
Multitarget Action of Xanthones from Garcinia mangostana against α-Amylase, α-Glucosidase and Pancreatic Lipase

 ,
,  , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
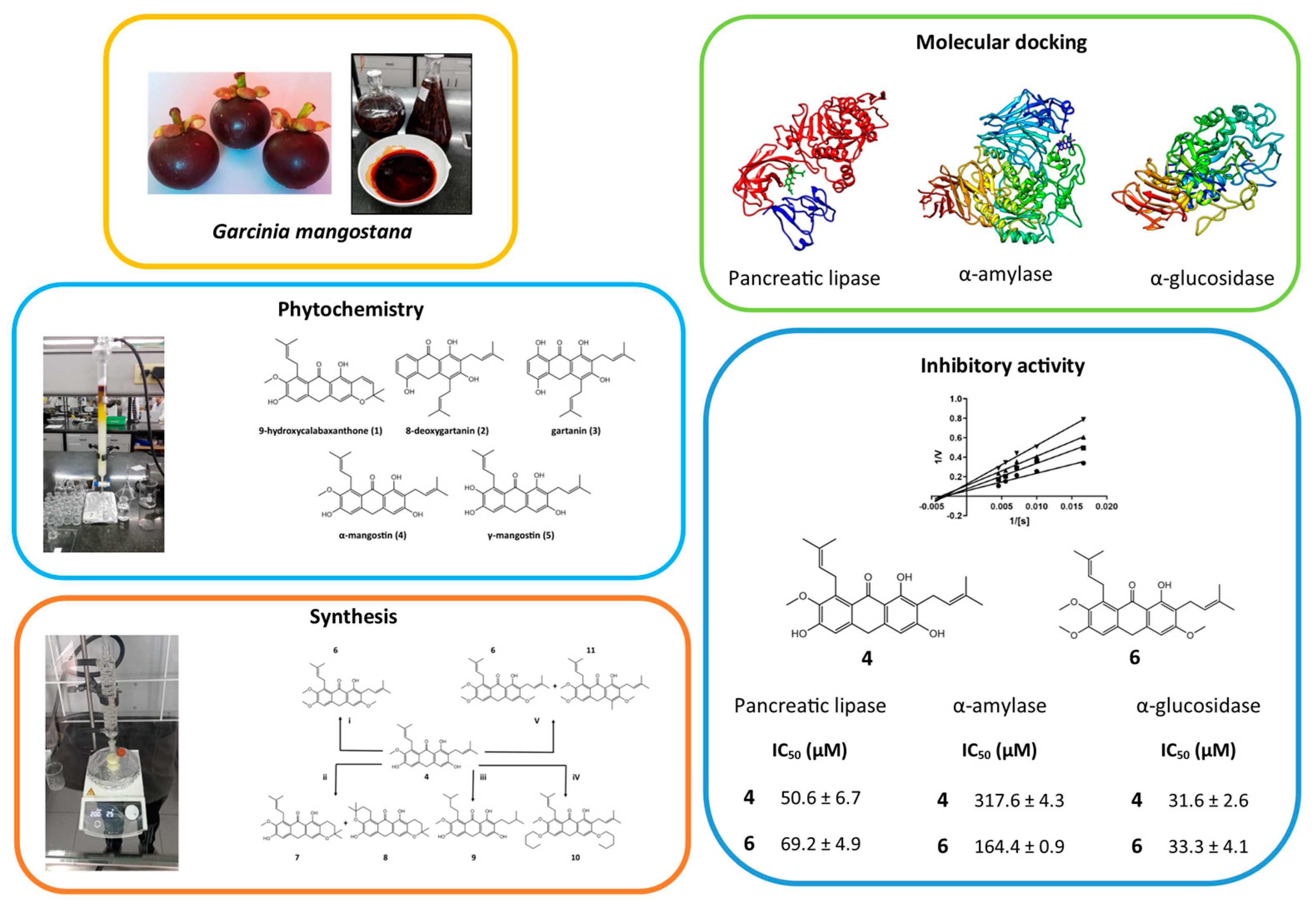
2.1. Phytochemical Study
2.2. Synthesis of Derivates
2.3. Determination of Enzymatic Inhibition against PL, AA and AG
2.4. Molecular Docking Studies
2.5. Compounds with Polypharmacological Action
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Isolation and Identification of Xanthones from G. mangostana
3.4. Preparation of Derivatives
3.5. Determination of Enzymatic Inhibition against PL, AA and AG
3.5.1. PL Inhibition Assay
3.5.2. AA Inhibition Assay
3.5.3. AG Inhibition Assay
3.6. Kinetic Study
3.7. Statistical Studies
3.8. Molecular Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Oguntibeju, O.O. Type 2 diabetes mellitus, oxidative stress and inflammation: Examining the links. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 45–63. [Google Scholar] [CrossRef] [PubMed]
- Piché, M.; Tchernof, A.; Després, J. Obesity Phenotypes, Diabetes, and Cardiovascular. Circ. Res. 2020, 126, 1477–1500. [Google Scholar] [CrossRef] [PubMed]
- Bullard, K.M.; Cowie, C.C.; Lessem, S.E.; Saydah, S.H.; Menke, A.; Geiss, L.S.; Orchard, T.J.; Rolka, D.B.; Imperatore, G. Prevalence of Diagnosed Diabetes in Adults by Diabetes Type. Morb. Mortal. Wkly. Rep. 2018, 7, 359–361. [Google Scholar] [CrossRef]
- Corbatón, A.; Cuervo, R.; Serrano, M. Diabetes mellitus. Concepto, clasificación y mecanismos etiopatogénicos. Med. Programa Form. Médica Contin. Acreditado 2004, 9, 963–970. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Diabetes. Available online: https://www.who.int/news-room/fact-sheets/detail/diabetes (accessed on 22 February 2022).
