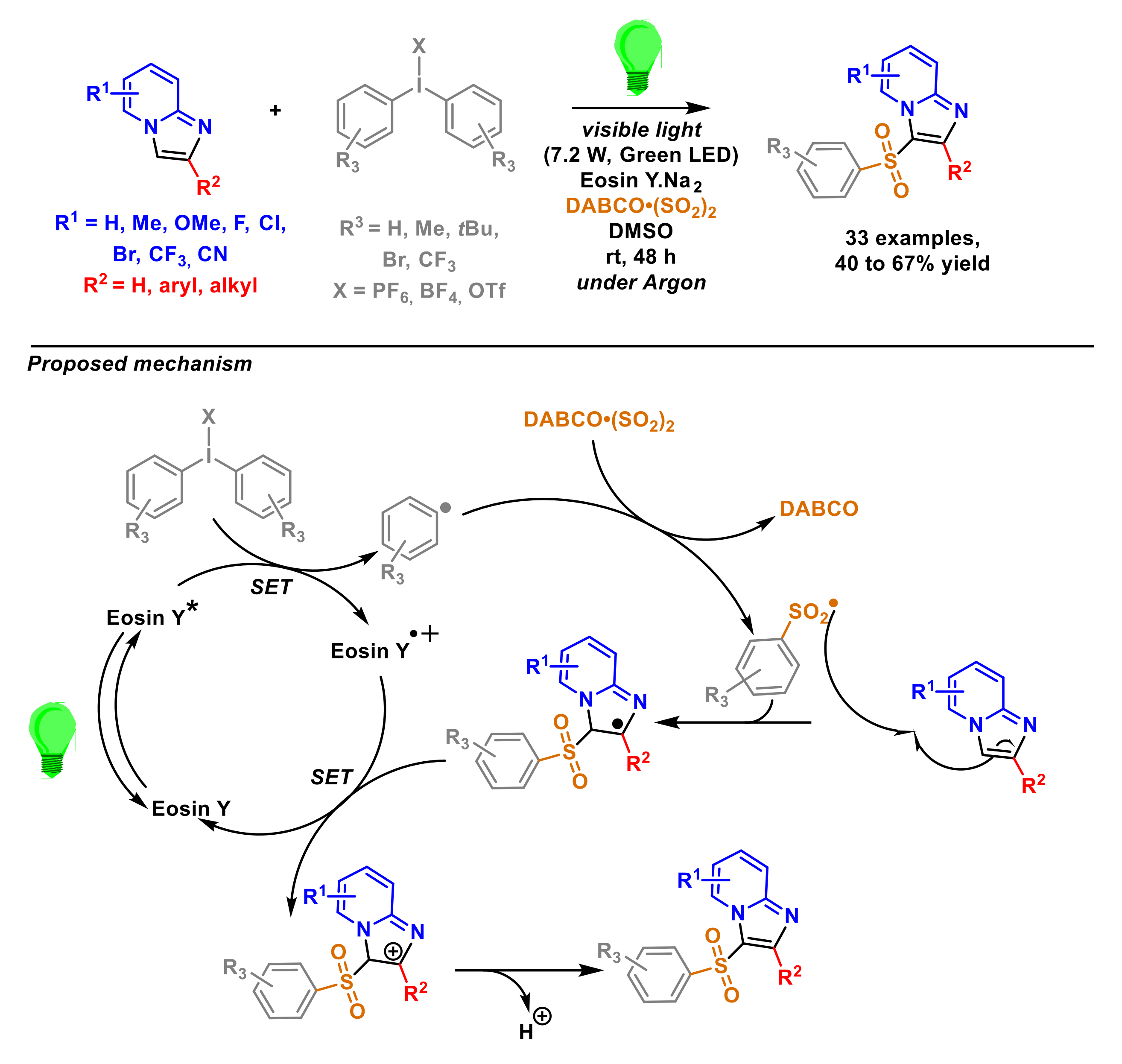
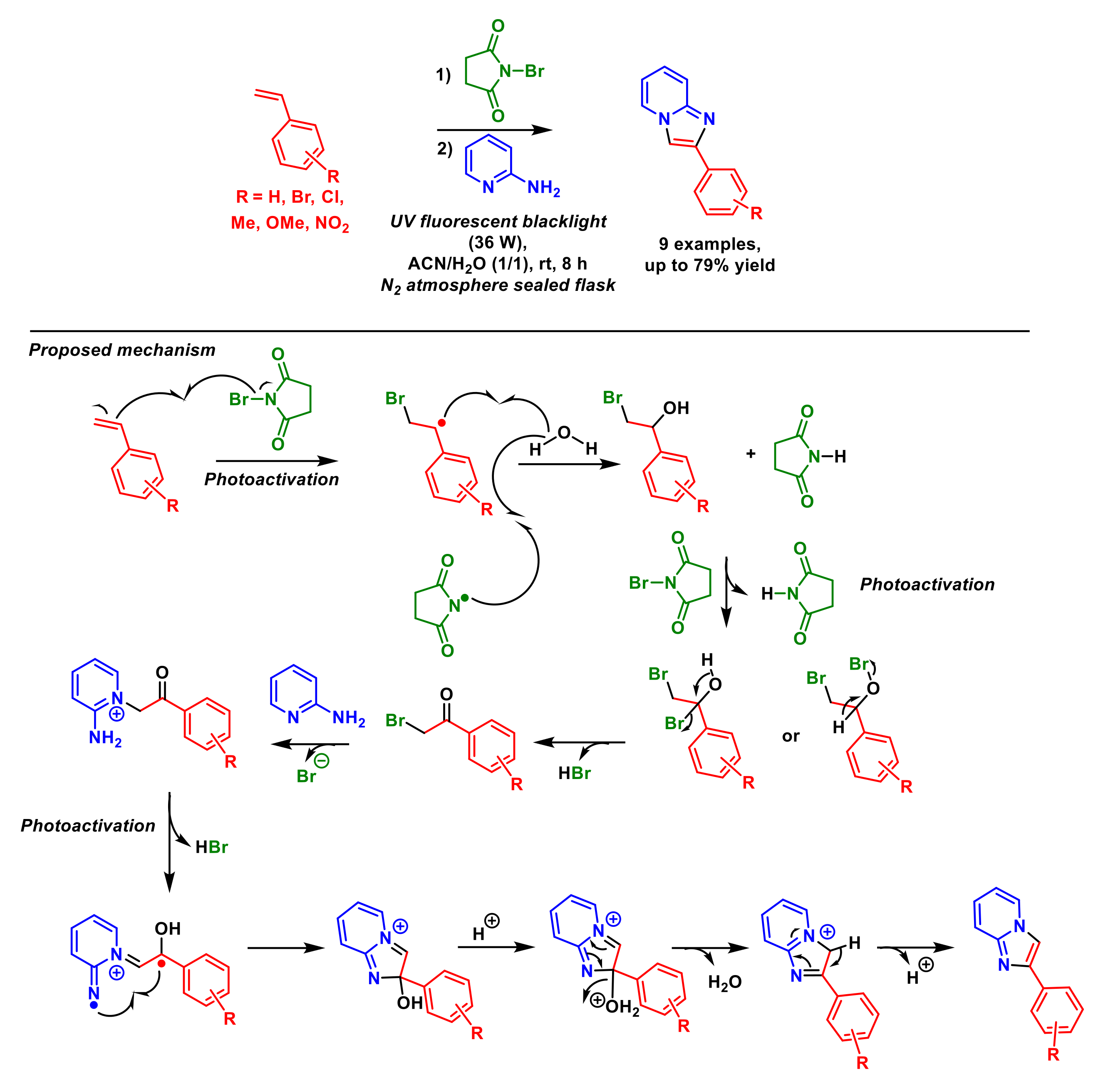
2.1.6. Arylation of Imidazopyridines
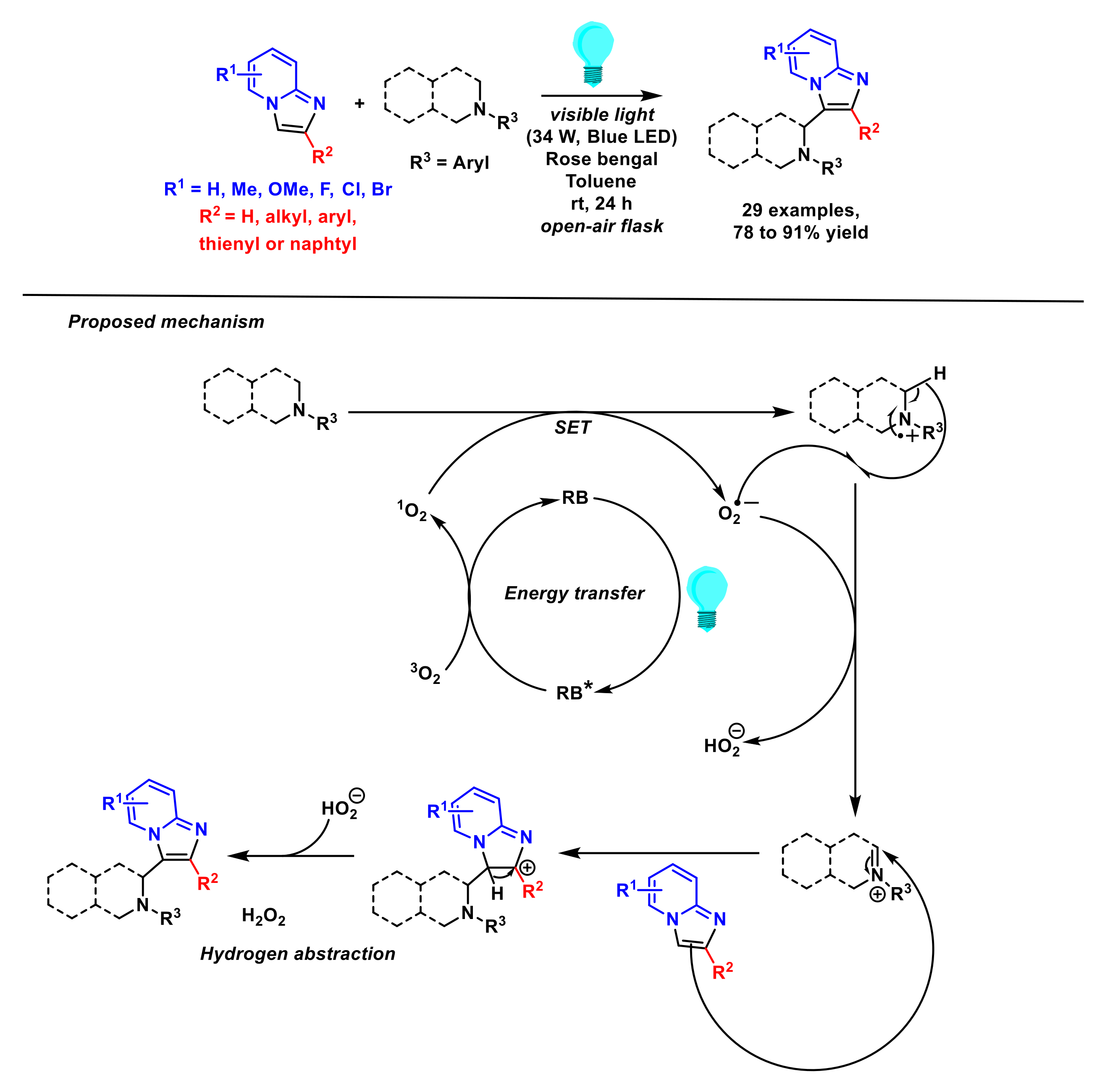
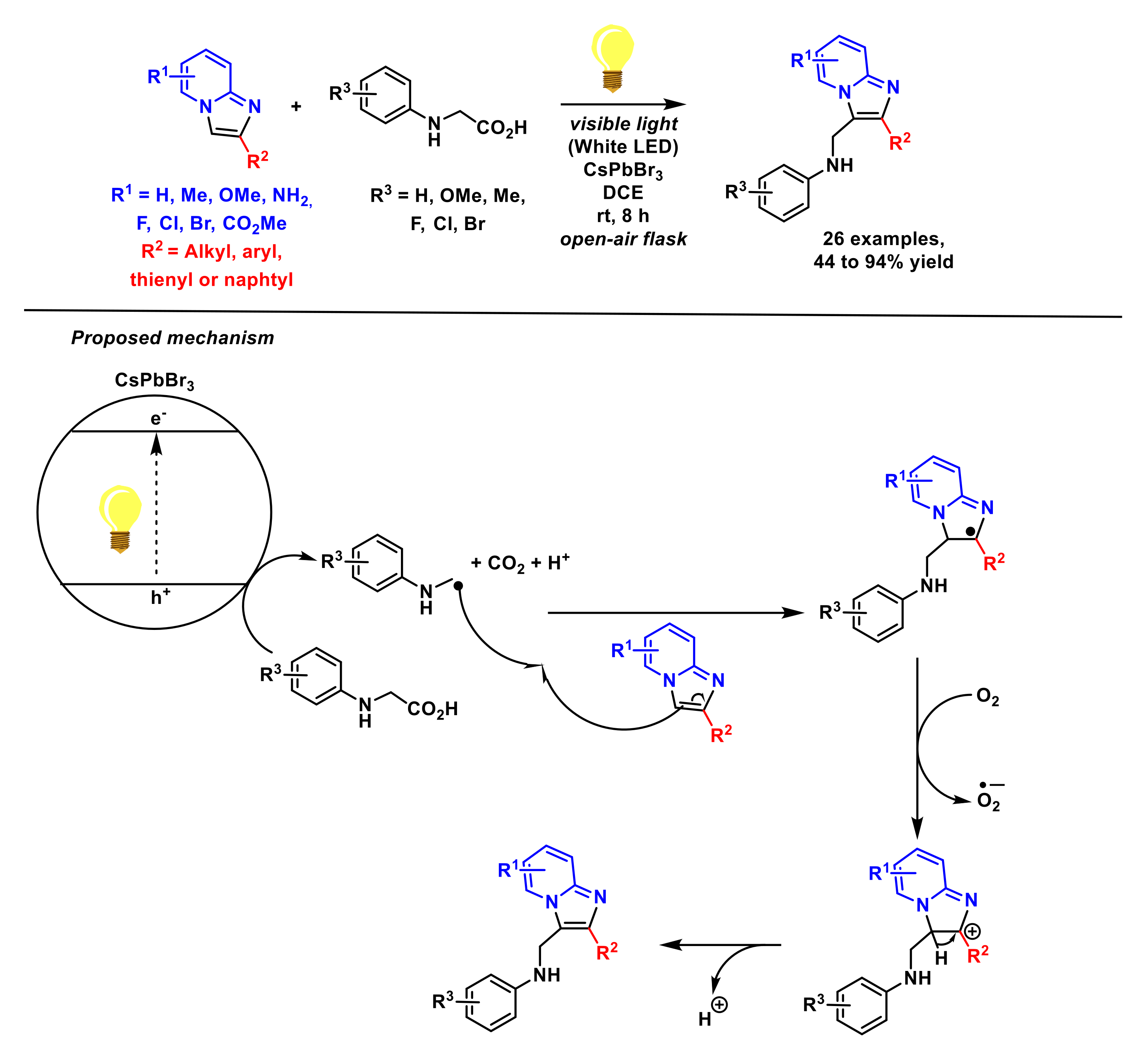
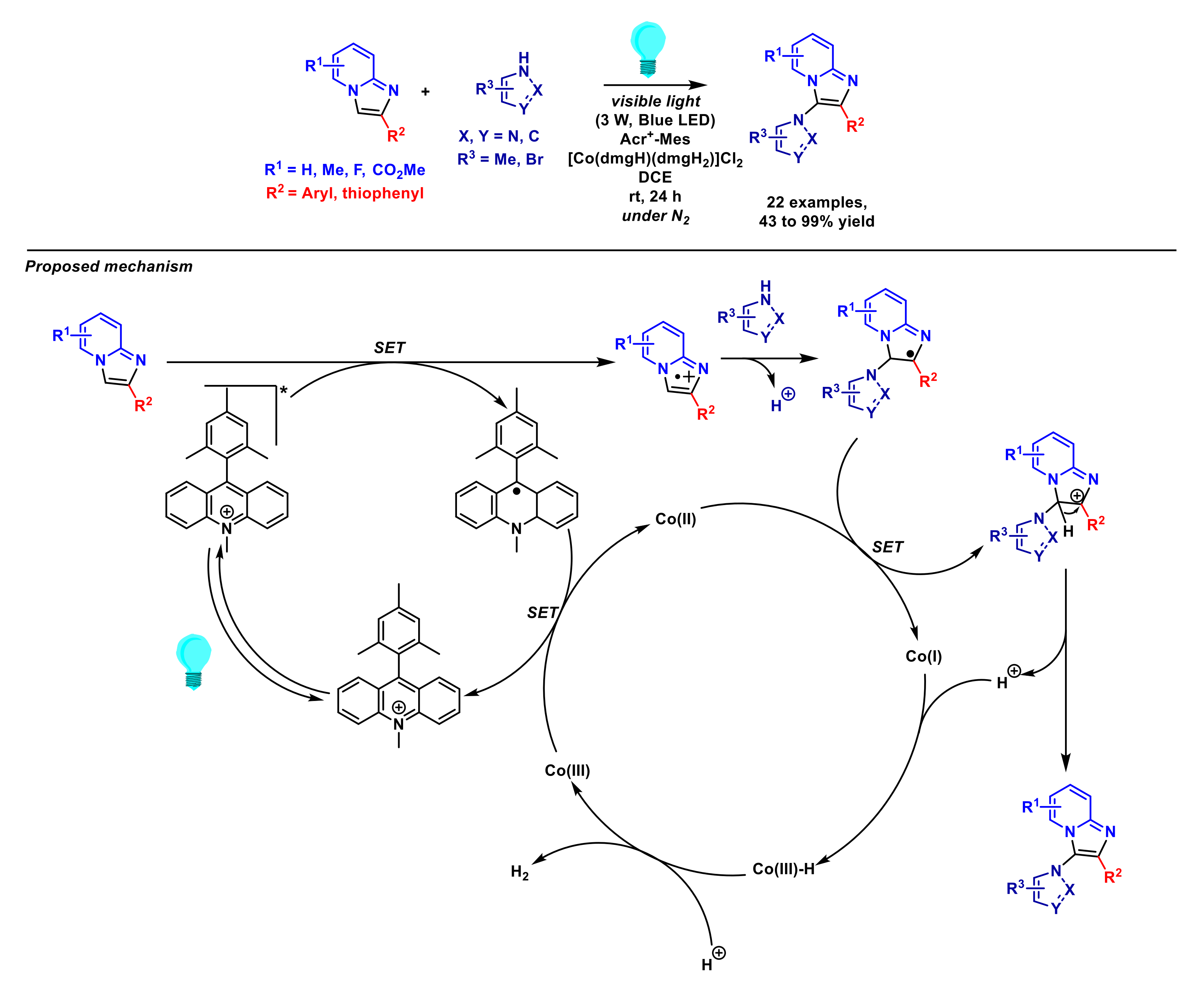
2.2. Formation of C–N Bonds
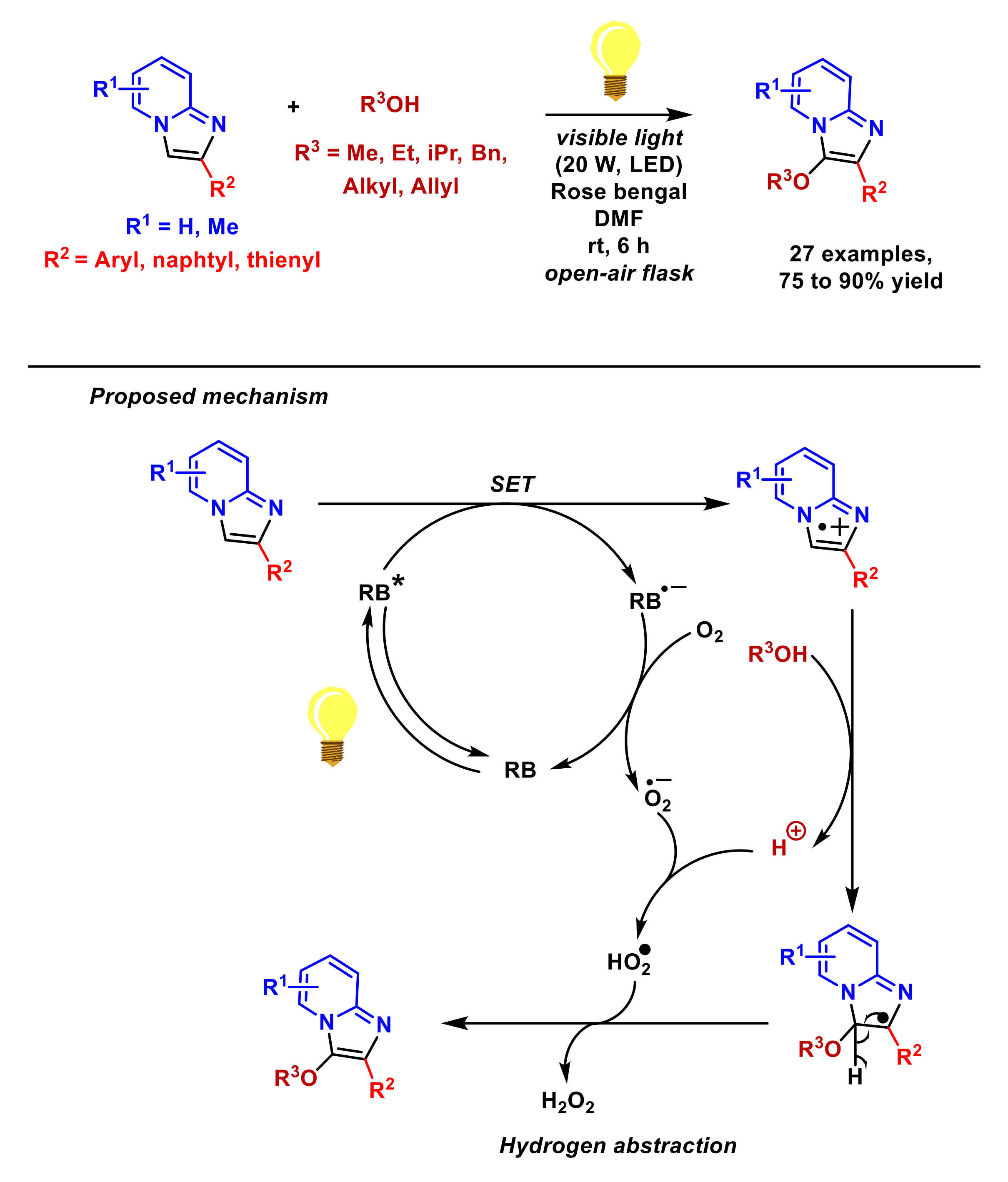
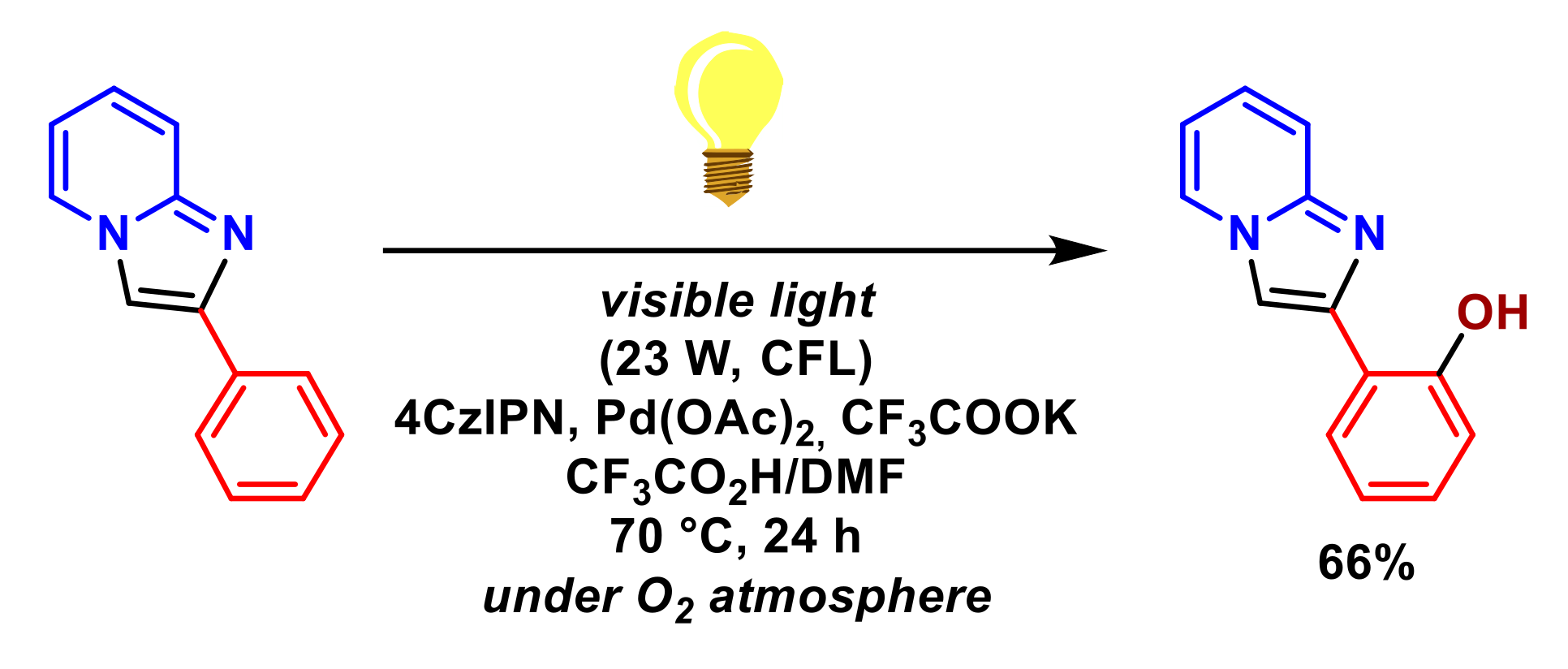
2.3. Formation of C–O Bonds
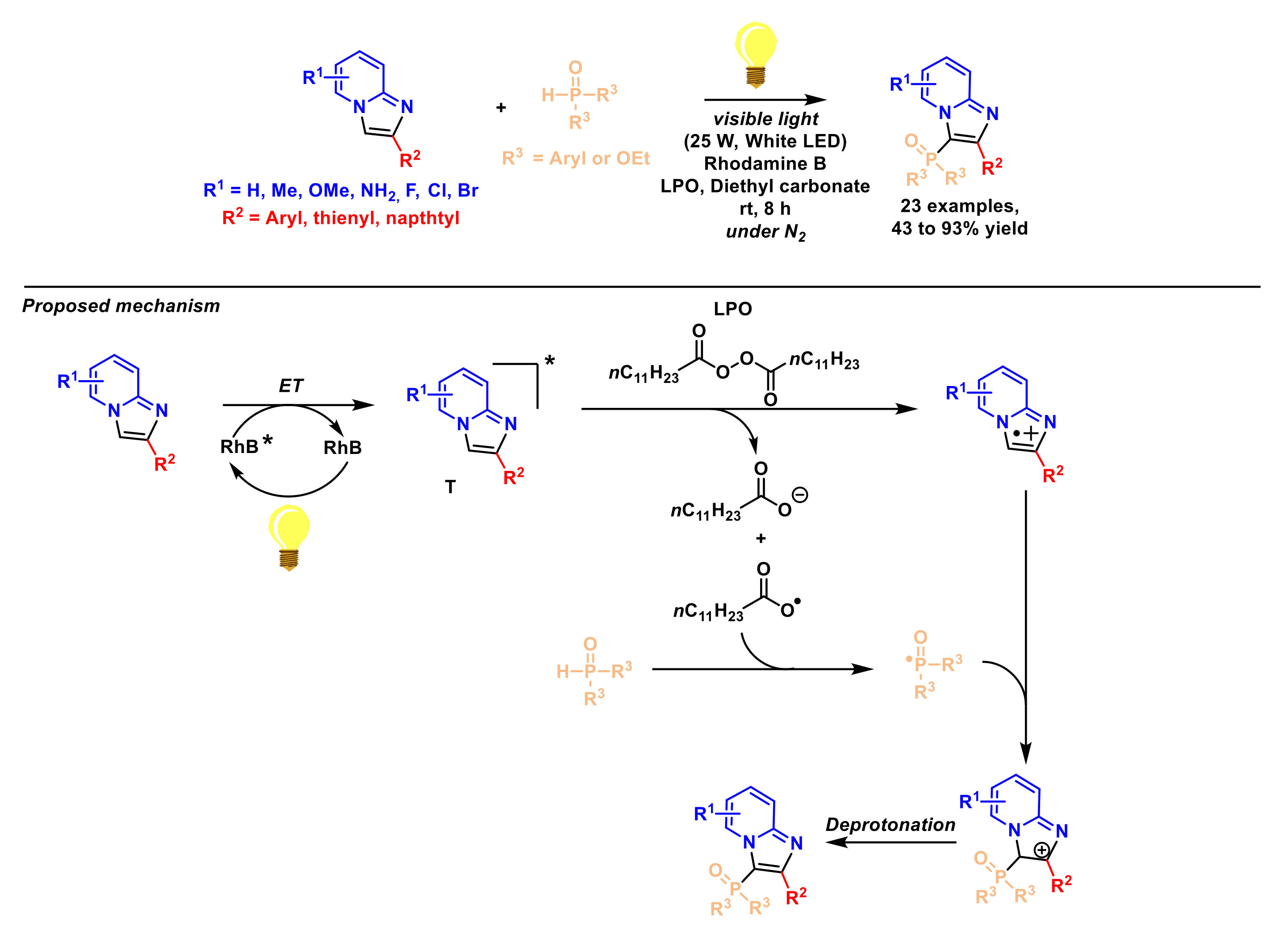
2.4. Formation of C–P Bonds
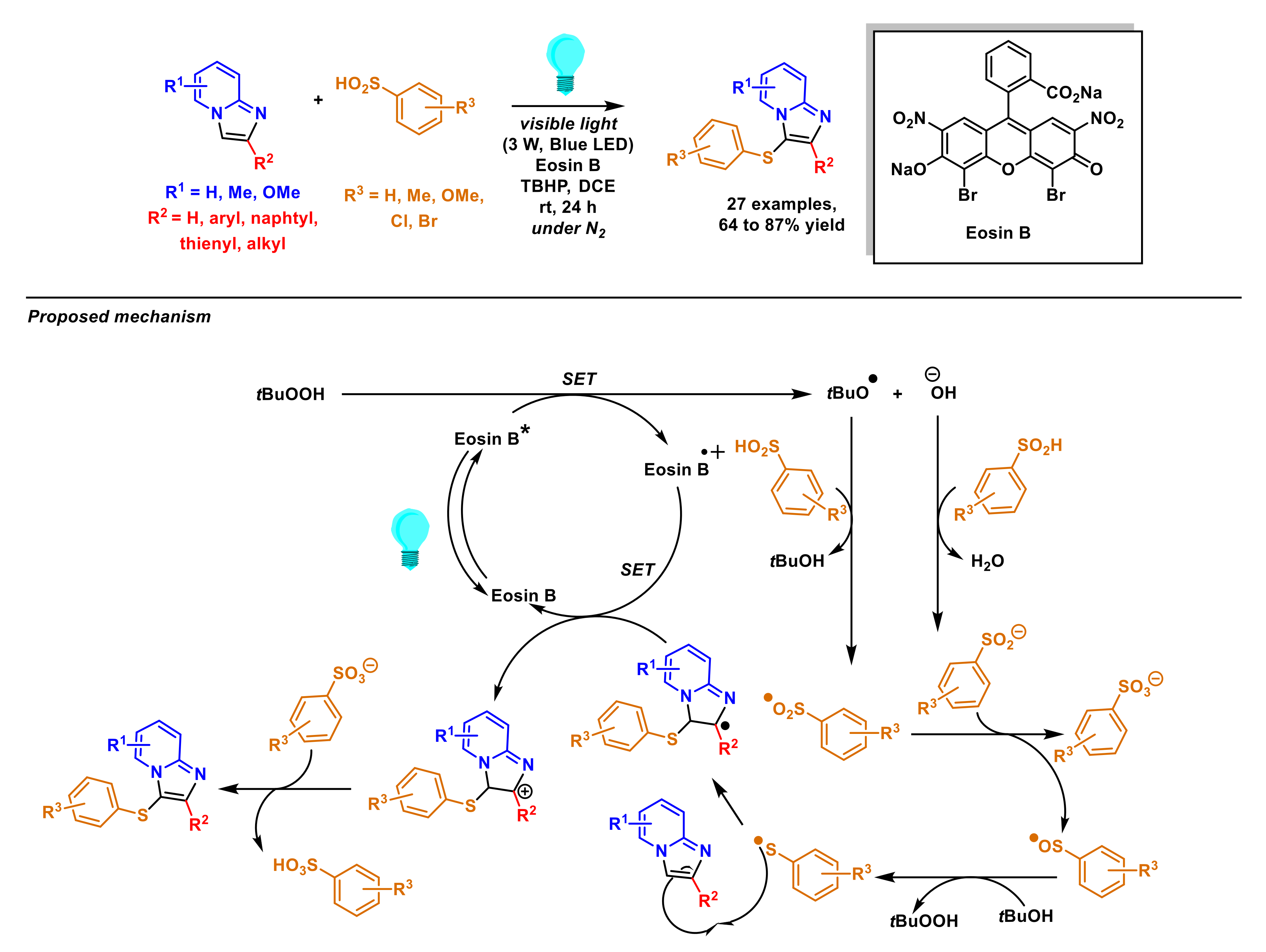
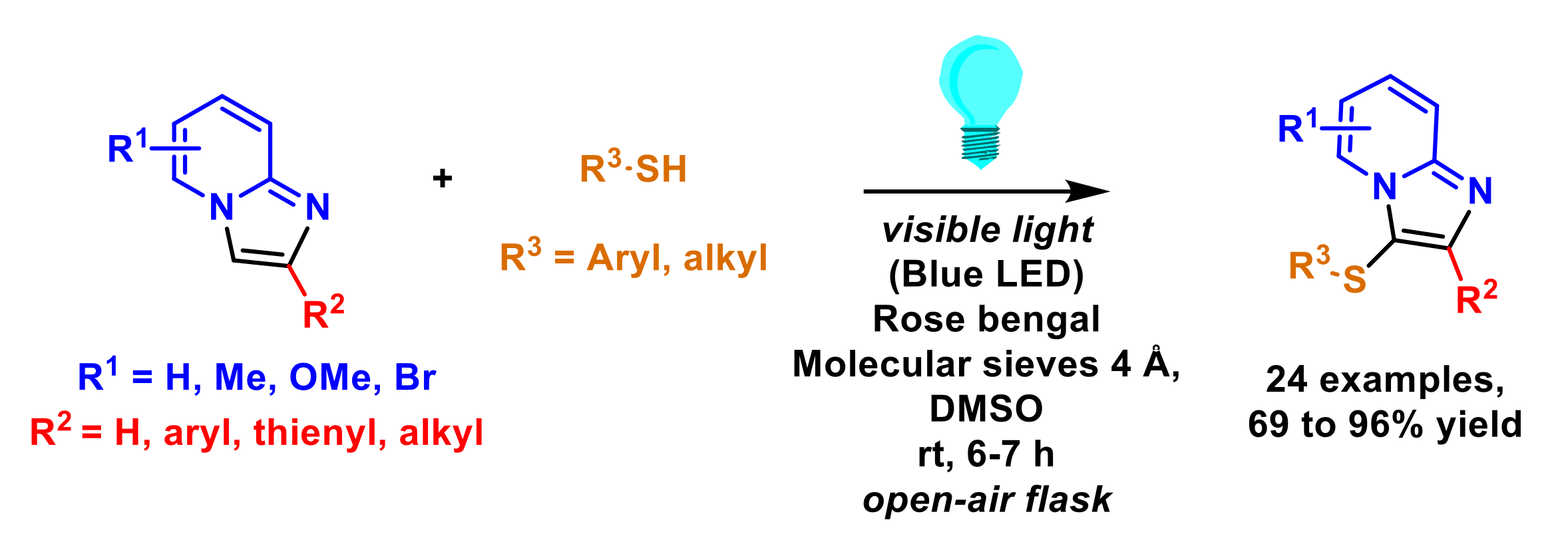
2.5. Formation of C–S Bonds
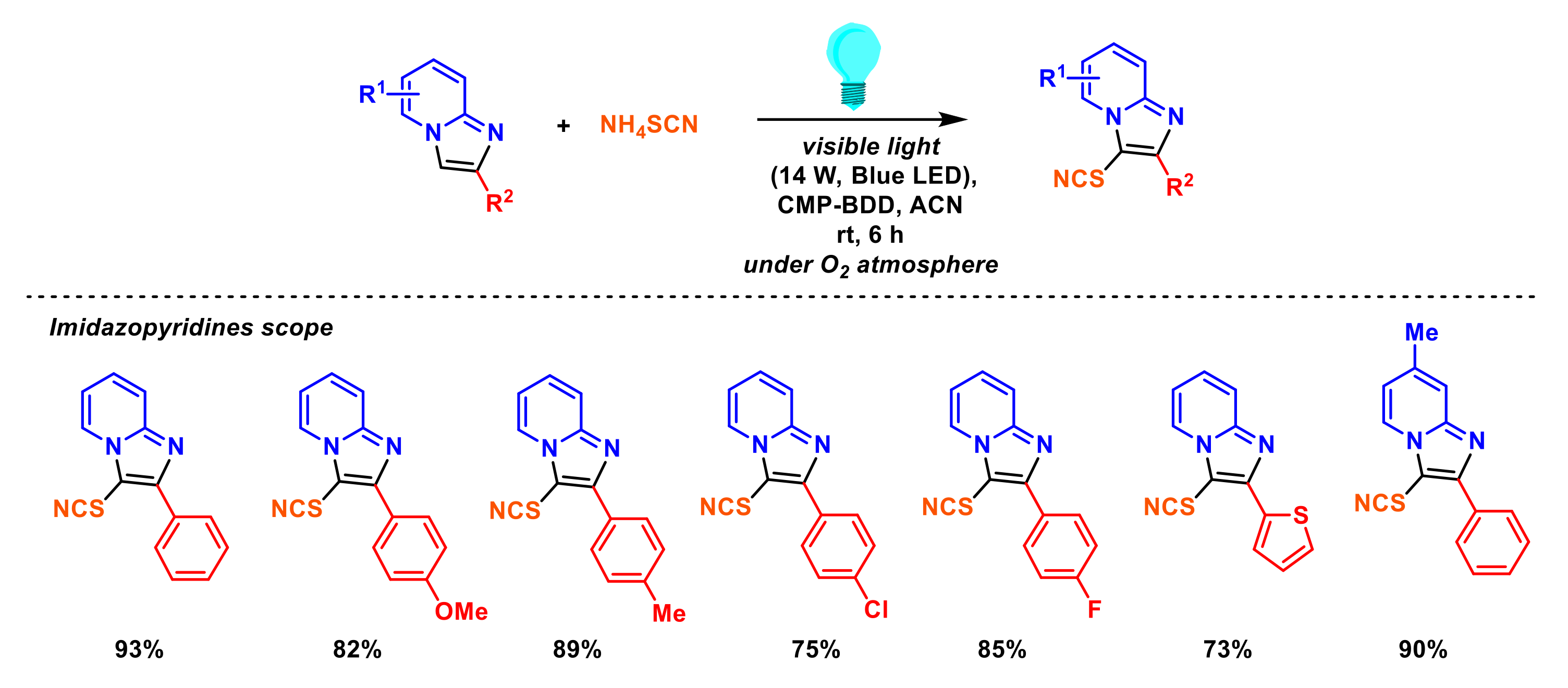
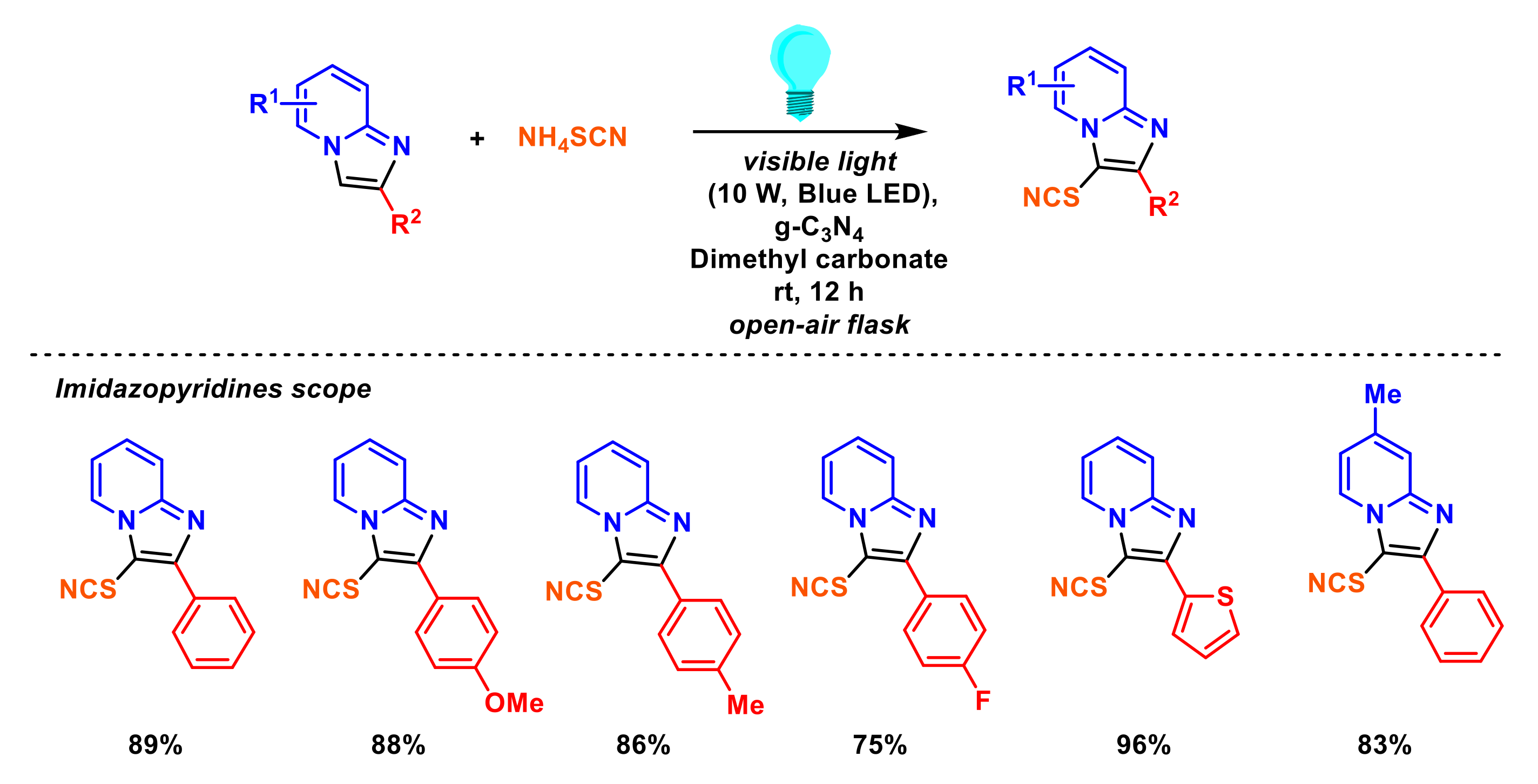
2.6. Formation of C–Se Bonds
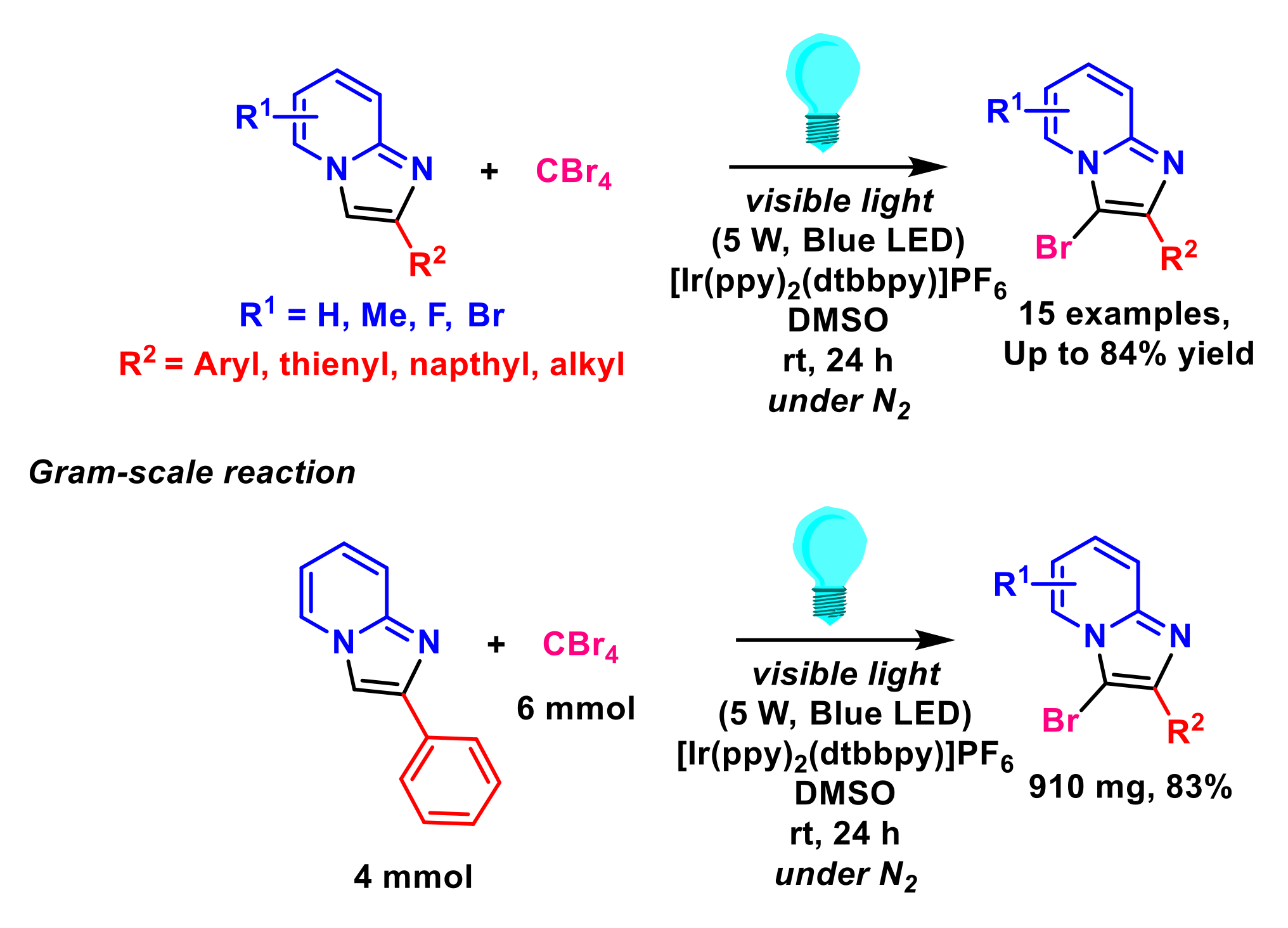
2.7. Formation of C–Br Bonds
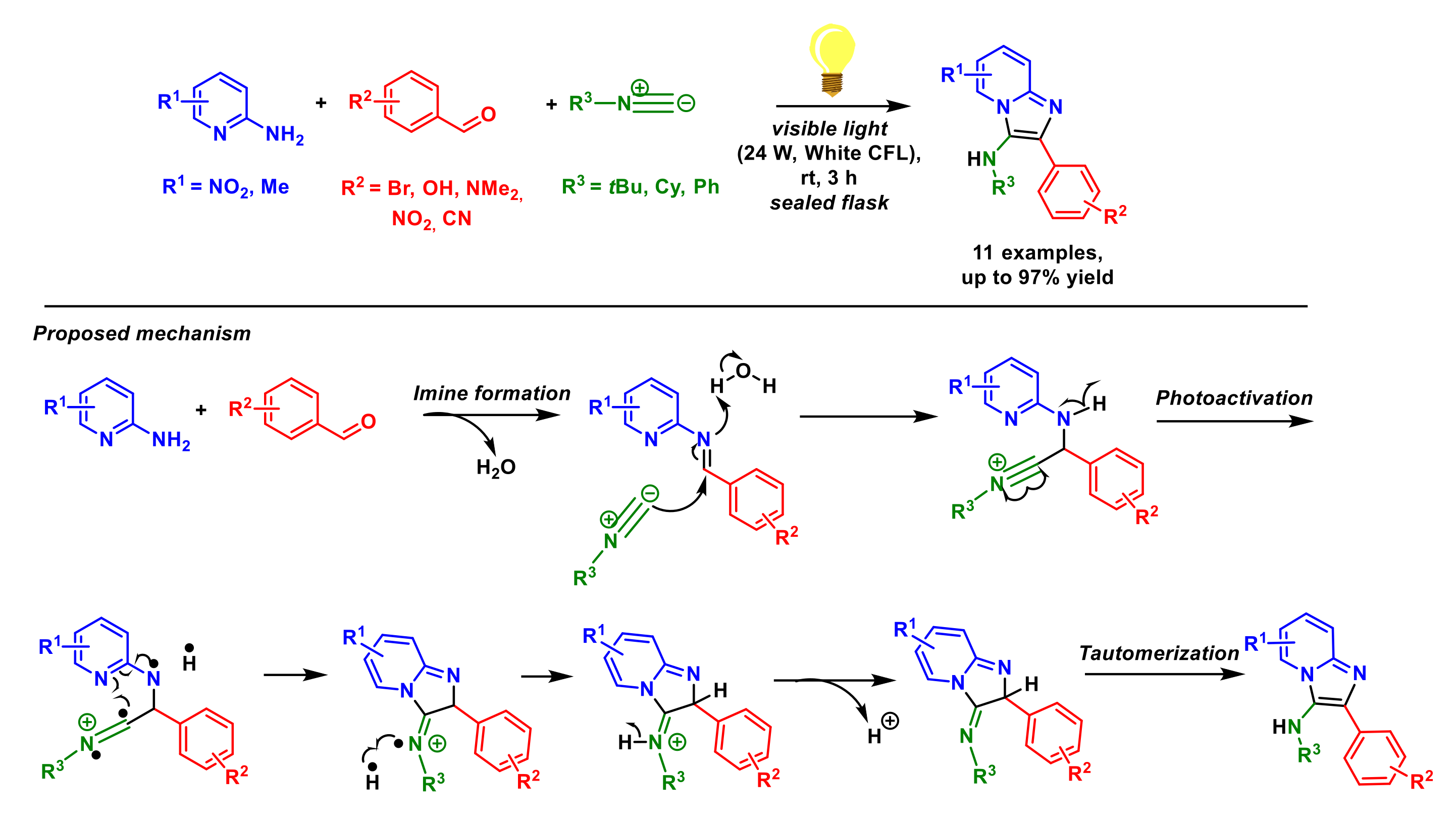
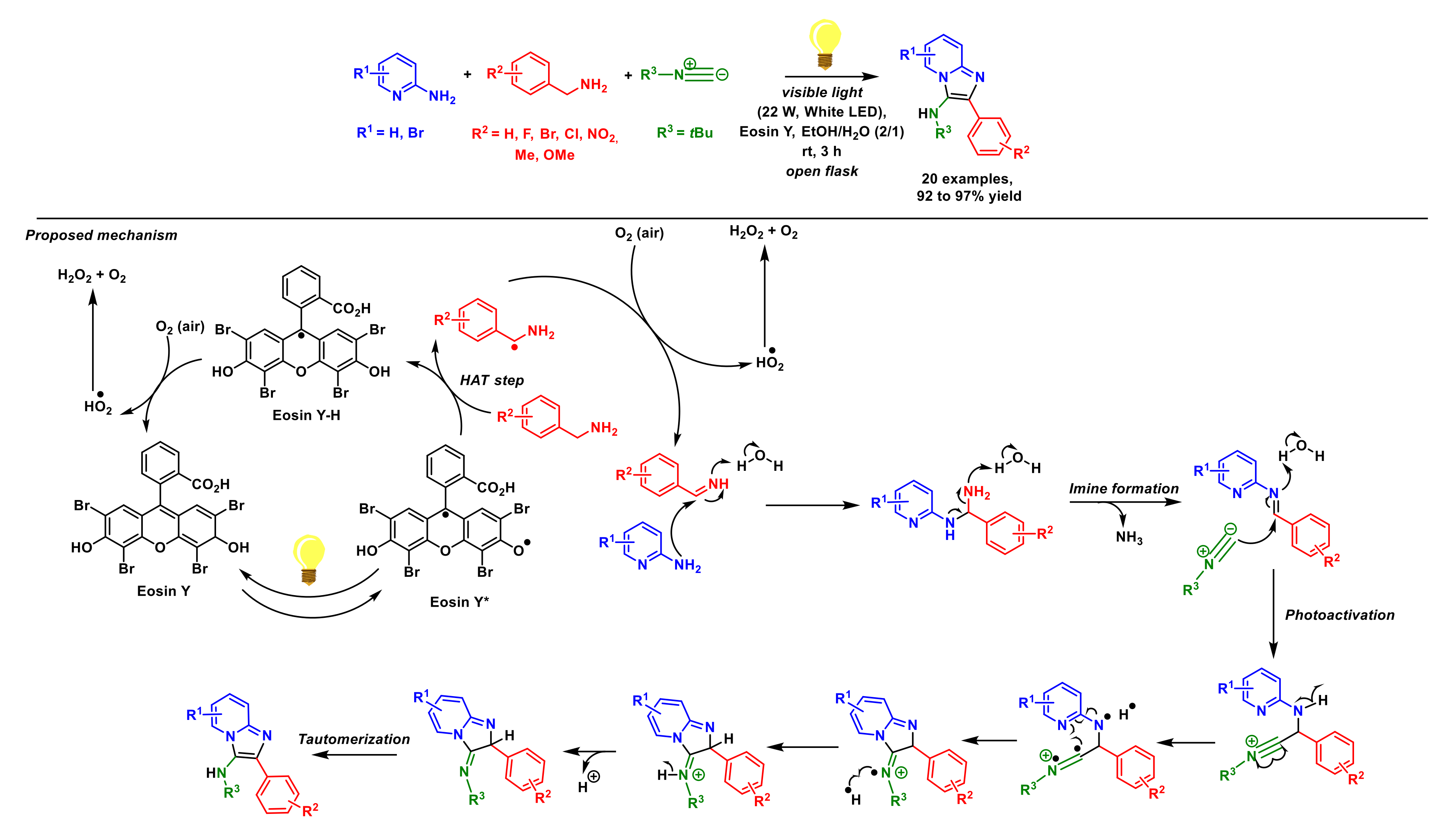
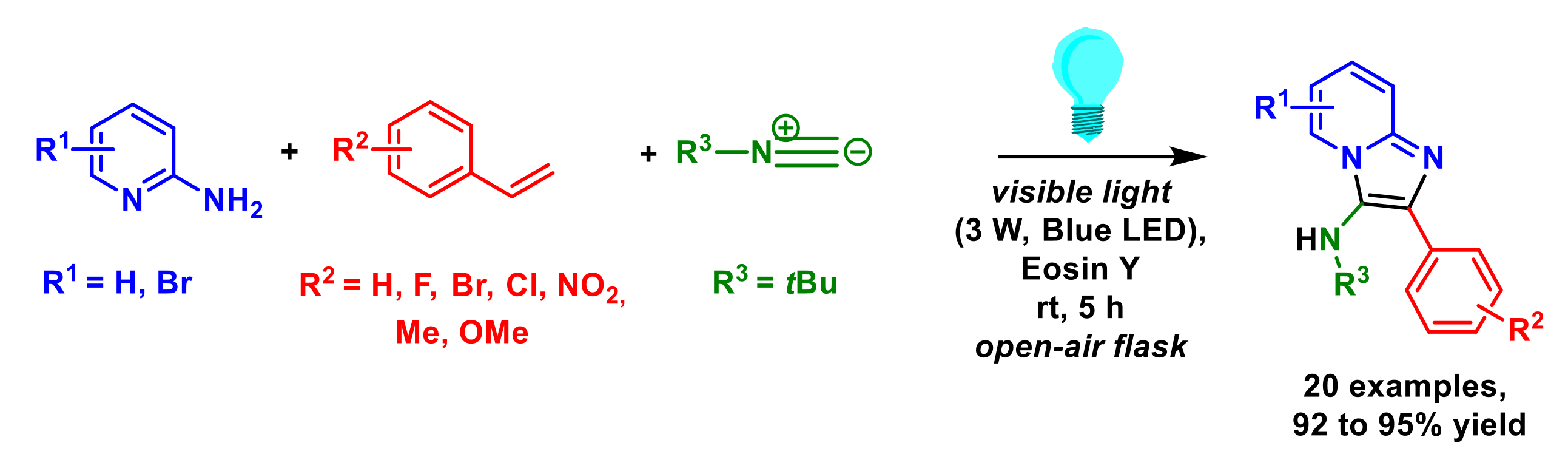
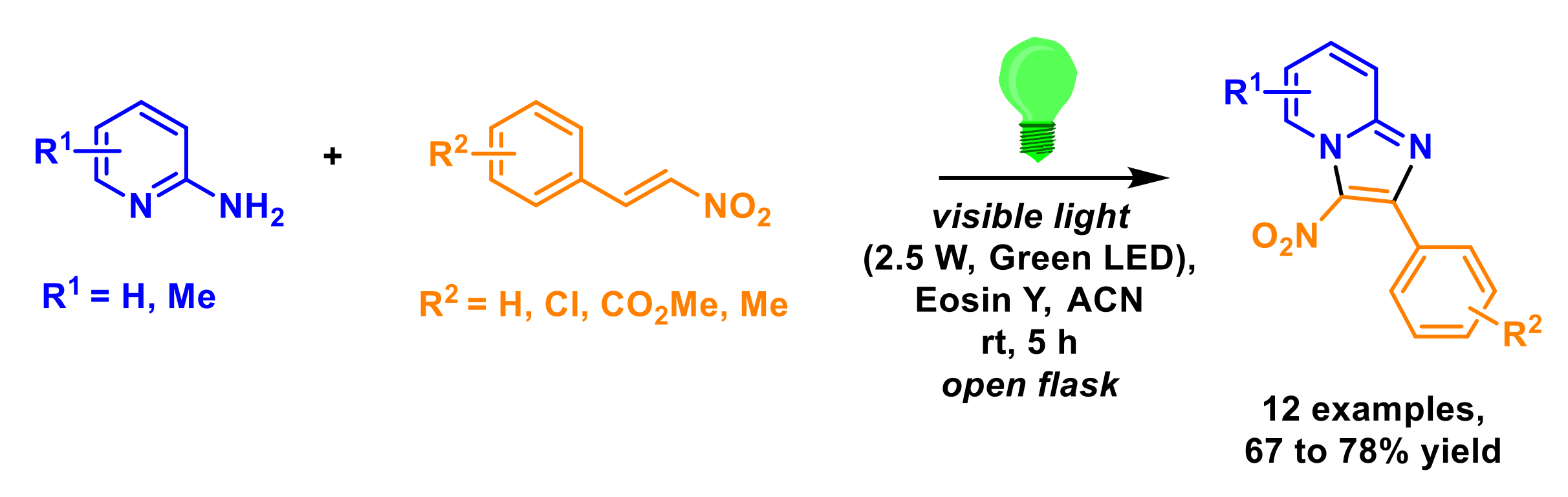
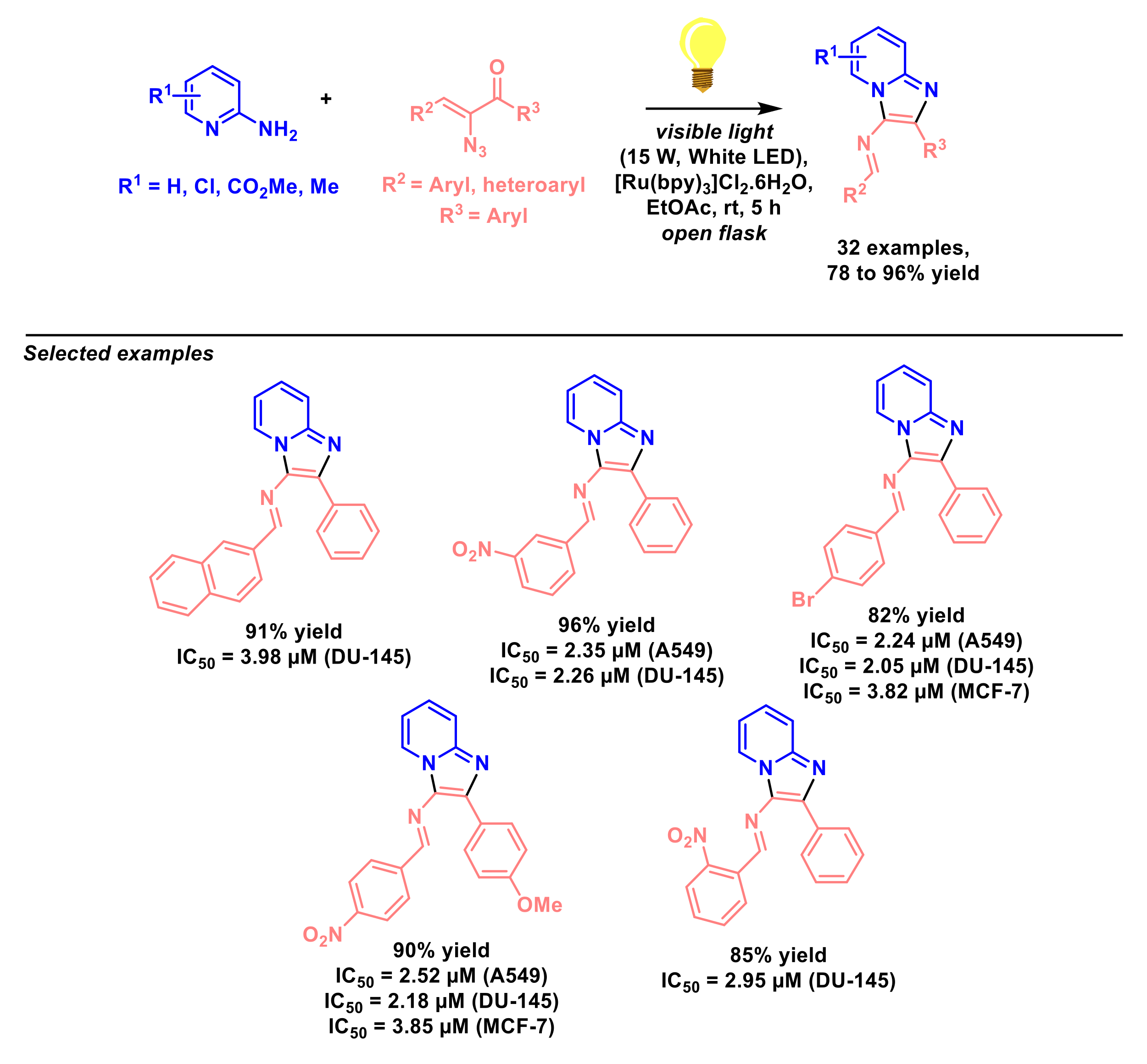
3. Multi-Component Reactions
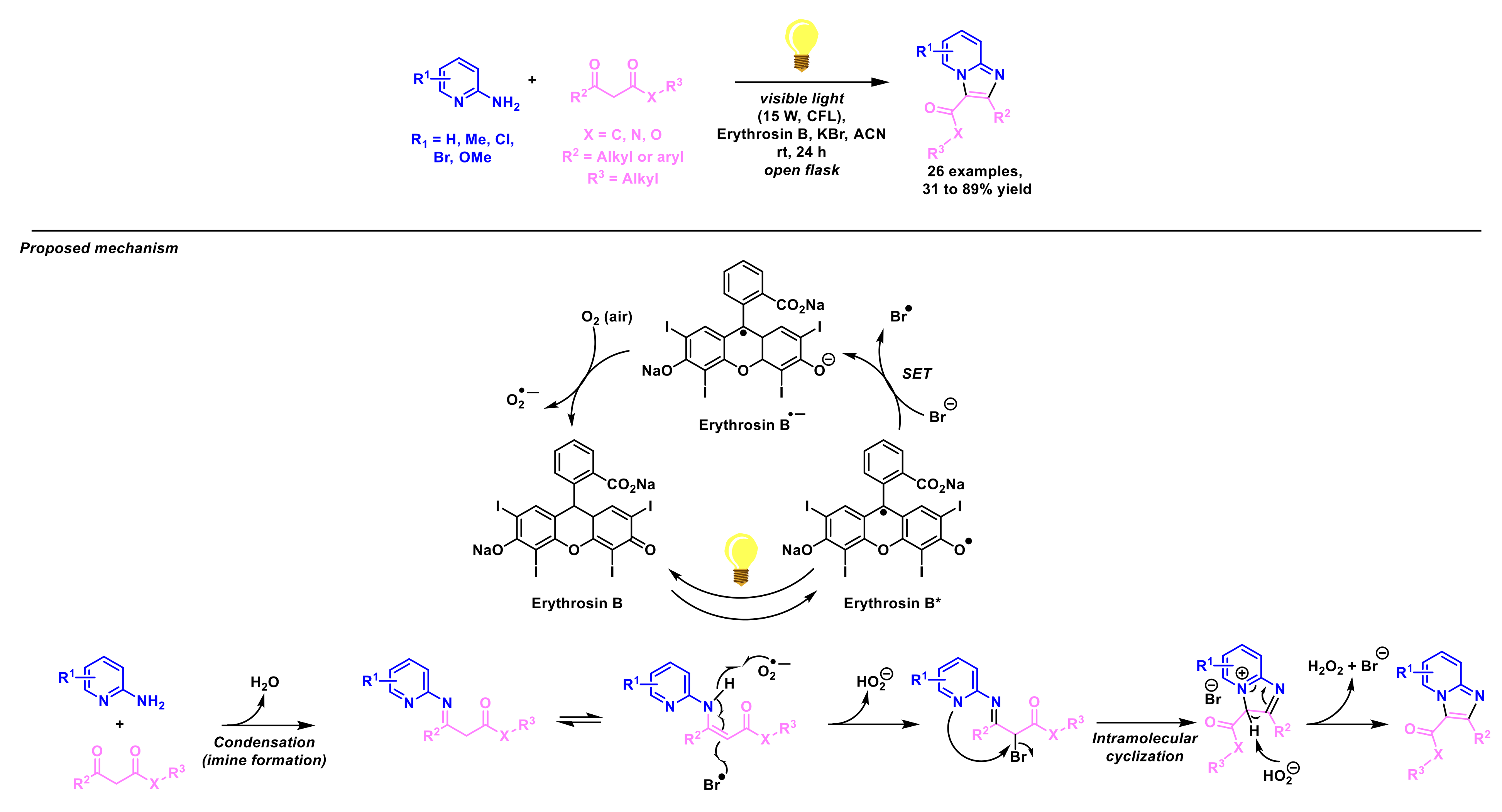
4. Miscellaneous Reactions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bagdi, A.K.; Santra, S.; Monir, K.; Hajra, A. Synthesis of imidazo[1,2-a] pyridines: A decade update. Chem. Chem. Commun. 2015, 51, 1555–1575. [Google Scholar] [CrossRef] [PubMed]
- Pericherla, K.; Kaswan, P.; Pandey, K.; Kumar, A. Recent developments in the synthesis of imidazo[1,2-a] pyridines. Synthesis 2015, 47, 887–912. [Google Scholar]
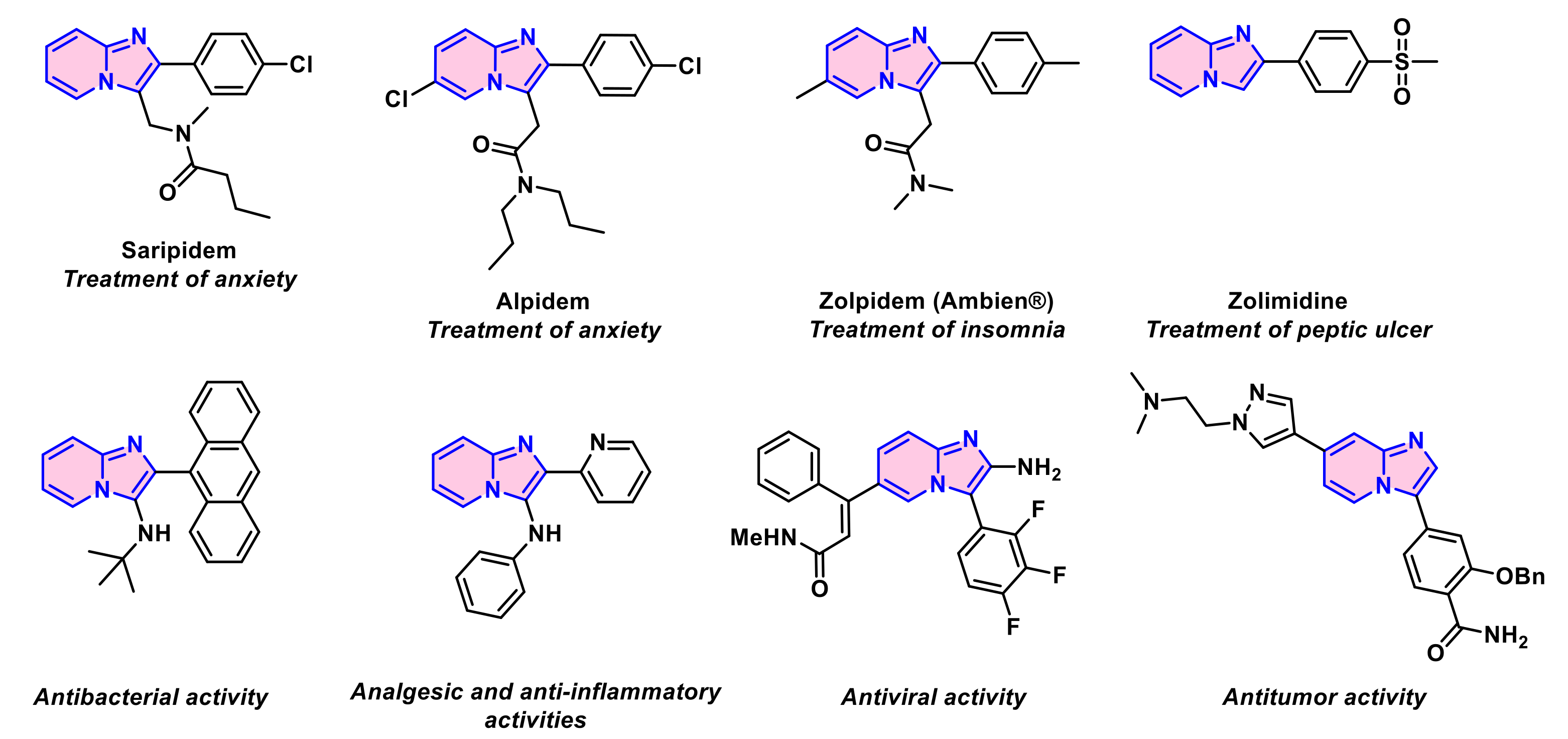
- Enguehard-Gueiffier, C.; Gueiffier, A. Recent progress in the pharmacology of imidazo[1,2-a] pyridines. Mini Rev. Med. Chem. 2007, 7, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Shukla, N.M.; Salunke, D.B.; Yoo, E.; Mutz, C.A.; Balakrishna, R.; David, S.A. Antibacterial activities of groebke–blackburn–bienaymé-derived imidazo[1,2-a] pyridin-3-amines. Bioorg. Med. Chem. 2012, 20, 5850–5863. [Google Scholar]
- Lacerda, R.B.; de Lima, C.K.F.; da Silva, L.L.; Romeiro, N.C.; Miranda, A.L.P.; Barreiro, E.J.; Fraga, C.A.M. Discovery of novel analgesic and anti-inflammatory 3-arylamine-imidazo[1,2-a] pyridine symbiotic prototypes. Bioorg. Med. Chem. 2009, 17, 74–84. [Google Scholar] [CrossRef]
- Hamdouchi, C.; de Blas, J.; del Prado, M.; Gruber, J.; Heinz, B.A.; Vance, L. 2-Amino-3-substituted-6-[(E)-1-phenyl-2-(N-methylcarbamoyl) vinyl] imidazo[1,2-a] pyridines as a novel class of inhibitors of human rhinovirus: Stereospecific synthesis and antiviral activity. J. Med. Chem. 1999, 42, 50–59. [Google Scholar] [CrossRef]
- Xi, J.-B.; Fang, Y.-F.; Frett, B.; Zhu, M.-L.; Zhu, T.; Kong, Y.-N.; Guan, F.-J.; Zhao, Y.; Zhang, X.-W.; Li, H.-y.; et al. Structure-based design and synthesis of imidazo[1,2-a] pyridine derivatives as novel and potent Nek2 inhibitors with in vitro and in vivo antitumor activities. Eur. J. Med. Chem. 2017, 126, 1083–1106. [Google Scholar] [CrossRef]
- Furukawa, S.; Shono, H.; Mutai, T.; Araki, K. Colorless, transparent, dye-doped polymer films exhibiting tunable luminescence color: Controlling the dual-color luminescence of 2-(2′-hydroxyphenyl) imidazo[1,2-a] pyridine derivatives with the surrounding matrix. ACS Appl. Mater. Interfaces 2014, 6, 16065–16070. [Google Scholar] [CrossRef]