- Arnold, C.T.; Victoria-Delgado, B.; Borlaug, J.; Jeroen, J. Diabesity: The combined burden of obesity and diabetes on heart disease and the role of imaging. Nat. Rev. Cardiol. 2021, 18, 291–304. [Google Scholar] [CrossRef]
- Tan, S.; Wong, J.; Sim, Y.; Wong, S.; Elhassan, S.; Tan, S.; Lim, G.; Tay, N.; Annan, N.; Bhattamisra, S.; et al. Type 1 and 2 diabetes mellitus: A review on current treatment approach and gene therapy as potential intervention. Diabetes Metab. Syndr. 2019, 13, 364–372. [Google Scholar] [CrossRef]
- Reed, J.; Bain, S.; Kanamarlapudi, V. A Review of Current Trends with Type 2 Diabetes Epidemiology, Aetiology, Pathogenesis, Treatments and Future Perspectives. Diabet. Metab. Synd. Obes. 2021, 14, 3567–3602. [Google Scholar] [CrossRef]
- Gong, L.; Feng, D.; Wang, T.; Ren, Y.; Liu, Y.; Wang, J. Inhibitors of α-amylase and α-glucosidase: Potential linkage for whole cereal foods on prevention of hyperglycemia. Food Sci. Nutr. 2020, 8, 6320–6337. [Google Scholar] [CrossRef]
- Li, X.; Bai, Y.; Jin, Z.; Svensson, B. Food-derived non-phenolic α-amylase and α-glucosidase inhibitors for controlling starch digestion rate and guiding diabetes-friendly recipes. LWT 2022, 153, 112455. [Google Scholar] [CrossRef]
- Klein, S.; Gastaldelli, A.; Yki-Järvinen, H.; Scherer, P.E. Why does obesity cause diabetes? Cell Metab. 2022, 34, 11–20. [Google Scholar] [CrossRef]
- Barrett, S.; Whytock, K.L.; Strauss, J.A.; Wagenmakers, A.J.M.; Shepherd, S.O. High intramuscular triglyceride turnover rates and the link to insulin sensitivity: Influence of obesity, type 2 diabetes and physical activity. Appl. Physiol. Nutr. Metab. 2022, 47, 343–356. [Google Scholar] [CrossRef] [PubMed]
- NCD Risk Factor Collaboration (NCD-RisC). Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19·2 million participants. Lancet 2016, 387, 1377–1396. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization (WHO). Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 22 February 2022).