- Chavana, K.H.; Kedar, N.A. Recent developments in synthesis of imidazo[1,2-a] pyridines (2016–2020). Chem. Biol. Interface 2021, 11, 34–39. [Google Scholar]
- Tschitschibabin, A.E.; Kirsanow, A.W. α, β′-diamino-pyridin und α, β-diamino-pyridin. Chem. Ber. 1927, 60, 766–776. [Google Scholar] [CrossRef]
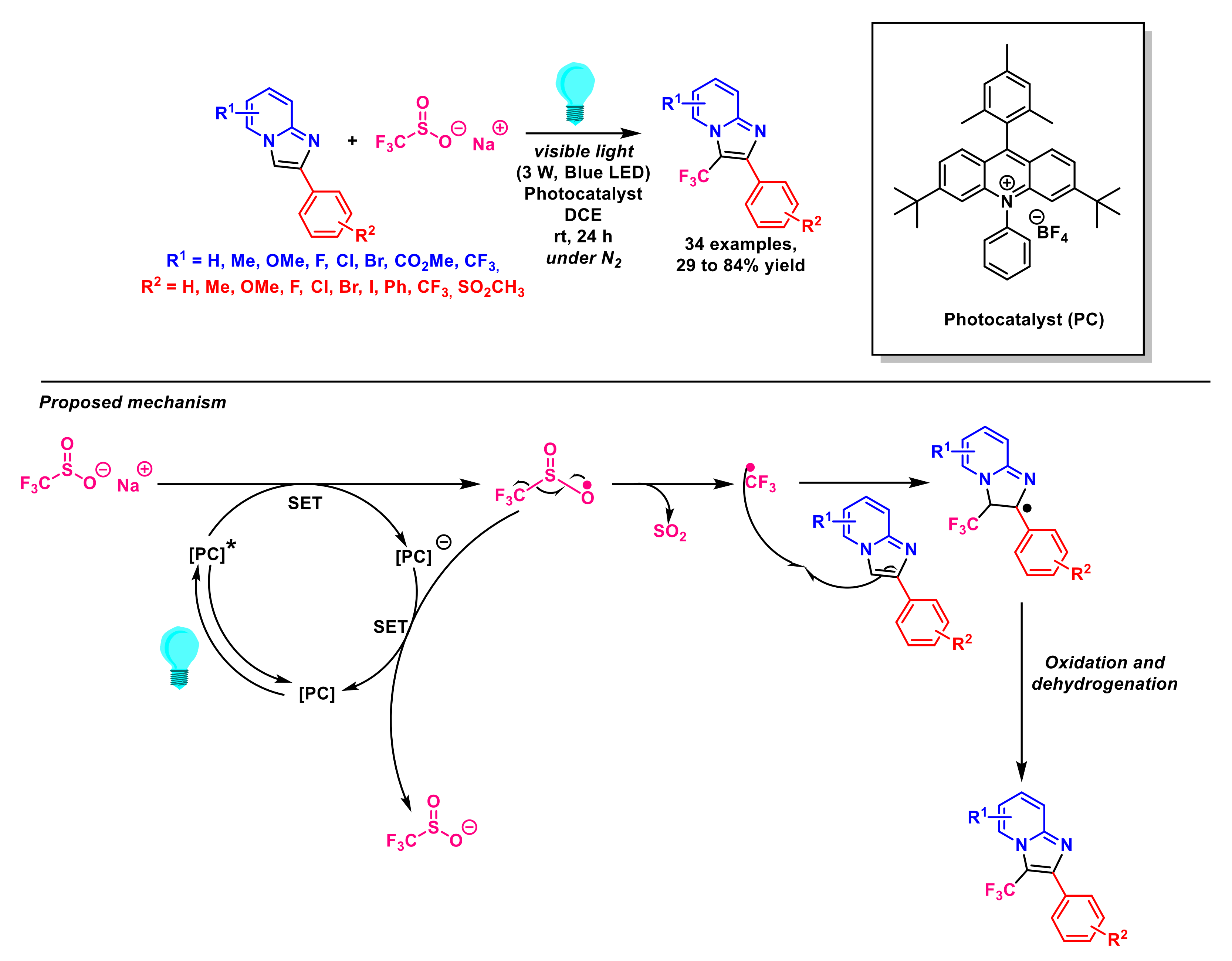
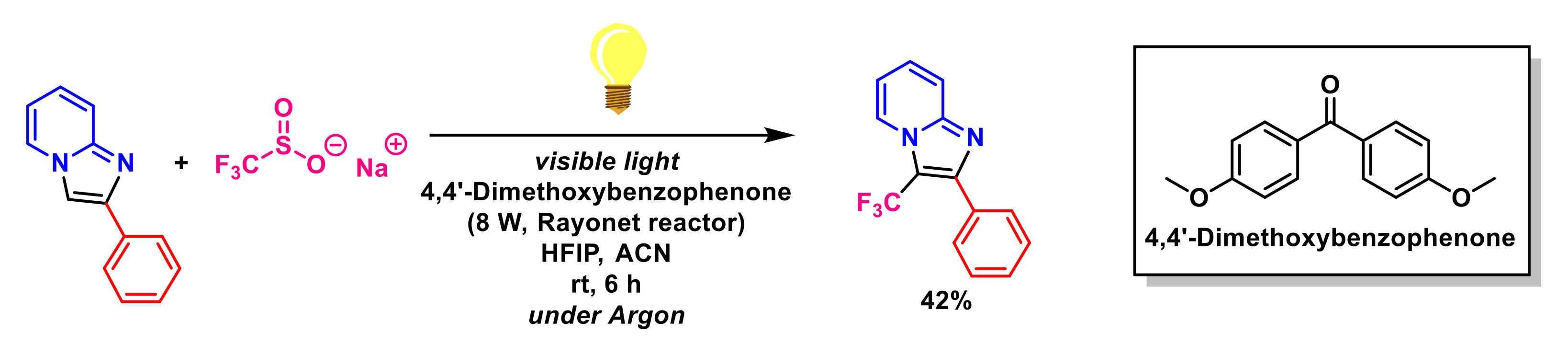
- Baishya, G.; Dutta, N.B. Recent Advances in direct C–H trifluoromethylation of n-heterocycles. ChemistrySelect 2021, 6, 13384–13408. [Google Scholar] [CrossRef]
- Ghosh, S.; Laru, S.; Hajra, A. Ortho C–H functionalization of 2-arylimidazo[1,2-a] pyridines. Chem. Rec. 2022, 22, e202100240. [Google Scholar] [CrossRef] [PubMed]
- Kaboudin, B.; Ghashghaee, M.; Bigdeli, A.; Farkhondeh, A.; Eskandari, M.; Esfandiari, H. Recent advances on the application of langlois’ reagent in organic transformations. ChemistrySelect 2021, 6, 12998–13014. [Google Scholar] [CrossRef]
- Patel, O.P.S.; Nandwana, N.K.; Legoabe, L.J.; Das, B.C.; Kumar, A. Recent advances in radical C–H bond functionalization of imidazoheterocycles. Adv. Synth. Catal. 2020, 362, 4226–4255. [Google Scholar] [CrossRef]
- Tashrifi, Z.; Mohammadi-Khanaposhtani, M.; Larijani, B.; Mahdavi, M. C3-functionalization of imidazo[1,2-a] pyridines. Eur. J. Org. Chem. 2020, 3, 269–284. [Google Scholar] [CrossRef]
- Mandlimath, T.R.; Sathiyanarayanan, K.I. Facile synthesis of ZnAl2O4 nanoparticles: Efficient and reusable porous nano ZnAl2O4 and copper supported on ZnAl2O4 catalysts for one pot green synthesis of propargylamines and imidazo[1,2-a] pyridines by A3 coupling reactions. RSC Adv. 2016, 6, 3117–3125. [Google Scholar] [CrossRef]
- Panda, J.; Raiguru, B.P.; Mishra, M.; Mohapatra, S.; Nayak, S. Recent advances in the synthesis of imidazo[1,2-a] pyridines: A brief review. ChemistrySelect 2022, 7, e202103987. [Google Scholar]
- Durka, J.; Turkowska, J.; Gryko, D. Lightening diazo compounds? ACS Sustain. Chem. Eng. 2021, 9, 8895–8918. [Google Scholar] [CrossRef]
- Bagdi, A.K.; Rahman, M.; Bhattacherjee, D.; Zyryanov, G.V.; Ghosh, S.; Chupakhin, O.N.; Hajra, A. Visible light promoted cross-dehydrogenative coupling: A decade update. Green Chem. 2020, 22, 6632–6681. [Google Scholar] [CrossRef]
- Bagdi, A.K.; Hajra, A. Visible light promoted C–H functionalization of imidazoheterocycles. Org. Biomol. Chem. 2020, 18, 2611–2631. [Google Scholar] [CrossRef]
- Uygur, M.; García Mancheño, O. Visible light-mediated organophotocatalyzed C–H bond functionalization reactions. Org. Biomol. Chem. 2019, 17, 5475–5489. [Google Scholar] [PubMed]
- Bogdos, M.K.; Pinard, E.; Murphy, J.A. Applications of organocatalysed visible-light photoredox reactions for medicinal chemistry. Beilstein J. Org. Chem. 2018, 14, 2035–2064. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.-H.; Chen, M.; Feng, Z.-W.; Zhang, Y.; Wang, J.; Jiang, Y.-Q.; Yu, B. Functionalization of imidazo[1,2-a] pyridines via radical reactions. New J. Chem. 2021, 45, 9302–9314. [Google Scholar] [CrossRef]
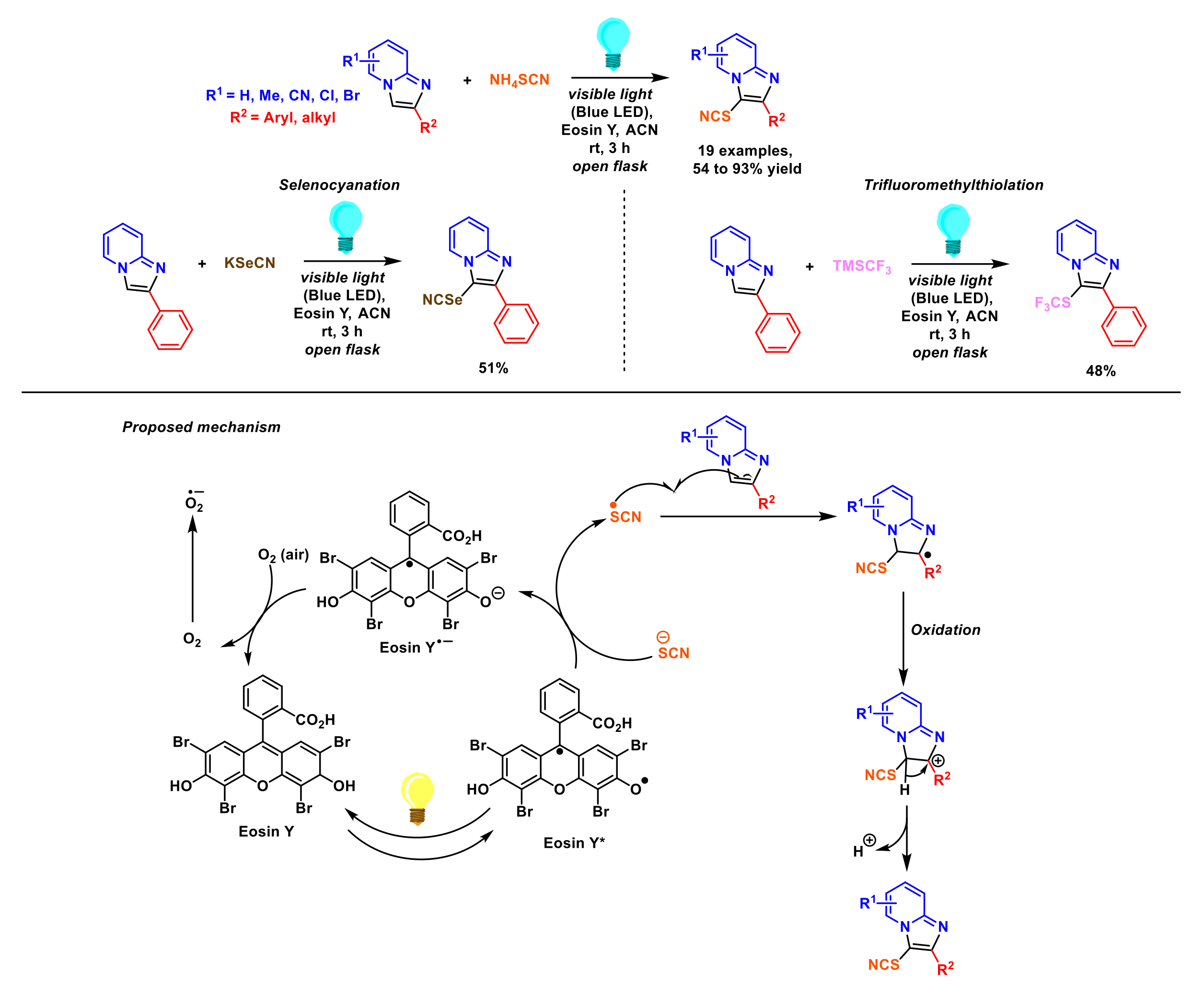
- Karmaker, P.G.; Alam, M.A.; Huo, F. Recent advances in photochemical and electrochemically induced thiocyanation: A greener approach for SCN-containing compound formation. RSC Adv. 2022, 12, 6214–6233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lei, A. Visible-light-induced C–H functionalization and C–C/C–X bond-forming oxidative cross-coupling reactions. Asian J. Org. Chem. 2018, 7, 1164–1177. [Google Scholar] [CrossRef]
- Srivastava, V.; Singh, P.K.; Singh, P.P. Recent advances of visible-light photocatalysis in the functionalization of organic compounds. J. Photochem. Photobiol. 2022, 50, 100488. [Google Scholar]
- Xiang Liu, W.L.; Liu, H.; Cao, H. Application on the construction of imidazo[1,2-a]pyridines C-3 canbon-hetero bonds by visible-light catalysis and electrochemistry. Chin. J. Org. Chem. 2021, 41, 1759–1773. [Google Scholar]
- Srivastava, A.; Singh, P.K.; Ali, A.; Singh, P.P.; Srivastava, V. Recent applications of rose bengal catalysis in N-heterocycles: A short review. RSC Adv. 2020, 10, 39495–39508. [Google Scholar] [CrossRef]
- Shi, L.; Li, T.; Mei, G.-J. Recent advances in transition-metal-free C–H functionalization of imidazo[1,2-a] pyridines. Green Synth. Catal. 2022; in press. [Google Scholar] [CrossRef]
- Kumar Hota, S.; Jinan, D.; Prakash Panda, S.; Pan, R.; Sahoo, B.; Murarka, S. Organophotoredox-catalyzed late-stage functionalization of heterocycles. Asian J. Org. Chem. 2021, 10, 1848–1860. [Google Scholar] [CrossRef]
- Kurteva, V. Recent progress in metal-free direct synthesis of imidazo[1,2-a] pyridines. ACS Omega 2021, 6, 35173–35185. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Sharma, A. Recent advances in photocatalytic manipulations of rose bengal in organic synthesis. Org. Biomol. Chem. 2019, 17, 4384–4405. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Ghosh, M.; Mishra, S.; Hajra, A. Metal-Free Thiocyanation of imidazoheterocycles through visible light photoredox catalysis. J. Org. Chem. 2015, 80, 8275–8281. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef]
- Barata-Vallejo, S.; Postigo, A. New visible-light-triggered photocatalytic trifluoromethylation reactions of carbon–carbon multiple bonds and (hetero)aromatic compounds. Chem. Eur. J. 2020, 26, 11065–11084. [Google Scholar] [CrossRef]
- Mi, X.; Kong, Y.; Yang, H.; Zhang, J.; Pi, C.; Cui, X. Visible-light-promoted metal-free C-H trifluoromethylation of imidazopyridines. Eur. J. Org. Chem. 2020, 8, 1019–1022. [Google Scholar] [CrossRef]
- Lefebvre, Q.; Hoffmann, N.; Rueping, M. Photoorganocatalysed and visible light photoredox catalysed trifluoromethylation of olefins and (hetero)aromatics in batch and continuous flow. Chem. Commun. 2016, 52, 2493–2496. [Google Scholar] [CrossRef]
- Zhou, Q.; Xu, S.; Zhang, R. Transition-metal-free, visible-light-mediated regioselective C–H trifluoromethylation of imidazo[1,2-a] pyridines. Tetrahedron Lett. 2019, 60, 734–738. [Google Scholar] [CrossRef]
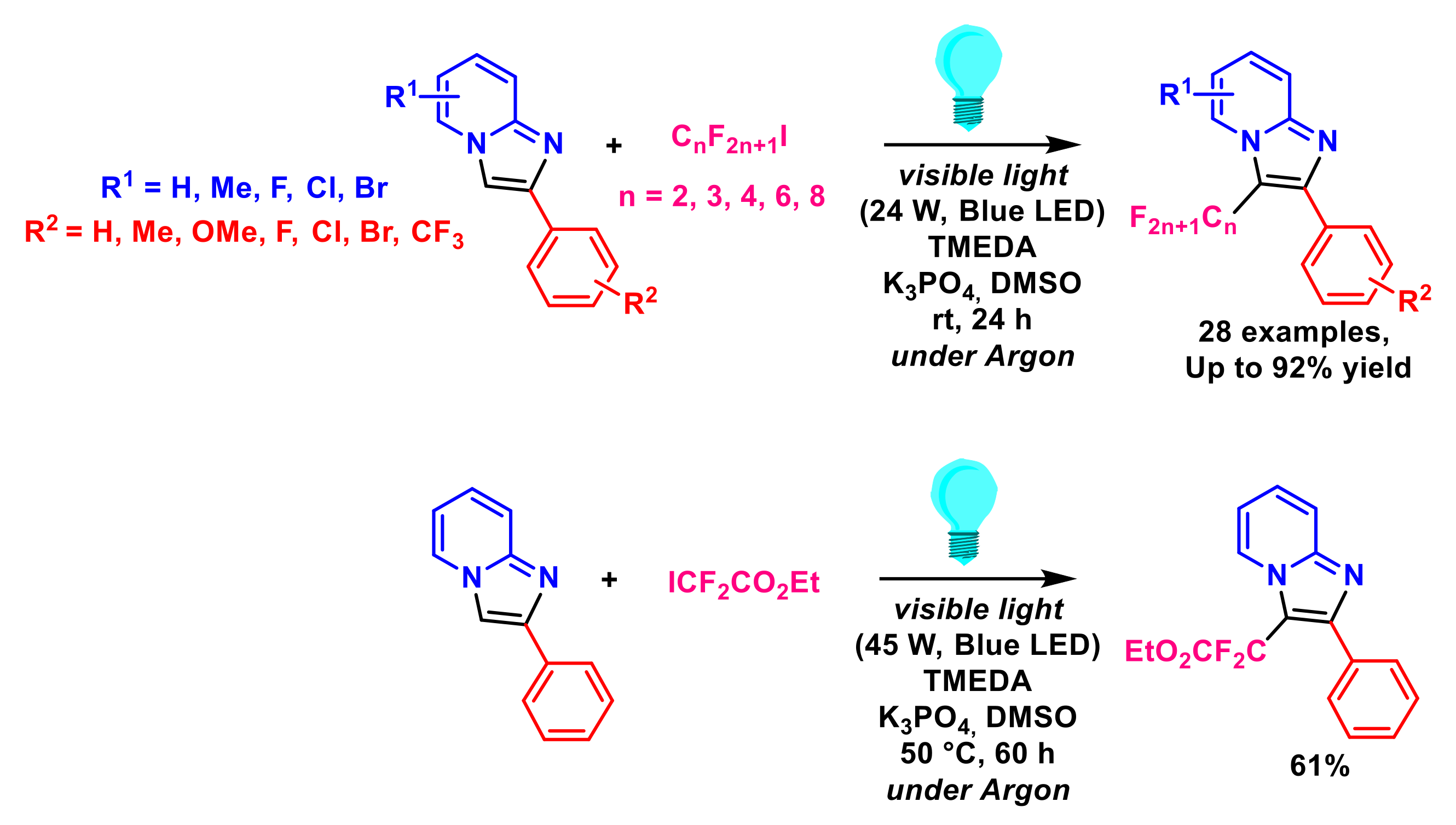
- Li, M.; Li, G.; Dai, C.; Zhou, W.; Zhan, W.; Gao, M.; Rong, Y.; Tan, Z.; Deng, W. Visible-light-promoted direct C3-trifluoromethylation and perfluoroalkylation of imidazopyridines. Org. Biomol. Chem. 2021, 19, 8301–8306. [Google Scholar] [CrossRef]
- Li, L.; Chen, X.; Pei, C.; Li, J.; Zou, D.; Wu, Y.; Wu, Y. Visible-light-mediated direct C-H perfluoroalkylation of imidazoheterocycles. Tetrahedron Lett. 2021, 83, 153407. [Google Scholar] [CrossRef]