- Tian-Tian, L.; Xiao-Tian, L.; Qing-Xi, C.; Shi, Y. Lipase Inhibitors for Obesity: A Review. Biomed. Pharmacother. 2020, 128, 110314. [Google Scholar] [CrossRef]
- Kumar, A.; Chauhan, S. Pancreatic lipase inhibitors: The road voyaged and successes. Life Sci. 2021, 271, 119115. [Google Scholar] [CrossRef]
- Urbizo-Reyes, U.; Liceaga, A.M.; Reddivari, L.; Kim, K.H.; Anderson, J.M. Enzyme kinetics, molecular docking, and in silico characterization of canary seed (Phalaris canariensis L.) peptides with ACE and pancreatic lipase inhibitory activity. J. Funct. Foods 2022, 88, 104892. [Google Scholar] [CrossRef]
- Lovino, P.; Bucci, C.; Tremolaterra, F.; Santonicola, A.; Chiarioni, G. Bloating and functional gastro-intestinal disorders: Where are we and where are we going? World J. Gastroenterol. 2014, 20, 14407–14419. [Google Scholar] [CrossRef]
- Artasensi, A.; Pedretti, A.; Vistoli, G.; Fumagalli, L. Type 2 diabetes mellitus: A review of multi-target drugs. Molecules 2020, 25, 1987. [Google Scholar] [CrossRef]
- Genovese, M.; Nesi, I.; Caselli, A.; Paoli, P. Natural α-glucosidase and protein tyrosine phosphatase 1B inhibitors: A source of scaffold molecules for synthesis of new multitarget antidiabetic drugs. Molecules 2021, 26, 4818. [Google Scholar] [CrossRef]
- Ting-Hsu, C.; May-Jywan, T.; Yaw-Syan, F.; Ching-Feng, W. The Exploration of Natural Compounds for Anti-Diabetes from Distinctive Species Garcinia linii with Comprehensive Review of the Garcinia Family. Biomolecules 2019, 9, 641. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Li, Y.; Dai, Y.; Peng, J. Natural products for the treatment of type 2 diabetes mellitus: Pharmacology and mechanisms. Pharmacol. Res. 2018, 130, 451–465. [Google Scholar] [CrossRef]
- Pereira, A.S.; Banegas-Luna, A.J.; Peña-García, J.; Pérez-Sánchez, H.; Apostolides, Z. Evaluation of the anti-diabetic activity of some common herbs and spices: Providing new insights with inverse virtual screening. Molecules 2019, 24, 4030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aizat, M.W.; Ahmad-Hashim, F.H.; Syed, S.N. Valorization of mangosteen, ‘‘The Queen of Fruits”, and new advances in postharvest and in food and engineering applications: A review. J. Adv. Res. 2019, 20, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Adnyana, I.; Abuzaid, A.; Iskandar, E.; Kurniati, N. Pancreatic lipase and α-amylase inhibitory potential of mangosteen (Garcinia Mangostana Linn.) pericarp extract. Int. J. Med. Res. Health Sci. 2016, 5, 23. [Google Scholar] [CrossRef]
- Santos, C.M.; Freitas, M.; Fernandes, E. A comprehensive review on xanthone derivatives as α-glucosidase inhibitors. Eur. J. Med. Chem. 2018, 157, 1460–1479. [Google Scholar] [CrossRef]
- Gunter, N.V.; Teh, S.S.; Lim, Y.M.; Mah, S.H. Natural Xanthones and Skin Inflammatory Diseases: Multitargeting Mechanisms of Action and Potential Application. Front. Pharmacol. 2020, 11, 1873. [Google Scholar] [CrossRef] [PubMed]