- Zhu, M.; Han, X.; Fu, W.; Wang, Z.; Ji, B.; Hao, X.-Q.; Song, M.-P.; Xu, C. Regioselective 2,2,2-trifluoroethylation of imidazopyridines by visible light photoredox catalysis. J. Org. Chem. 2016, 81, 7282–7287. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wu, D.; Bai, X.; Cai, P.; Zhu, W.-G. Catalyst-free, visible-light-induced direct radical cross-coupling perfluoroalkylation of the imidazo[1,2-a] pyridines with perfluoroalkyl iodides. New J. Chem. 2021, 45, 4925–4929. [Google Scholar] [CrossRef]
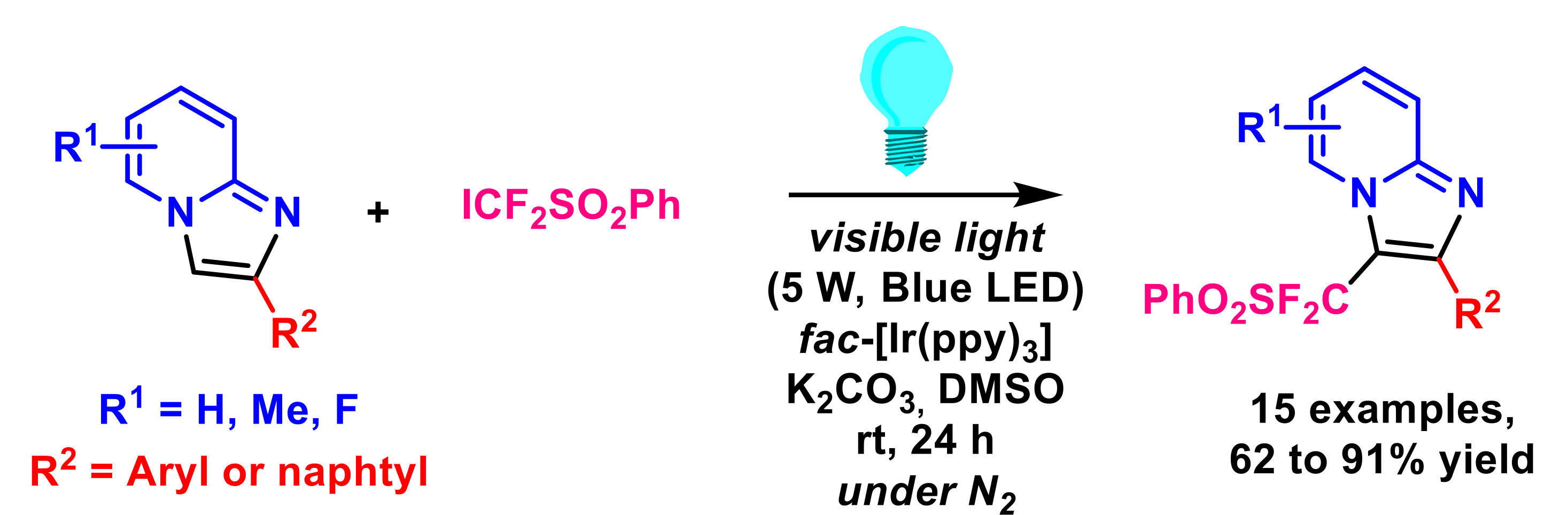
- Yin, G.; Zhu, M.; Fu, W. Visible-light mediated regioselective (phenylsulfonyl)difluoromethylation of fused imidazoles with iododifluoromethyl phenyl sulfone. Heterocycl. Commun. 2017, 23, 275–279. [Google Scholar] [CrossRef]
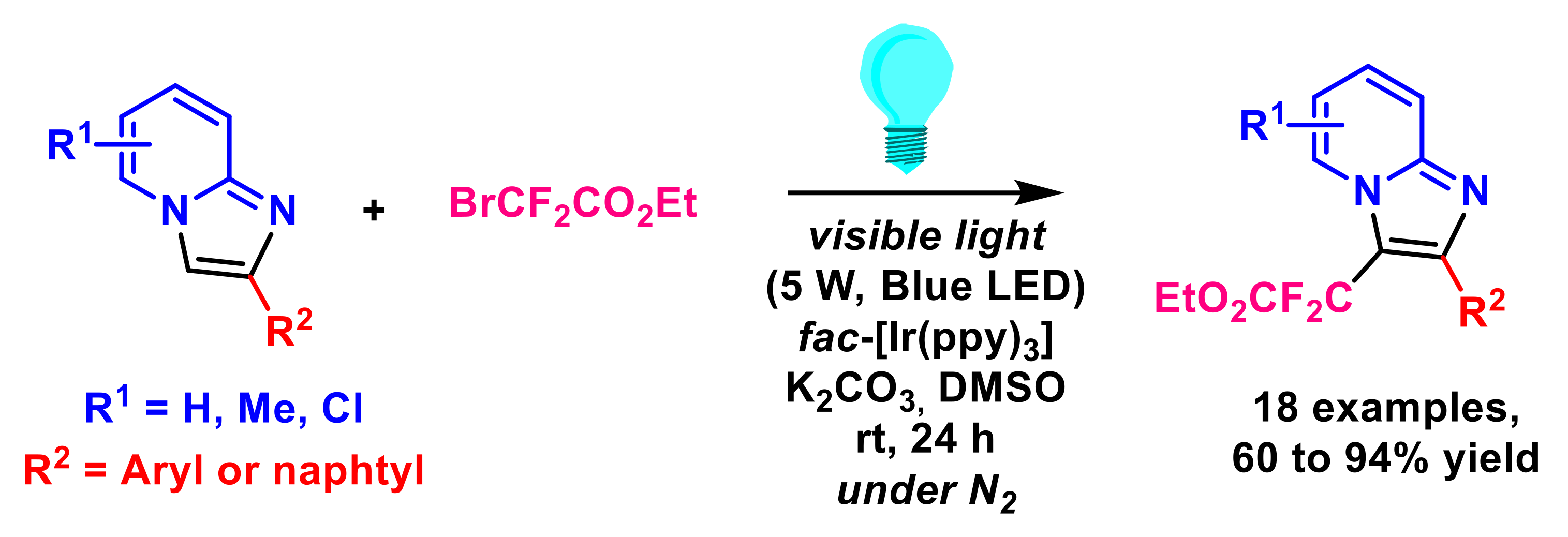
- Yin, G.; Zhu, M.; Fu, W. Visible-light-induced photocatalytic difluoroacetylation of imidazopyridines via direct and regioselective CH functionalization. J. Fluor. Chem. 2017, 199, 14–19. [Google Scholar] [CrossRef]
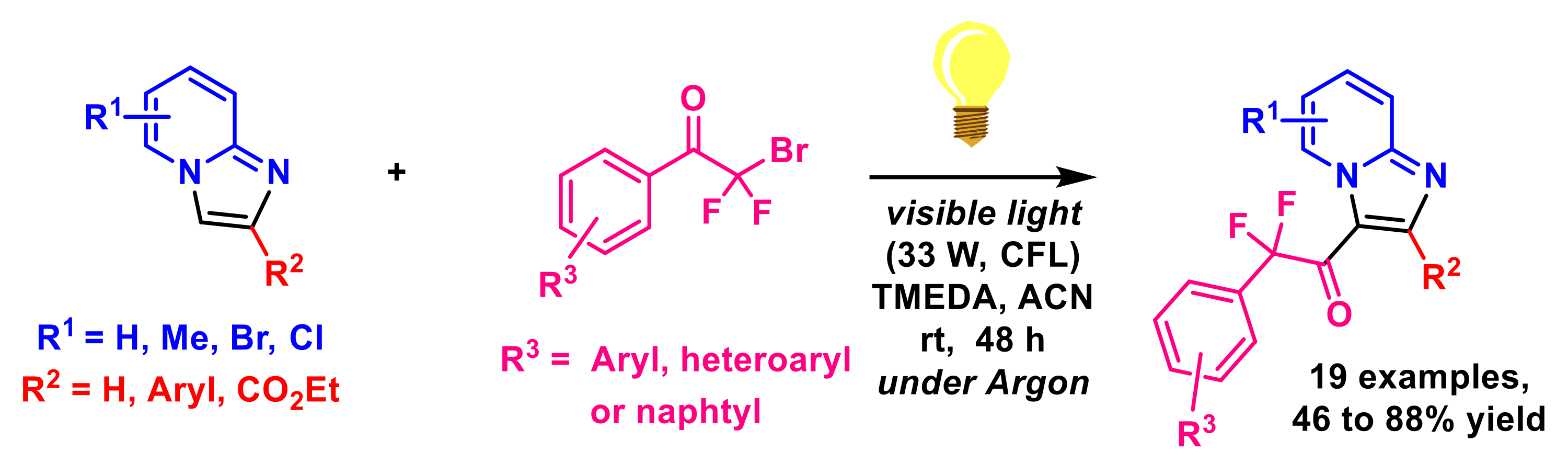
- Qu, C.-H.; Song, G.-T.; Xu, J.; Yan, W.; Zhou, C.-H.; Li, H.-Y.; Chen, Z.-Z.; Xu, Z.-G. Merging visible light with cross-coupling: The photochemical direct C–H difluoroalkylation of imidazopyridines. Org. Lett. 2019, 21, 8169–8173. [Google Scholar] [CrossRef]
- Singsardar, M.; Mondal, S.; Laru, S.; Hajra, A. Organophotoredox-catalyzed C(sp2)–H difluoromethylenephosphonation of imidazoheterocycles. Org. Lett. 2019, 21, 5606–5610. [Google Scholar] [CrossRef]
- Chang, Q.; Liu, Z.; Liu, P.; Yu, L.; Sun, P. Visible-light-induced regioselective cyanomethylation of imidazopyridines and its application in drug synthesis. J. Org. Chem. 2017, 82, 5391–5397. [Google Scholar] [CrossRef]
- Nair, D.K.; Mobin, S.M.; Namboothiri, I.N.N. Synthesis of imidazopyridines from the morita–baylis–hillman acetates of nitroalkenes and convenient access to alpidem and zolpidem. Org. Lett. 2012, 14, 4580–4583. [Google Scholar] [CrossRef]
- Kibriya, G.; Bagdi, A.K.; Hajra, A. Visible-light-promoted C(sp3)–C(sp2) cross-dehydrogenative coupling of tertiary amine with imidazopyridine. J. Org. Chem. 2018, 83, 10619–10626. [Google Scholar] [CrossRef]
- Shi, T.; Sun, K.; Chen, X.-L.; Zhang, Z.-X.; Huang, X.-Q.; Peng, Y.-Y.; Qu, L.-B.; Yu, B. Recyclable perovskite as heterogeneous photocatalyst for aminomethylation of imidazo-fused heterocycles. Adv. Synth. Catal. 2020, 362, 2143–2149. [Google Scholar] [CrossRef]
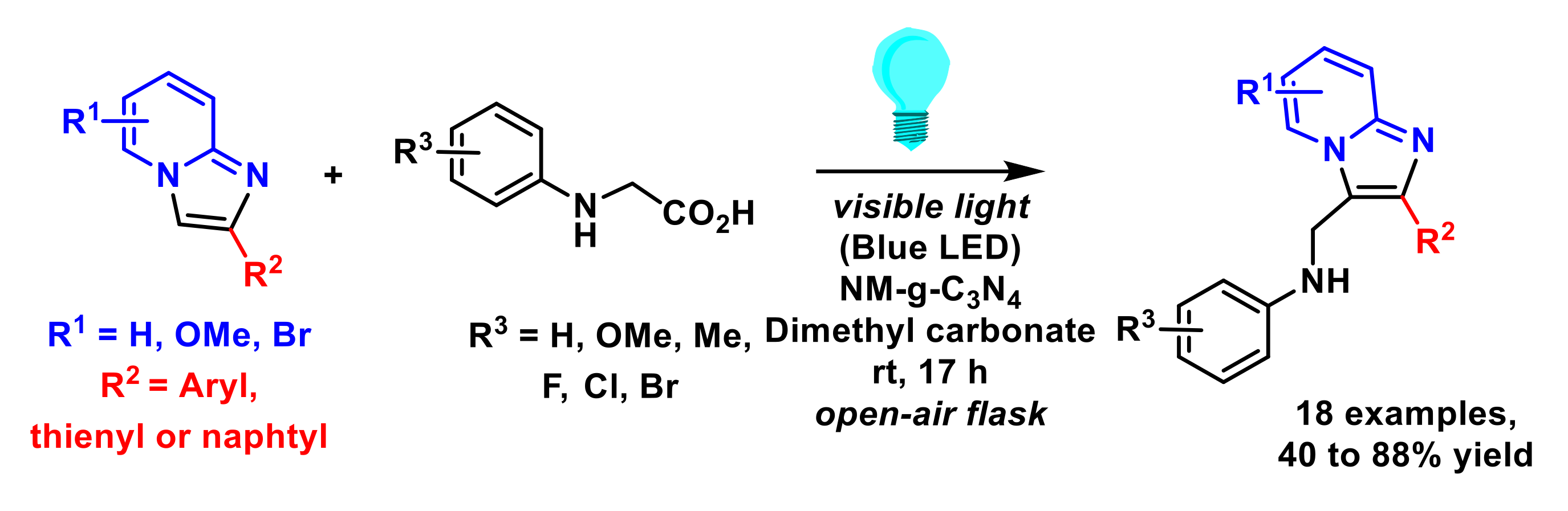
- Shi, T.; Liu, Y.-T.; Wang, S.-S.; Lv, Q.-Y.; Yu, B. Recyclable carbon nitride nanosheet-photocatalyzed aminomethylation of imidazo[1,2-a] pyridines in green solvent. Chin. J. Chem. 2022, 40, 97–103. [Google Scholar] [CrossRef]
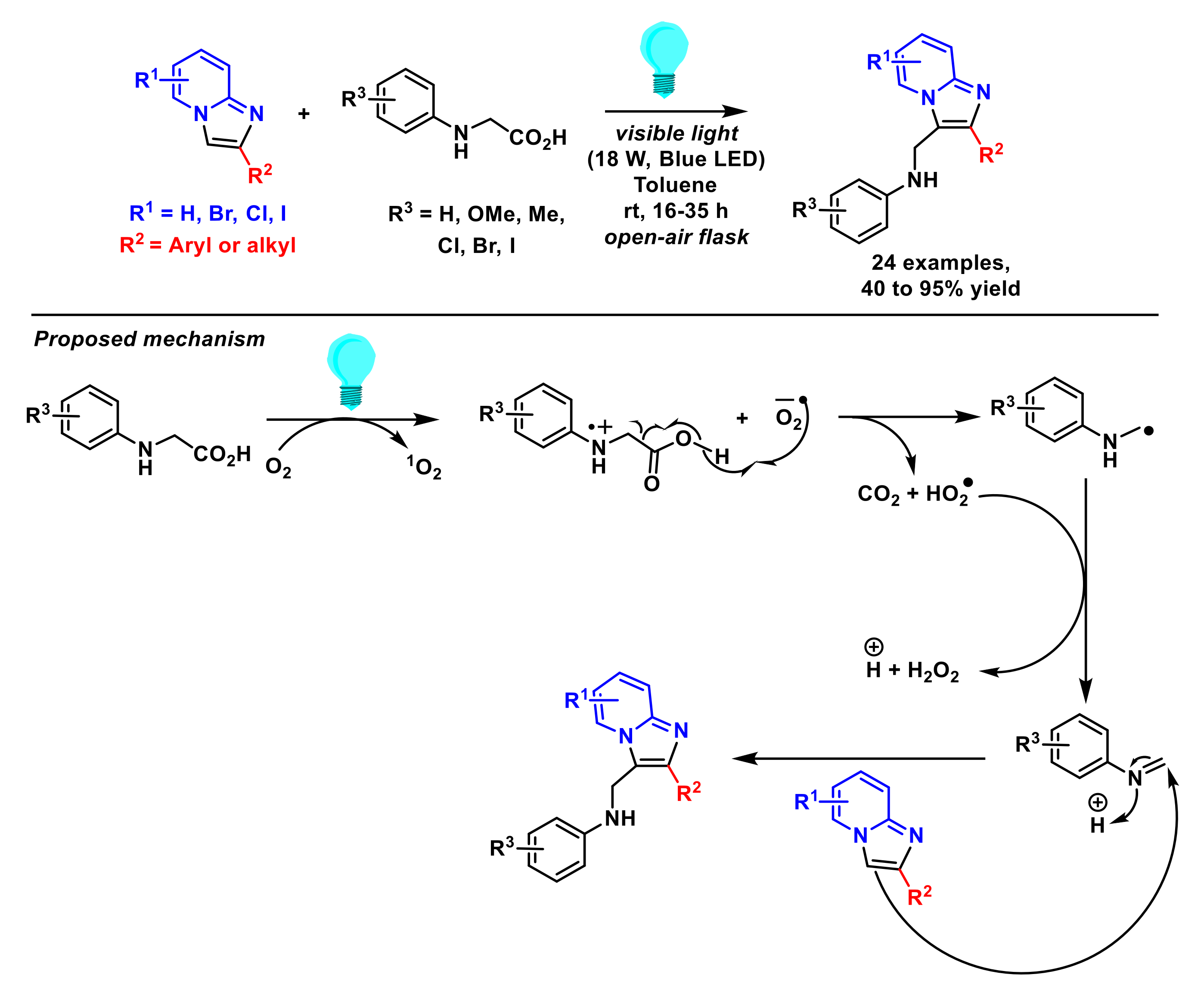
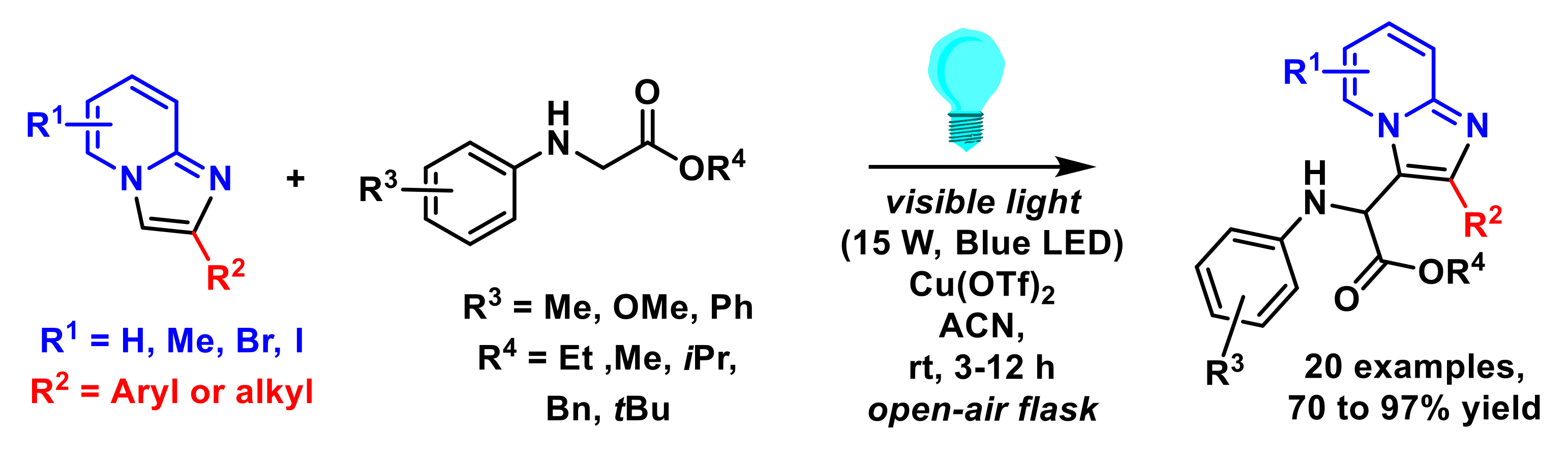
- Ji, J.-J.; Zhu, Z.-Q.; Xiao, L.-J.; Guo, D.; Zhu, X.; Tang, J.; Wu, J.; Xie, Z.-B.; Le, Z.-G. Photocatalyst-free decarboxylative aminoalkylation of imidazo[1,2-a] pyridines with N-aryl glycines enabled by visible light. Org. Chem. Front. 2019, 6, 3693–3697. [Google Scholar] [CrossRef]
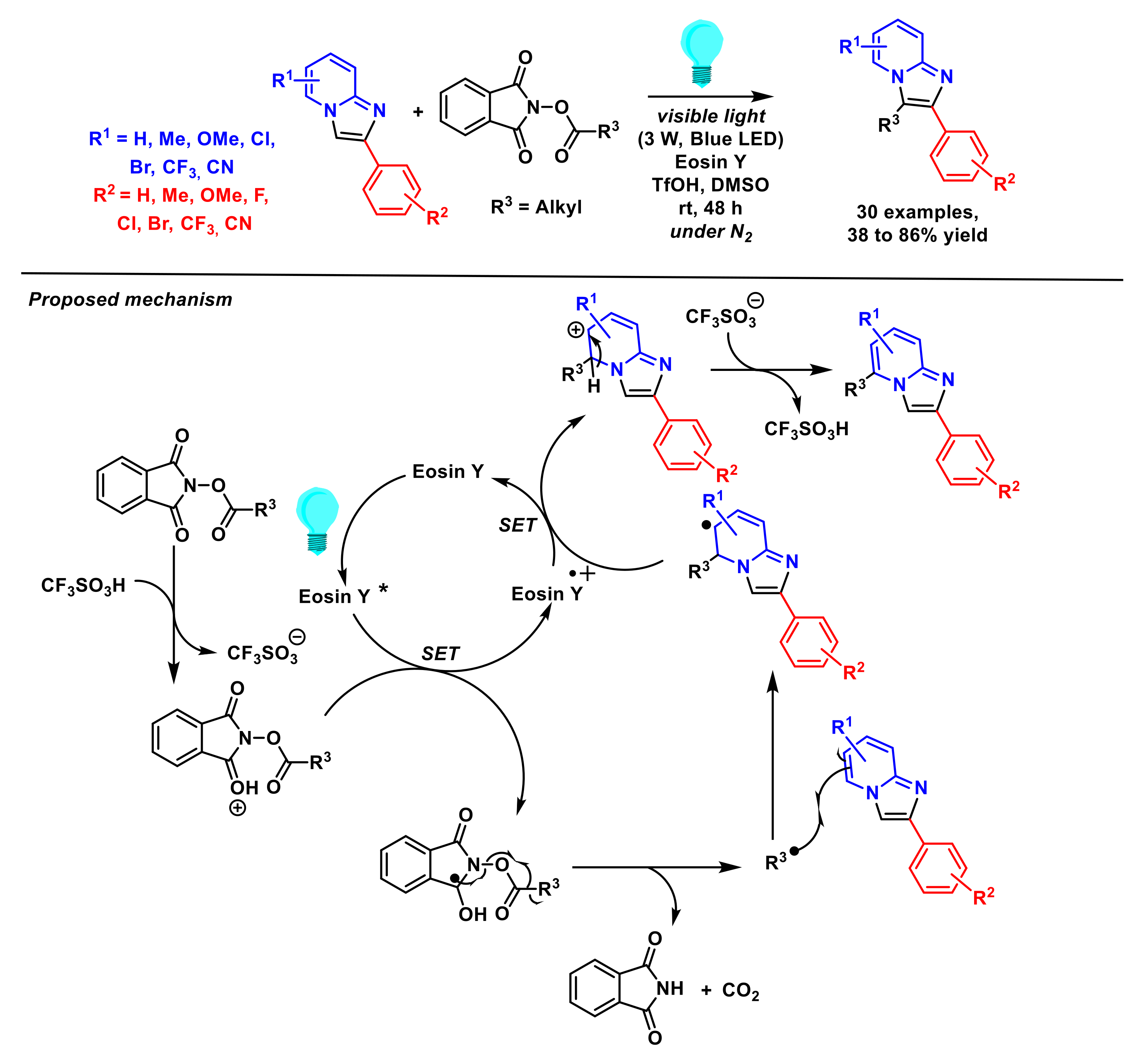
- Sun, B.; Xu, T.; Zhang, L.; Zhu, R.; Yang, J.; Xu, M.; Jin, C. Metal-free regioselective alkylation of imidazo[1,2-a] pyridines with N-hydroxyphthalimide esters under organic photoredox catalysis. Synlett 2020, 31, 363–368. [Google Scholar]
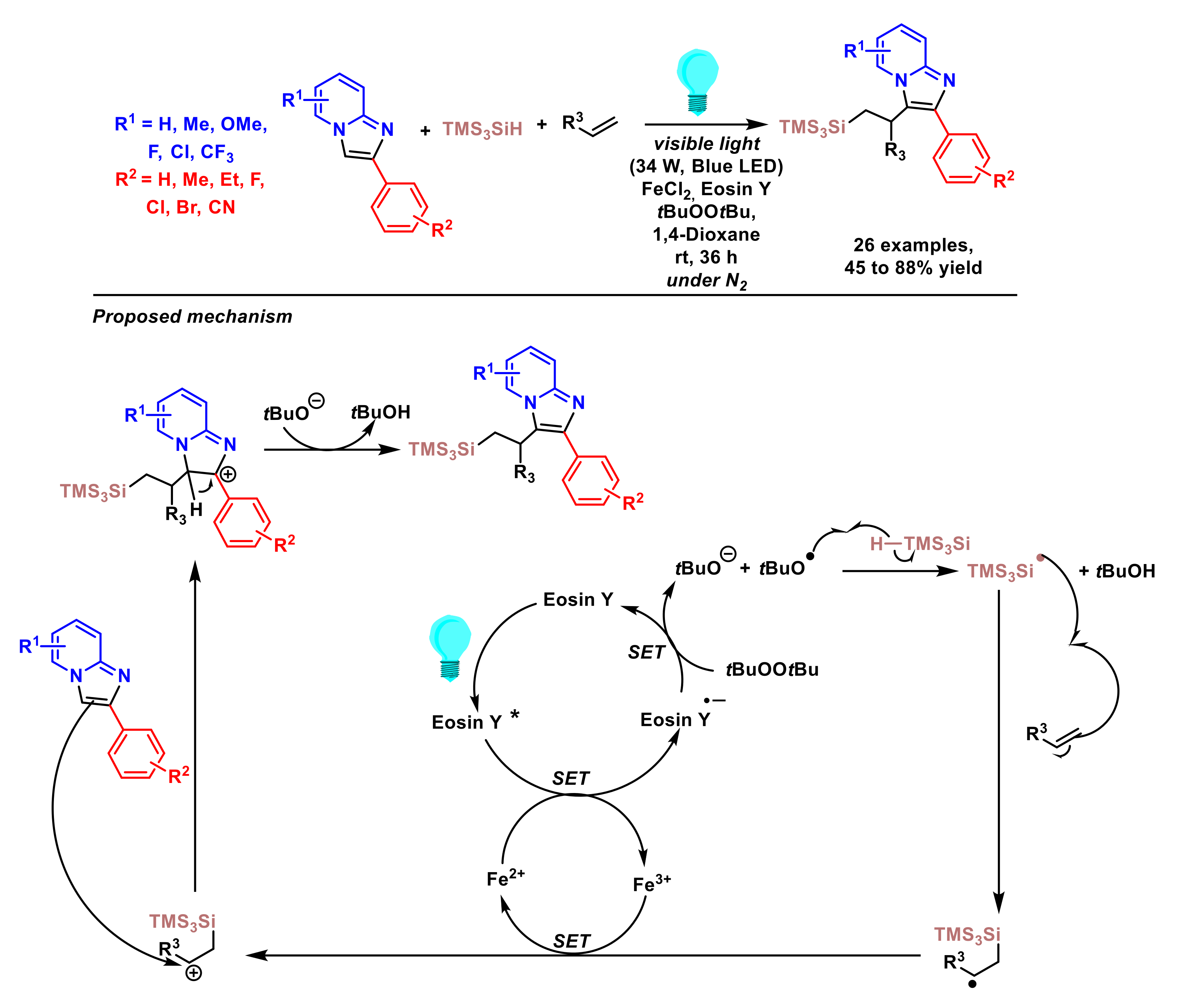
- Neogi, S.; Kumar Ghosh, A.; Mandal, S.; Ghosh, D.; Ghosh, S.; Hajra, A. Three-component carbosilylation of alkenes by merging iron and visible-light photocatalysis. Org. Lett. 2021, 23, 6510–6514. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.-Q.; Xiao, L.-J.; Zhou, C.-C.; Song, H.-L.; Xie, Z.-B.; Le, Z.-G. A visible-light-promoted cross-dehydrogenative-coupling reaction of N-arylglycine esters with imidazo[1,2-a] pyridines. Tetrahedron Lett. 2018, 59, 3326–3331. [Google Scholar] [CrossRef]
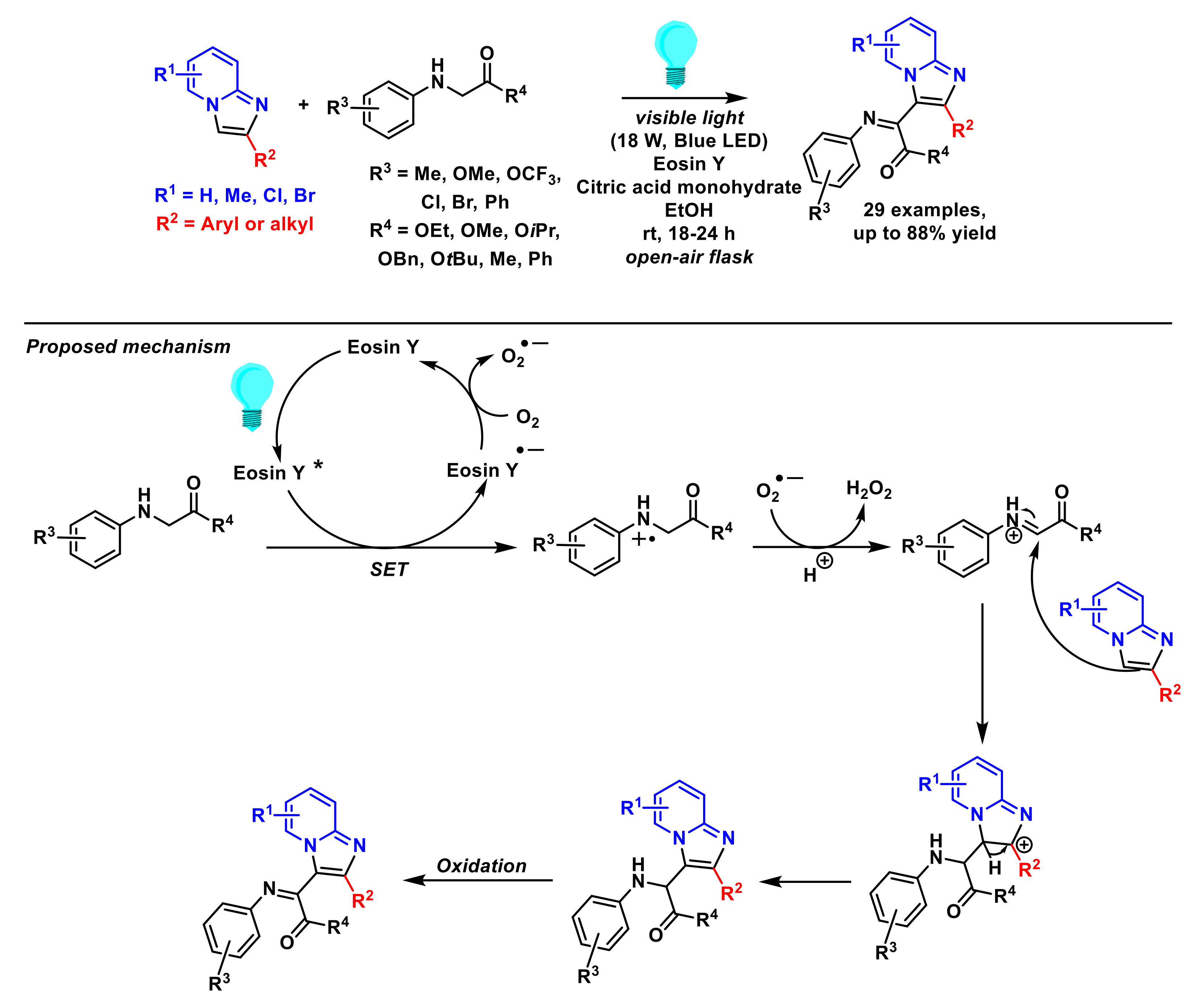
- Zhu, Z.-Q.; Guo, D.; Ji, J.-J.; Zhu, X.; Tang, J.; Xie, Z.-B.; Le, Z.-G. Visible-light-induced dehydrogenative imidoylation of imidazo[1,2-a] pyridines with α-amino acid derivatives and α-amino ketones. J. Org. Chem. 2020, 85, 15062–15071. [Google Scholar] [CrossRef]
- Ma, C.-H.; Zhao, L.; He, X.; Jiang, Y.-Q.; Yu, B. Visible-light-induced direct 3-ethoxycarbonylmethylation of 2-aryl-2H-indazoles in water. Org. Chem. Front. 2022, 9, 1445–1450. [Google Scholar] [CrossRef]
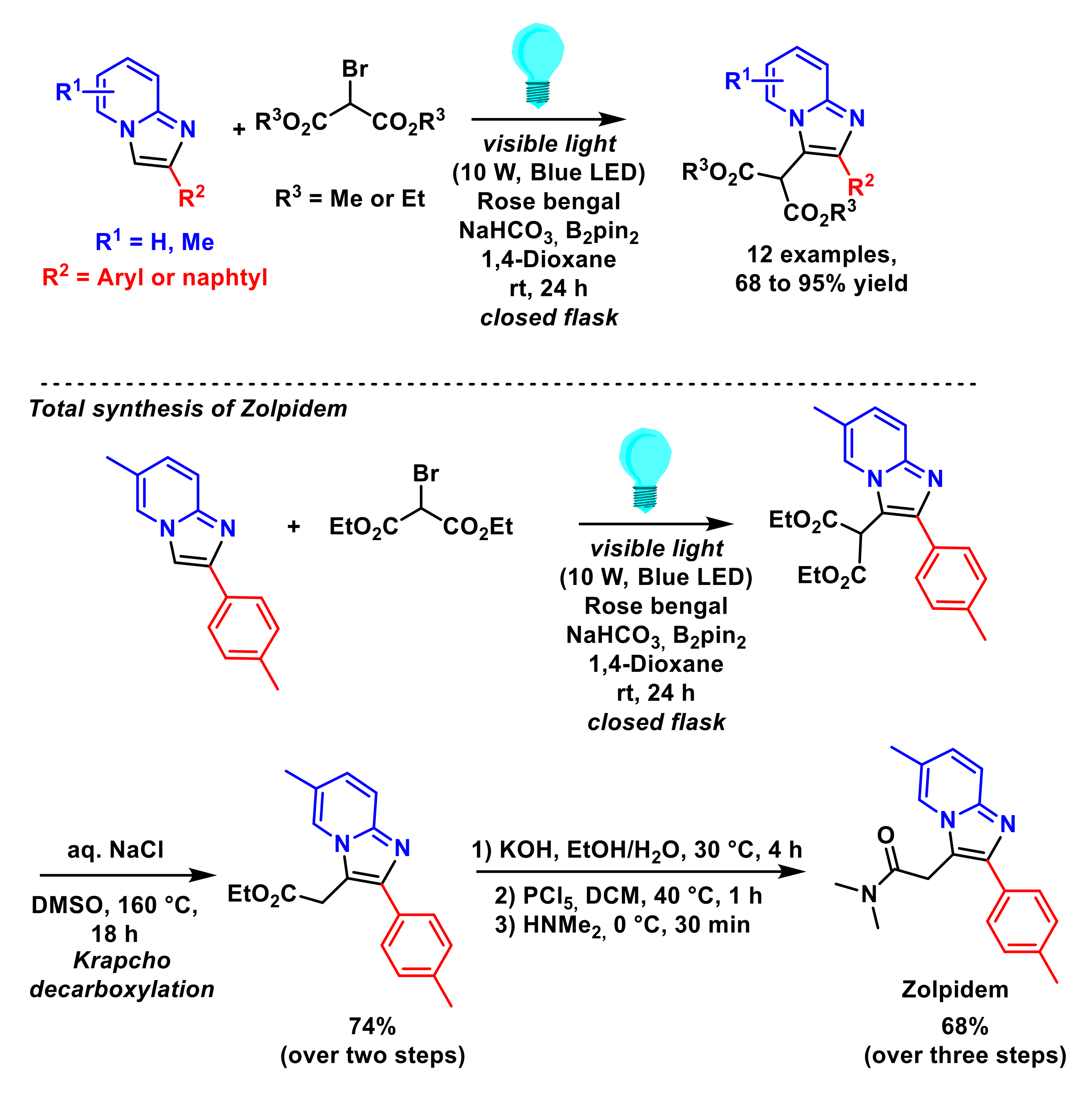
- Chaubey, N.R.; Kapdi, A.R.; Maity, B. Organophotoredox-catalyzed C–H alkylation of imidazoheterocycles with malonates: Total synthesis of zolpidem. Synthesis 2021, 53, 1524–1530. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Laru, S.; Samanta, S.; Singsardar, M.; Hajra, A. Visible light-induced photocatalytic C–H ethoxycarbonylmethylation of imidazoheterocycles with ethyl diazoacetate. RSC Adv. 2020, 10, 27984–27988. [Google Scholar] [CrossRef]
- Xiao, Y.; Yu, L.; Yu, Y.; Tan, Z.; Deng, W. Visible-light-mediated C3-ethoxycarbonylmethylation of imidazo[1,2-a] pyridines and convenient access to Zolpidem. Tetrahedron Lett. 2020, 61, 152606. [Google Scholar] [CrossRef]
- Tang, F.; Guan, Z.; He, Y.-H. Metal-free regioselective carbonylation of imidazo[1,2-a] pyridines via photoredox catalysis using nitrones. Asian J. Org. Chem. 2019, 8, 867–872. [Google Scholar] [CrossRef]
- Mi, X.; Kong, Y.; Zhang, J.; Pi, C.; Cui, X. Visible-light-promoted sulfonylmethylation of imidazopyridines. Chin. Chem. Lett. 2019, 30, 2295–2298. [Google Scholar] [CrossRef]
- Kibriya, G.; Bagdi, A.K.; Hajra, A. Visible light induced tetramethylethylenediamine assisted formylation of imidazopyridines. Org. Biomol. Chem. 2018, 16, 3473–3478. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Pi, C.; Cui, X.; Wu, Y. Divergent C(sp2)–H arylation of heterocycles via organic photoredox catalysis. Green Chem. 2022, 24, 3017–3022. [Google Scholar] [CrossRef]
- Chang, Q.; Wu, Z.; Yu, L.; Liu, P.; Sun, P. Visible-light-mediated C3-azolylation of imidazo[1,2-a] pyridines with 2-bromoazoles. Org. Biomol. Chem. 2017, 15, 5318–5324. [Google Scholar] [CrossRef]
- Hili, R.; Yudin, A.K. Making carbon-nitrogen bonds in biological and chemical synthesis. Nat. Chem. Biol. 2006, 2, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Arockiam, P.B.; Guillemard, L.; Wencel-Delord, J. Regiodivergent visible light-induced C–H functionalization of quinolines at c-5 and c-8 under metal-, photosensitizer- and oxidant-free conditions. Adv. Synth. Catal. 2017, 359, 2571–2579. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Yamaguchi, E.; Itoh, A. Cross-dehydrogenative C–H amination of indoles under aerobic photo-oxidative conditions. Org. Lett. 2017, 19, 1282–1285. [Google Scholar] [CrossRef]