- Ovalle-Magallanes, B.; Eugenio-Pérez, D.; Pedraza-Chaverri, J. Medicinal properties of mangosteen (Garcinia mangostana L.): A comprehensive update. Food Chem. Toxicol. 2017, 109, 102–122. [Google Scholar] [CrossRef]
- Rohman, A.; Arifah, F.H.; Irnawati, A.G.; Muchtaridi, M.R. A review on phytochemical constituents, role on metabolic diseases, and toxicological assessments of underutilized part of Garcinia mangostana L. Fruit. J. Appl. Pharm. Sci. 2020, 10, 127–146. [Google Scholar] [CrossRef]
- Rizaldy, D.; Hartati, R.; Nadhifa, T.; Fidrianny, I. Chemical Compounds and Pharmacological Activities of Mangosteen (Garcinia mangostana L.)—Updated Review. Biointerface Res. Appl. Chem. 2021, 12, 2503–2516. [Google Scholar] [CrossRef]
- Alhakamy, N.A.; Mohamed, G.A.; Fahmy, U.A.; Eid, B.G.; Ahmed, O.A.A.; Al-Rabia, M.W.; Ibrahim, S.R.M. New Alpha-Amylase Inhibitory Metabolites from Pericarps of Garcinia mangostana. Life 2022, 12, 384. [Google Scholar] [CrossRef]
- Ibrahim, S.R.M.; Mohamed, G.A.; Khayat, M.T.A.; Ahmed, S.; Abo-Haded, H. Garcixanthone D, a New Xanthone, and Other Xanthone Derivatives from Garcinia mangostana Pericarps: Their α-Amylase Inhibitory Potential and Molecular Docking Studies. Starke 2019, 71, 1800354. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, D.; Li, J.B.; Zhang, X.; Zhou, N.; Zhang, W.Y.; Lu, H. Prenylated xanthones with α-glucosidase and α-amylase inhibitory effects from the pericarp of Garcinia mangostana. J. Asian Nat. Prod. Res. 2021, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.S.; Kim, E.Y.; Han, L.; Kim, N.R.; Lam, B.; Paik, J.H.; Yoon, K.D.; Choi, Y.H.; Chin, Y.W. Xanthones with pancreatic lipase inhibitory activity from the pericarps of Garcinia mangostana L. (Guttiferae). Eur. J. Lipid Sci. Technol. 2016, 118, 1416–1421. [Google Scholar] [CrossRef]
- Bisswanger, H. Enzyme Assays. Perspect. Sci. 2014, 1, 41–55. [Google Scholar] [CrossRef] [Green Version]
- Al-Massarani, S.M.; El-Gamal, A.A.; Al-Musayei, N.M.; Mothana, R.A.; Basudan, O.A.; Al-Rehaily, A.J.; Farag, F.; Assaf, M.H.; El-Tahir, H.; Maes, L. Phytochemical, Antimicrobial and Antiprotozoal Evaluation of Garcinia mangostana Pericarp and α-Mangostin, Its Major Xanthone Derivative. Molecules 2013, 18, 599. [Google Scholar] [CrossRef] [Green Version]
- Han, A.R.; Kim, J.A.; Lantvit, D.D.; Kardono, L.B.S.; Riswam, S.; Chai, H.; Carcache de Blanco, E.J.; Farnsworth, N.R.; Swanson, S.M.; Kinghorn, A.D. Cytotoxic xanthone constituents of the stem bark of Garcinia mangostana (mangosteen). J. Nat. Prod. 2009, 72, 2028–2031. [Google Scholar] [CrossRef] [Green Version]
- Assemian, I.C.; Chadon, A.; Bouyahya, A.; Dakka, N.; Bakri, Y. Garcinia mangostana leaf extracts from ivory coast possess remarkable antioxidant, anti-inflammatory and cytotoxicological properties. Biomed. Pharmacol. J. 2019, 12, 571–578. [Google Scholar] [CrossRef]