- Samanta, S.; Ravi, C.; Rao, S.N.; Joshi, A.; Adimurthy, S. Visible-light-promoted selective C–H amination of heteroarenes with heteroaromatic amines under metal-free conditions. Org. Biomol. Chem. 2017, 15, 9590–9594. [Google Scholar] [CrossRef]
- Chen, H.; Yi, H.; Tang, Z.; Bian, C.; Zhang, H.; Lei, A. External oxidant-free regioselective cross dehydrogenative coupling of 2-arylimidazoheterocycles and azoles with H2 evolution via photoredox catalysis. Adv. Synth. Catal. 2018, 360, 3220–3227. [Google Scholar] [CrossRef]
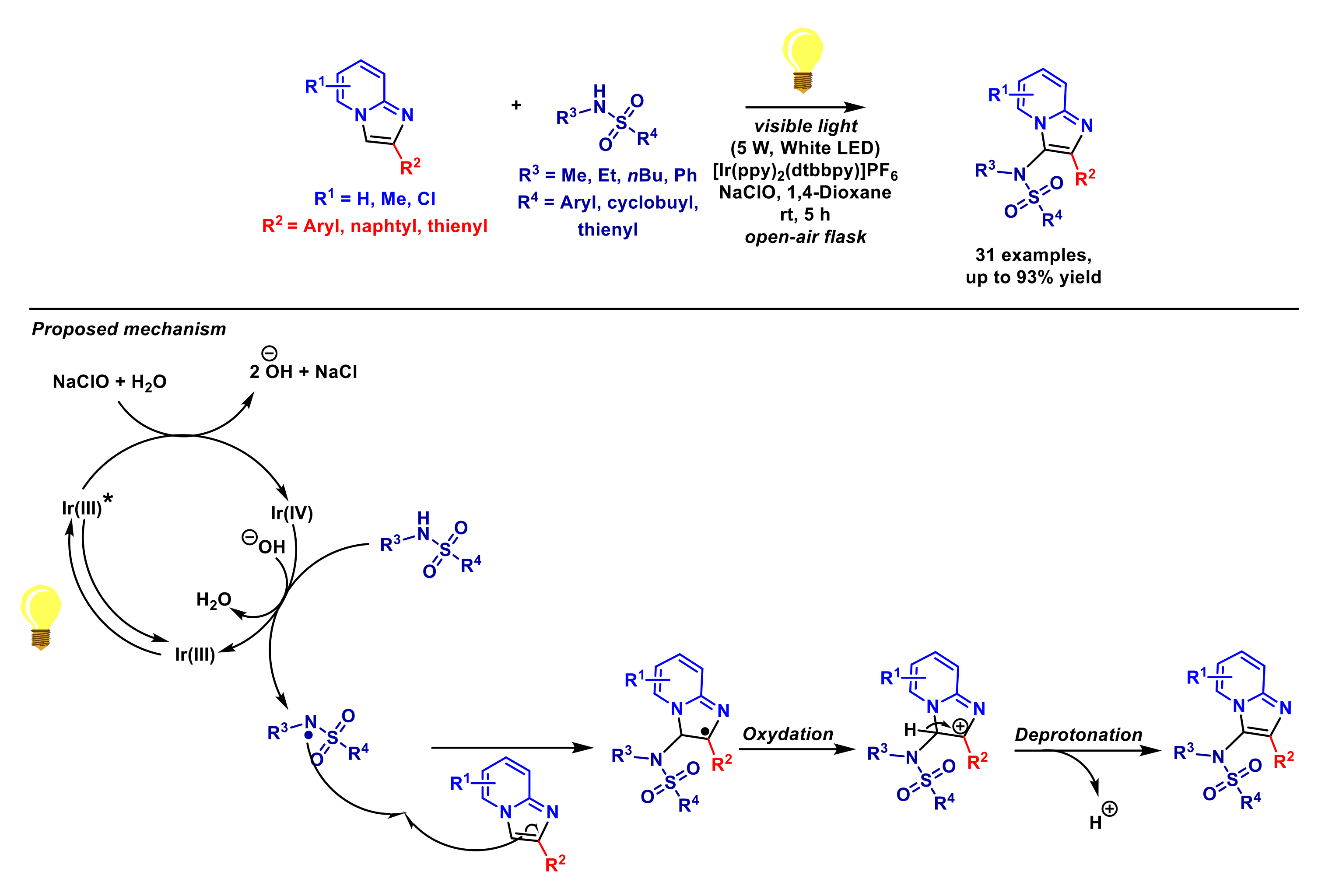
- Gao, Y.; Chen, S.; Lu, W.; Gu, W.; Liu, P.; Sun, P. Visible light-induced C3-sulfonamidation of imidazopyridines with sulfamides. Org. Biomol. Chem. 2017, 15, 8102–8109. [Google Scholar] [CrossRef] [PubMed]
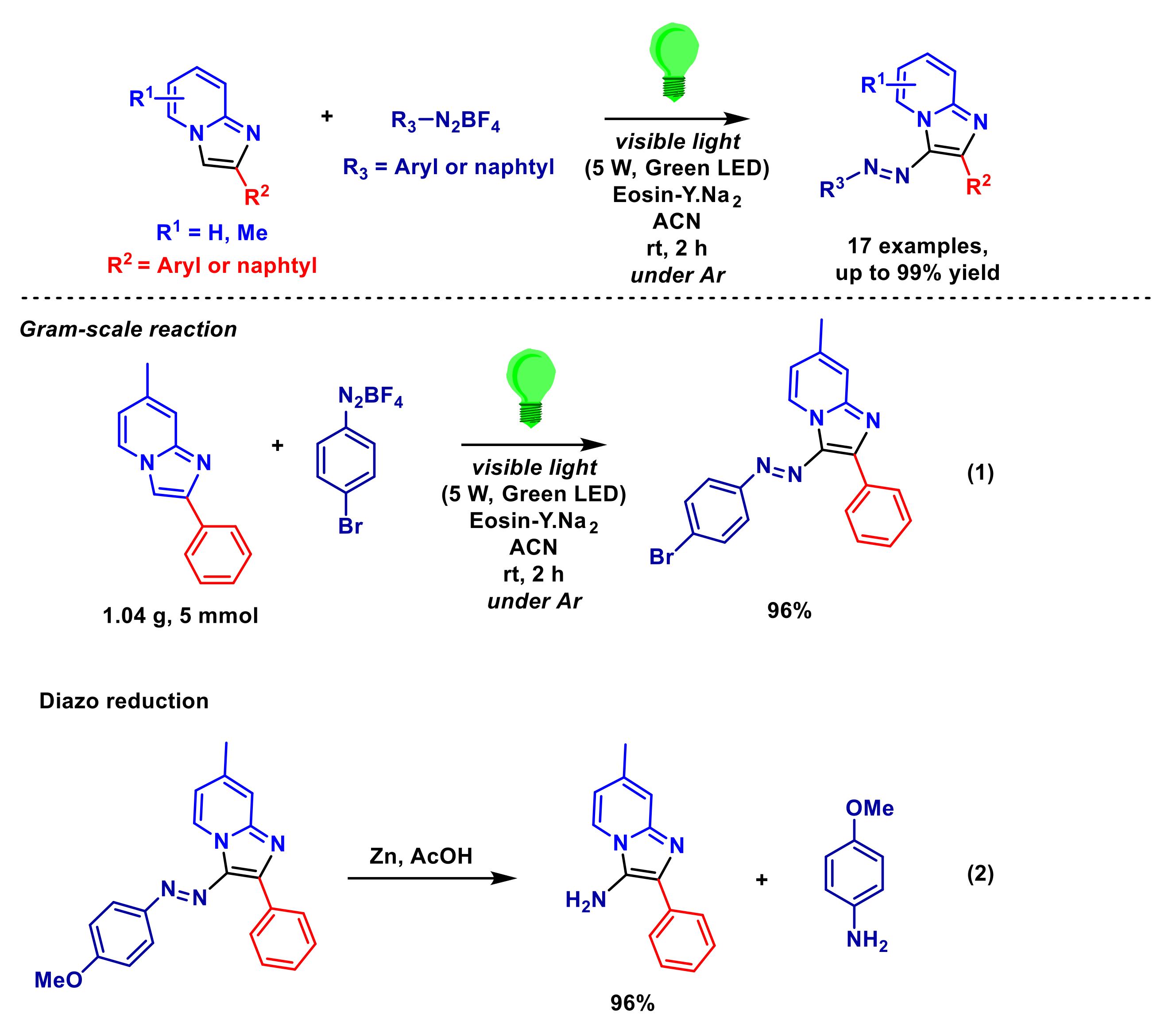
- Saba, S.; Dos Santos, C.R.; Zavarise, B.R.; Naujorks, A.A.S.; Franco, M.S.; Schneider, A.R.; Scheide, M.R.; Affeldt, R.F.; Rafique, J.; Braga, A.L. Photoinduced, direct C(sp2)–H bond azo coupling of imidazoheteroarenes and imidazoanilines with aryl diazonium salts catalyzed by Eosin Y. Chem. Eur. J. 2020, 26, 4461–4466. [Google Scholar] [CrossRef] [PubMed]
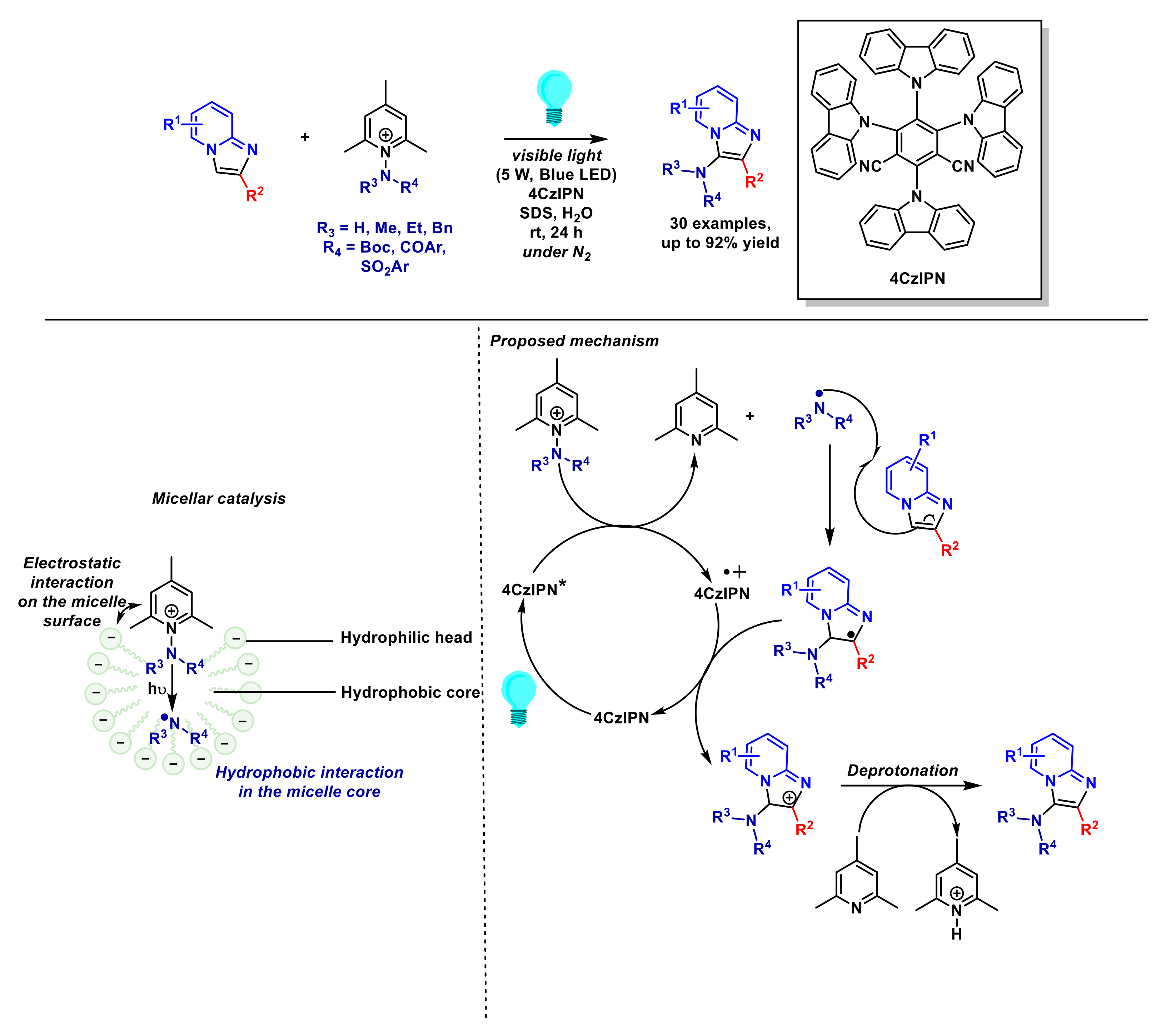
- Yang, Z.; Cao, K.; Peng, X.; Lin, L.; Fan, D.; Li, J.-L.; Wang, J.; Zhang, X.; Jiang, H.; Li, J. Micellar catalysis: Visible-light mediated imidazo[1,2-a] pyridine C–H amination with N-aminopyridinium salt accelerated by surfactant in water. Chin. J. Chem. 2021, 39, 3347–3352. [Google Scholar] [CrossRef]
- Kibriya, G.; Samanta, S.; Jana, S.; Mondal, S.; Hajra, A. Visible light organic photoredox-catalyzed C–H alkoxylation of imidazopyridine with alcohol. J. Org. Chem. 2017, 82, 13722–13727. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.S.; Shee, M.; Singh, A.K.; Paul, A.; Singh, N.D.P. Direct oxygenation of C–H bonds through photoredox and palladium catalysis. J. Org. Chem. 2020, 85, 3426–3439. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Sun, K.; Chen, X.-L.; Shi, T.; Li, X.-Y.; Qu, L.-B.; Zhao, Y.-F.; Yu, B. Visible-light-induced phosphorylation of imidazo-fused heterocycles under metal-free conditions. J. Org. Chem. 2020, 85, 14744–14752. [Google Scholar] [CrossRef]
- Jiang, C.-S.; Müller, W.E.G.; Schröder, H.C.; Guo, Y.-W. Disulfide- and multisulfide-containing metabolites from marine organisms. Chem. Rev. 2012, 112, 2179–2207. [Google Scholar] [CrossRef]
- Mann, G.; Baranano, D.; Hartwig, J.F.; Rheingold, A.L.; Guzei, I.A. Carbon–sulfur bond-forming reductive elimination involving sp-, sp2-, and sp3-hybridized carbon. Mechanism, steric effects, and electronic effects on sulfide formation. J. Am. Chem. Soc. 1998, 120, 9205–9219. [Google Scholar] [CrossRef]
- Arisawa, M.; Suzuki, T.; Ishikawa, T.; Yamaguchi, M. Rhodium-catalyzed substitution reaction of aryl fluorides with disulfides: P-orientation in the polyarylthiolation of polyfluorobenzenes. J. Am. Chem. Soc. 2008, 130, 12214–12215. [Google Scholar] [CrossRef]
- Correa, A.; Carril, M.; Bolm, C. Iron-catalyzed s-arylation of thiols with aryl iodides. Angew. Chem. Int. Ed. 2008, 47, 2880–2883. [Google Scholar] [CrossRef]
- Guo, W.; Tao, K.; Tan, W.; Zhao, M.; Zheng, L.; Fan, X. Recent advances in photocatalytic C–S/P–S bond formation via the generation of sulfur centered radicals and functionalization. Org. Chem. Front. 2019, 6, 2048–2066. [Google Scholar] [CrossRef]
- Sun, P.; Yang, D.; Wei, W.; Jiang, M.; Wang, Z.; Zhang, L.; Zhang, H.; Zhang, Z.; Wang, Y.; Wang, H. Visible light-induced C–H sulfenylation using sulfinic acids. Green Chem. 2017, 19, 4785–4791. [Google Scholar] [CrossRef]
- Rahaman, R.; Das, S.; Barman, P. Visible-light-induced regioselective sulfenylation of imidazopyridines with thiols under transition metal-free conditions. Green Chem. 2018, 20, 141–147. [Google Scholar] [CrossRef]
- Zhang, H.; Huang, Q.; Zhang, W.; Pan, C.; Wang, J.; Ai, C.; Tang, J.; Yu, G. Benzodithiophenedione-based conjugated microporous polymer catalysts for aerobic oxidation reactions driven by visible-light. ChemPhotoChem 2019, 3, 645–651. [Google Scholar] [CrossRef]
- Zeng, F.-L.; Zhu, H.-L.; Chen, X.-L.; Qu, L.-B.; Yu, B. Visible light-induced recyclable g-C3N4 catalyzed thiocyanation of C(sp2)–H bonds in sustainable solvents. Green Chem. 2021, 23, 3677–3682. [Google Scholar] [CrossRef]
- Breton-Patient, C.; Naud-Martin, D.; Mahuteau-Betzer, F.; Piguel, S. Three-component C–H bond sulfonylation of imidazoheterocycles by visible-light organophotoredox catalysis. Eur. J. Org. Chem. 2020, 42, 6653–6660. [Google Scholar] [CrossRef]
- Rafique, J.; Rampon, D.S.; Azeredo, J.B.; Coelho, F.L.; Schneider, P.H.; Braga, A.L. Light-mediated seleno-functionalization of organic molecules: Recent advances. Chem. Rec. 2021, 21, 2739–2761. [Google Scholar] [CrossRef]
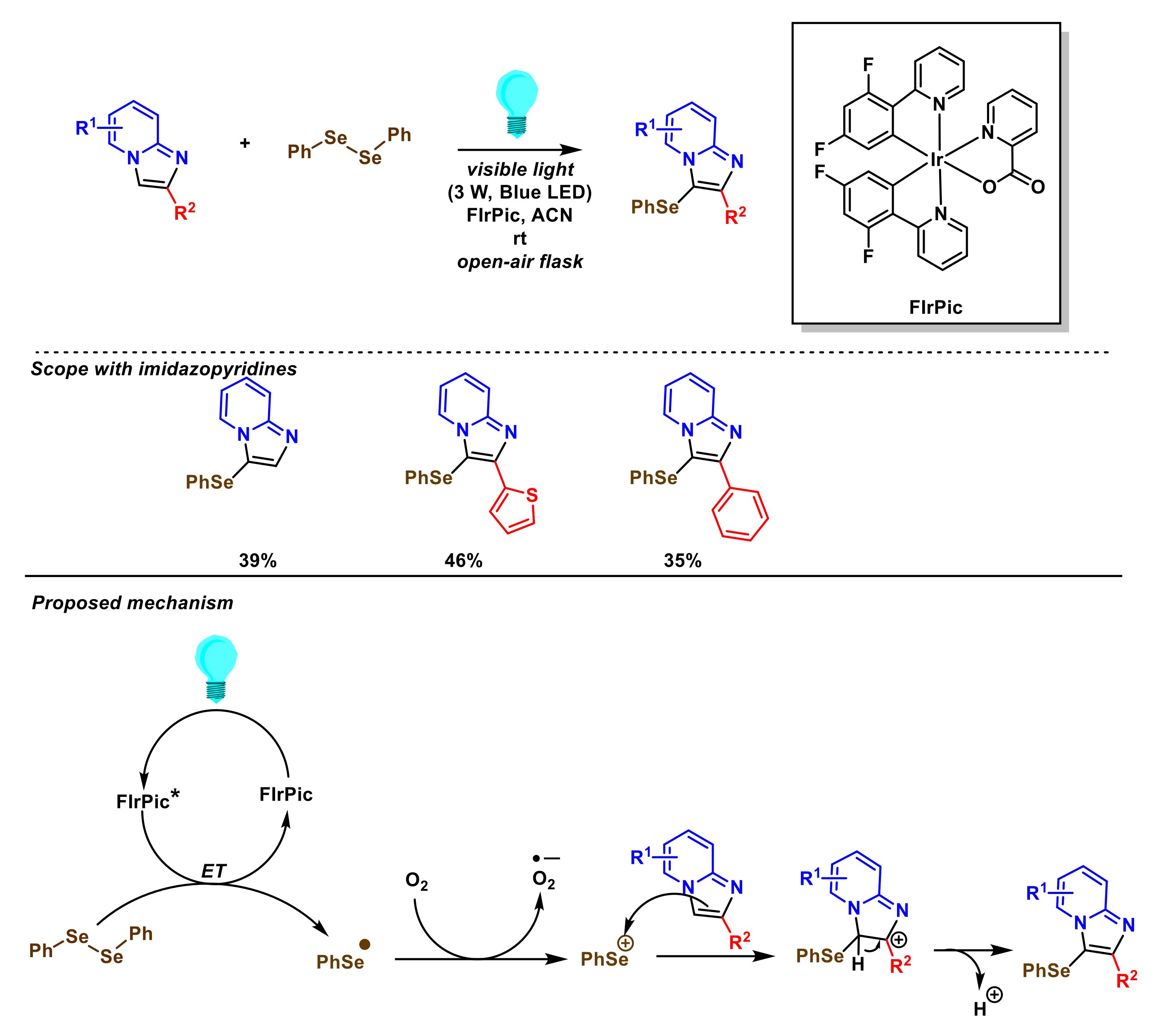
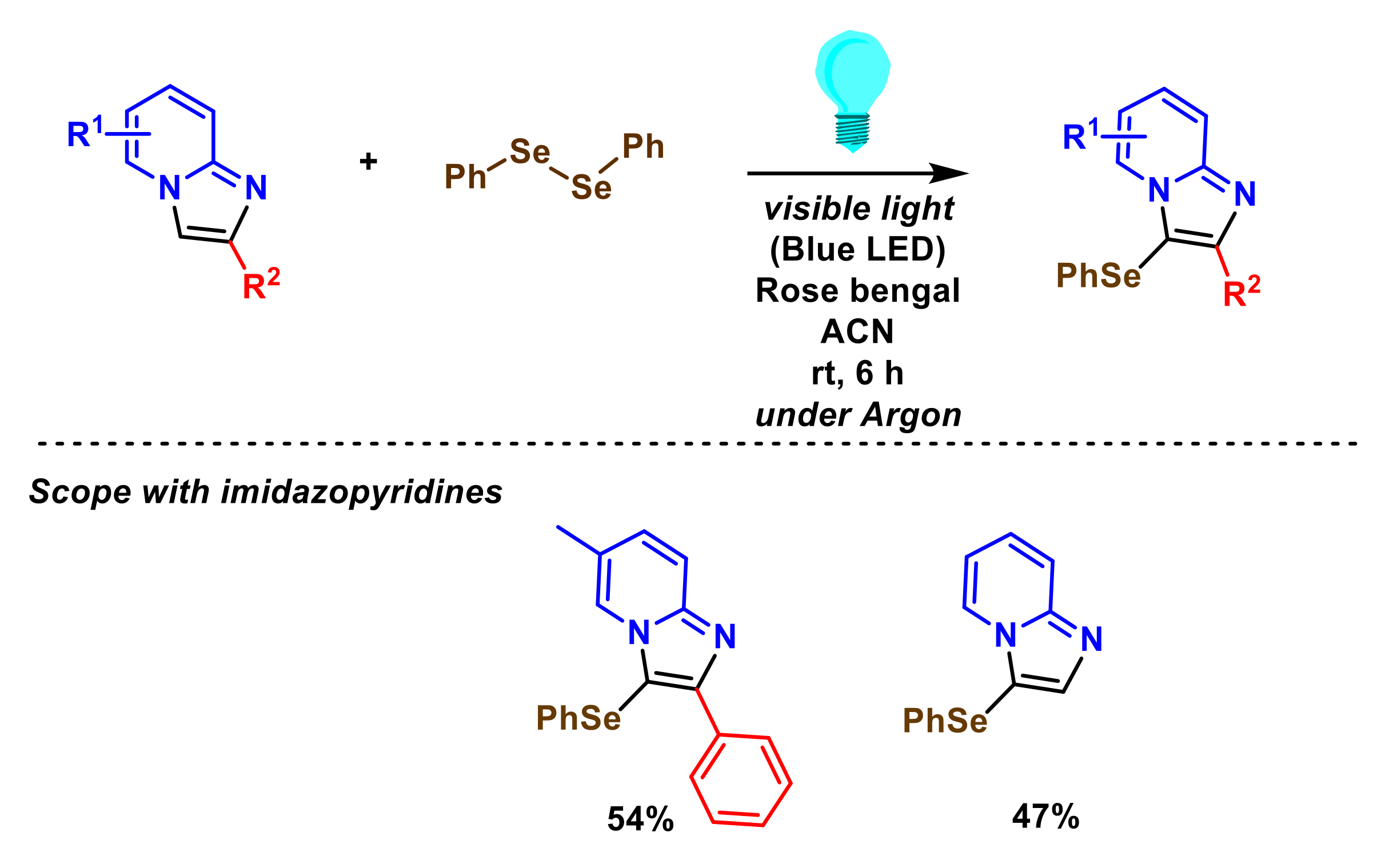
- Zhang, Q.-B.; Ban, Y.-L.; Yuan, P.-F.; Peng, S.-J.; Fang, J.-G.; Wu, L.-Z.; Liu, Q. Visible-light-mediated aerobic selenation of (hetero)arenes with diselenides. Green Chem. 2017, 19, 5559–5563. [Google Scholar] [CrossRef]
- Saba, S.; Rafique, J.; Franco, M.S.; Schneider, A.R.; Espíndola, L.; Silva, D.O.; Braga, A.L. Rose Bengal catalysed photo-induced selenylation of indoles, imidazoles and arenes: A metal free approach. Org. Biomol. Chem. 2018, 16, 880–885. [Google Scholar] [CrossRef]
- Kumaraswamy, G.; Ramesh, V.; Gangadhar, M.; Vijaykumar, S. Catalyst and sensitizer-free visible-light-induced C(sp2)–H chalcogenation of arenes/heteroarenes with dichalcogenides. Asian J. Org. Chem. 2018, 7, 1689–1697. [Google Scholar] [CrossRef]
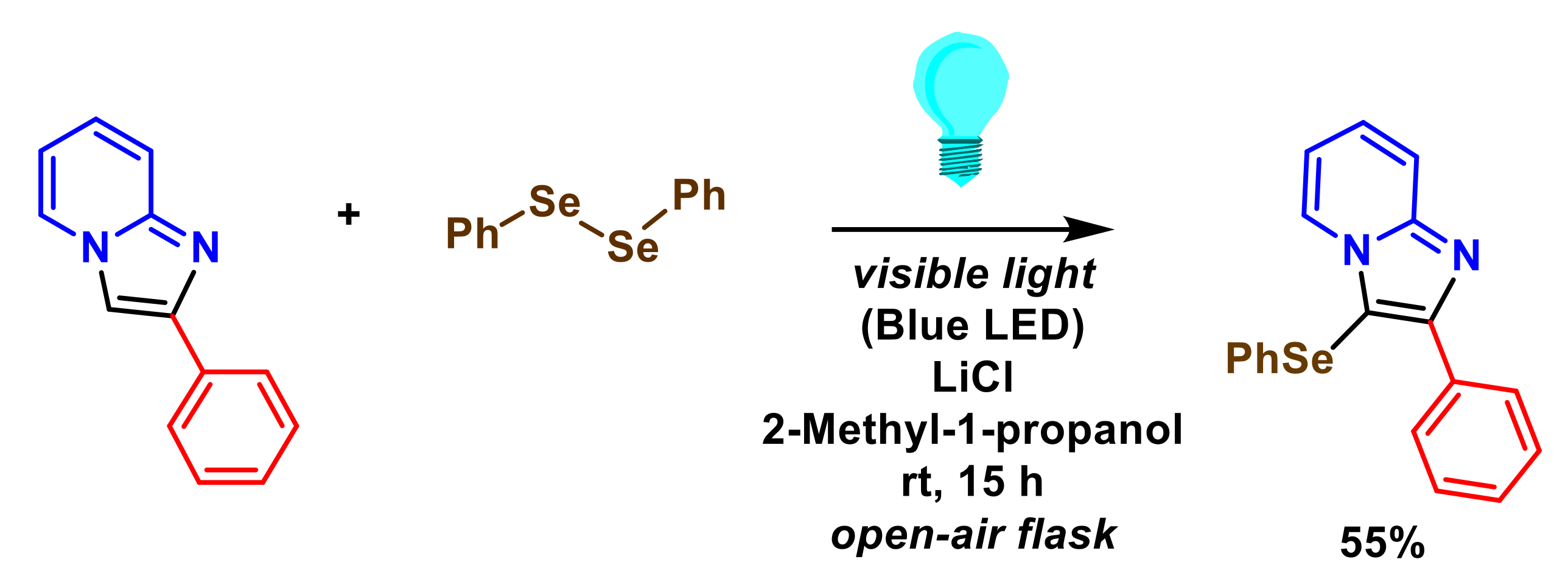
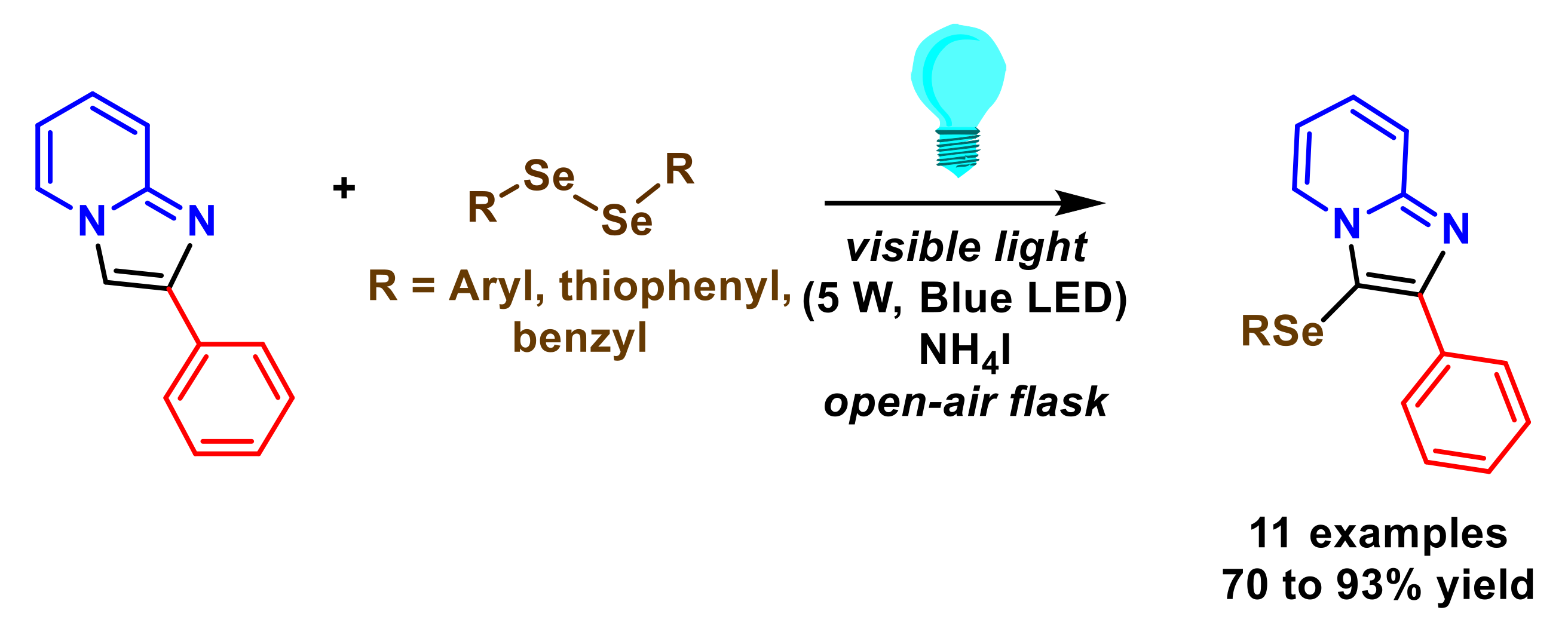
- Yasuike, S.; Kanasaki, K.; Murata, Y.; Kondo, K.; Kakusawa, N.; Matsumura, M. Visible-light-promoted se-arylation of diaryl diselenides with 2-phenylimidazopyridines in the presence of ammonium iodide: Synthesis of 2-phenyl-3-(arylselanyl)imidazo[1,2-±]pyridines. Heterocycles 2019, 99, 596–603. [Google Scholar] [CrossRef]
- Lee, J.H.; Jung, H.I.; Kim, D.Y. Visible light-mediated photocatalytic bromination of 2-arylimidazo[1,2-a] pyridines using CBr4 as bromine source. Synth. Commun. 2020, 50, 197–206. [Google Scholar] [CrossRef]
- Shivhare, K.N.; Jaiswal, M.K.; Srivastava, A.; Tiwari, S.K.; Siddiqui, I.R. Visible-light-activated C–C and C–N bond formation in the synthesis of imidazo[1,2-a] pyridines and imidazo[2,1-b] thiazoles under catalyst and solvent-free conditions. New J. Chem. 2018, 42, 16591–16601. [Google Scholar] [CrossRef]
- Singh, H.K.; Kamal, A.; Kumari, S.; Kumar, D.; Maury, S.K.; Srivastava, V.; Singh, S. Eosin Y-catalyzed synthesis of 3-aminoimidazo[1,2-a] pyridines via the HAT process under visible light through formation of the C–N bond. ACS Omega 2020, 5, 29854–29863. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.K.; Kamal, A.; Kumari, S.; Maury, S.K.; Kushwaha, A.K.; Srivastava, V.; Singh, S. Visible-light-promoted synthesis of fusesd imidazoheterocycle by eosin y under metal-free and solvent-free conditions. ChemistrySelect 2021, 6, 13982–13991. [Google Scholar] [CrossRef]
- Das, A.; Thomas, K.R.J. Light promoted synthesis of quinoxalines and imidazo[1,2-a] pyridines via oxybromination from alkynes and alkenes. Asian J. Org. Chem. 2020, 9, 1820–1825. [Google Scholar] [CrossRef]
- Yadav, S.; Srivastava, M.; Rai, P.; Tripathi, B.P.; Mishra, A.; Singh, J.; Singh, J. Oxidative organophotoredox catalysis: A regioselective synthesis of 2-nitro substituted imidazopyridines and 3-substituted indoles, initiated by visible light. New J. Chem. 2016, 40, 9694–9701. [Google Scholar] [CrossRef]
- Adiyala, P.R.; Sastry, K.N.V.; Kovvuri, J.; Nagarajan, A.; Reddy, V.G.; Sayeed, I.B.; Nayak, V.L.; Maurya, R.A.; Kamal, A. Visible light driven coupling of 2-aminopyridines and α-keto vinyl azides for the synthesis of imidazo[1,2-a] pyridines and their cytotoxicity. ChemistrySelect 2017, 2, 8158–8161. [Google Scholar] [CrossRef]
- Roslan, I.I.; Ng, K.-H.; Jaenicke, S.; Chuah, G.-K. Photocatalytic regeneration of brominating agent in the visible light-mediated synthesis of imidazo[1,2-a] pyridines. Catal. Sci. Technol. 2019, 9, 1528–1534. [Google Scholar] [CrossRef]
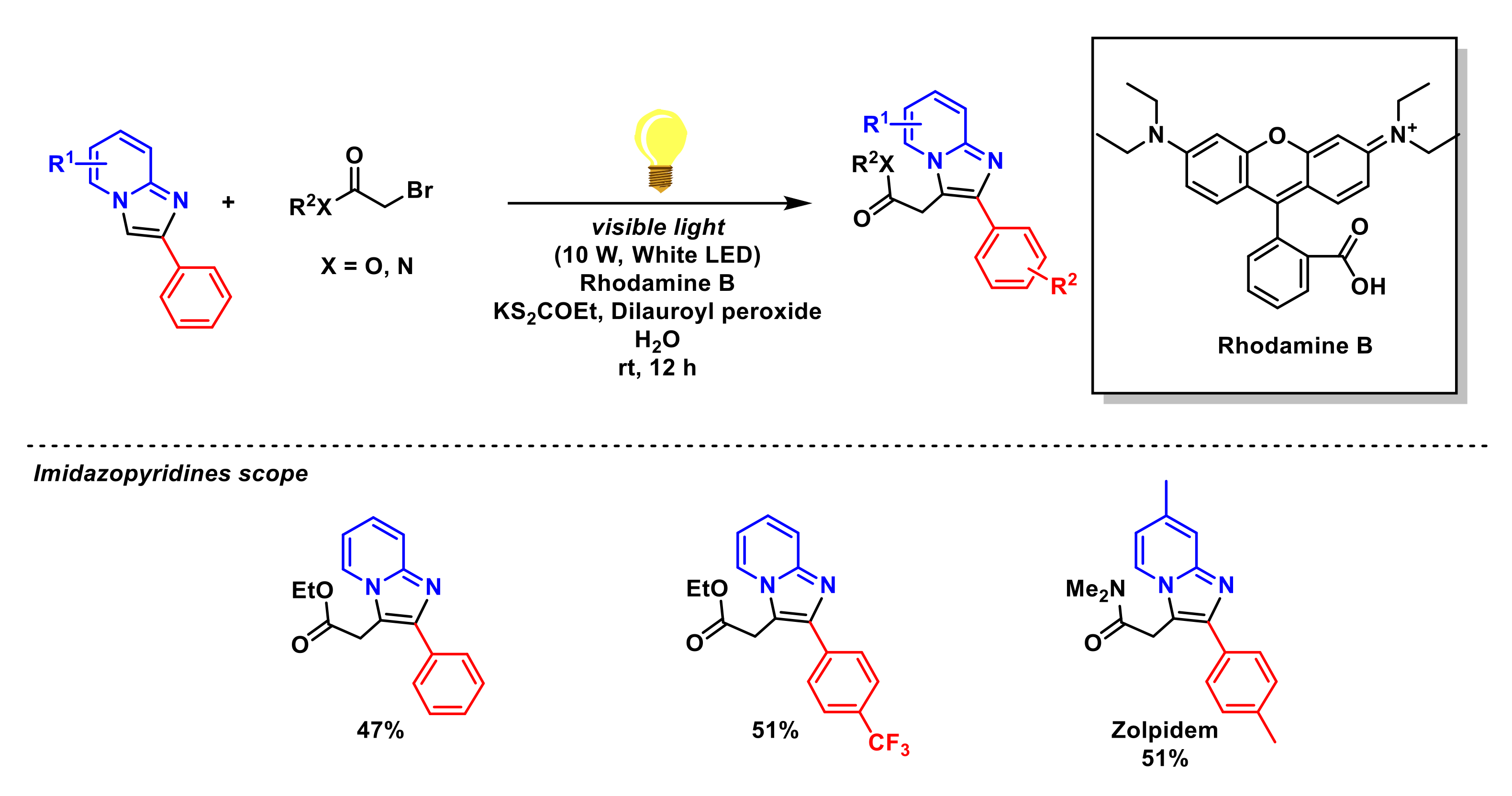
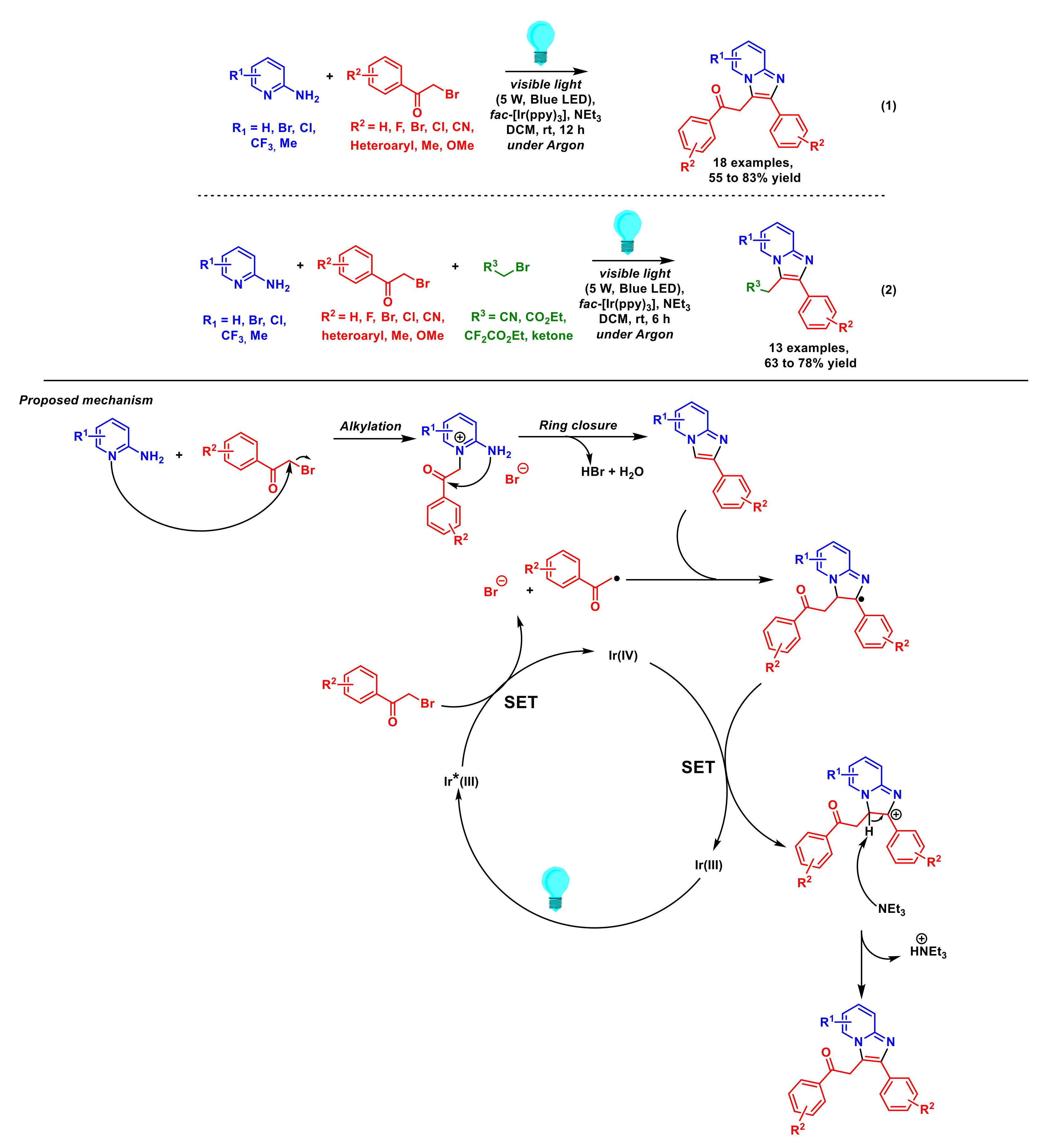
- Tong, J.; Zhan, Y.; Li, J.; Liu, P.; Sun, P. One-pot synthesis of C3-alkylated imidazopyridines from α-bromocarbonyls under photoredox conditions. Eur. J. Org. Chem. 2021, 32, 4541–4545. [Google Scholar] [CrossRef]
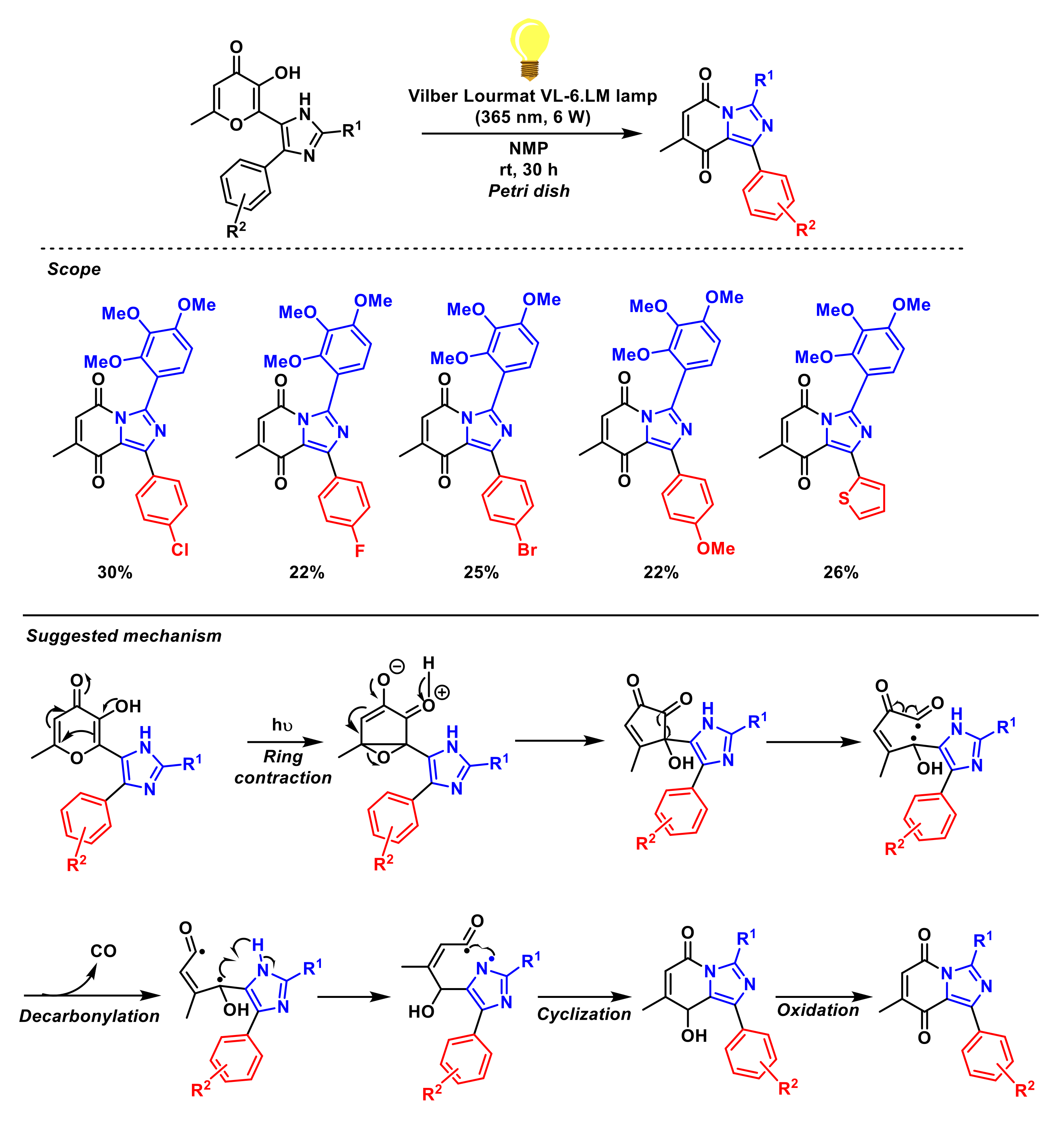
- Melekhina, V.G.; Komogortsev, A.N.; Lichitsky, B.V.; Mityanov, V.S.; Fakhrutdinov, A.N.; Dudinov, A.A.; Migulin, V.A.; Nelyubina, Y.V.; Melnikova, E.K.; Krayushkin, M.M. Unexpected photochemical transformation of imidazole derivatives containing the 5-hydroxy-2-methyl-4H-pyran-4-one moiety. Environmentally friendly method for the synthesis of substituted imidazo[1,5-a] pyridine-5,8-diones. Tetrahedron Lett. 2019, 60, 151080. [Google Scholar] [CrossRef]


























































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, C.; Hamze, A. Recent Developments in the Photochemical Synthesis of Functionalized Imidazopyridines. Molecules 2022, 27, 3461. https://doi.org/10.3390/molecules27113461
Tran C, Hamze A. Recent Developments in the Photochemical Synthesis of Functionalized Imidazopyridines. Molecules. 2022; 27(11):3461. https://doi.org/10.3390/molecules27113461
Chicago/Turabian StyleTran, Christine, and Abdallah Hamze. 2022. "Recent Developments in the Photochemical Synthesis of Functionalized Imidazopyridines" Molecules 27, no. 11: 3461. https://doi.org/10.3390/molecules27113461