- Ryu, H.W.; Cho, J.K.; Curtis-Long, M.J.; Yuk, H.J.; Kim, K.S.; Jung, S.; Kim, Y.S.; Lee, B.W.; Park, K.H. α-Glucosidase inhibition and antihyperglycemic activity of prenylated xanthones from Garcinia mangostana. Phytochemistry 2011, 72, 2148–2154. [Google Scholar] [CrossRef]
- Raksat, A.; Sripisut, T.; Maneerat, W. Bioactive xanthones from Cratoxylum cochinchinense. Nat. Prod. Commun. 2015, 10, 1969–1972. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.J.; Fu, W.W.; Wu, R.; Yang, J.L.; Yao, C.Y.; Yan, B.X.; Tan, H.S.; Zheng, C.W.; Song, Z.J.; Xu, H.X. Cytotoxic prenylated xanthones from the leaves of Garcinia bracteata. Planta Med. 2019, 85, 444–452. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, S.R.M.; Mohamed, C.A.; Khayat, M.T.; Ahmed, S.; Abo-Haded, H.; Alshali, K.Z. Mangostanaxanthone VIIII, a new xanthone from Garcinia mangostana pericarps, α-amylase inhibitory activity, and molecular docking studies. Braz. J. Pharmacogn. 2019, 29, 206–212. [Google Scholar] [CrossRef]
- Peres, V.; Nagem, T.J.; De Oliveira, F.F. Tetraoxygenated naturally occurring xanthones. Phytochemistry 2000, 55, 683–710. [Google Scholar] [CrossRef] [Green Version]
- Sultanbawa, M.U.S. Xanthonoids of tropical plants. Tetrahedron 1980, 36, 1465–1506. [Google Scholar] [CrossRef]
- Aravindakshanpillai, P.; Aravind, P.; Pandey, R.; Kumar, B.; Asha, K.R.T.; Rameshkumar, K.B. Phytochemical screening of garcinia travancorica by HPLC-ESI-QTOF mass spectrometry and cytotoxicity studies of the major biflavonoid fukugiside. Nat. Prod. Commun. 2016, 11, 1839–1842. [Google Scholar] [CrossRef] [Green Version]
- Ito, C.; Itoigawa, M.; Takakura, T.; Ruangrungsi, N.; Tokuda, H.; Nishino, H.; Furukawa, H. Chemical constituents of Garcinia fusca: Structure elucidation of eight new xanthones and their cancer chemopreventive activity. J. Nat. Prod. 2003, 66, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Che-Hassan, N.K.N.; Taher, M.; Susanti, D. Phytochemical constituents and pharmacological properties of Garcinia xanthochymus- a review. Biomed. Pharmacother. 2018, 106, 1378–1389. [Google Scholar] [CrossRef] [PubMed]
- Brito, L.C.; Berenger, A.L.R.; Figueiredo, M.R. An overview of anticancer activity of Garcinia and Hypericum. Food Chem. Toxicol. 2017, 109, 847–862. [Google Scholar] [CrossRef]
- Balasubramanian, K.; Rajagopalan, K. Novel xanthones from Garcinia mangostana, structures of BR-xanthone-A and BR-xanthone-B. Phytochemistry 1988, 27, 1552–1554. [Google Scholar] [CrossRef]
- Sudta, P.; Jiarawapi, P.; Suksamrarn, A.; Hongmanee, P.; Suksamrarn, S. Potent activity against multidrug-resistant Mycobacterium tuberculosis of α-mangostin analogs. Chem. Pharm. Bull. 2013, 61, 194–203. [Google Scholar] [CrossRef] [Green Version]
- Ha, L.D.; Hansen, P.E.; Vang, O.; Duus, F.; Pham, H.D.; Nguyen, L.H.D. Cytotoxic geranylated xanthones and O-alkylated derivatives of α-mangostin. Chem. Pharm. Bull. 2009, 57, 830–834. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Susan, M.; Lantvit, D.D.; Ninh, T.N.; Chai, H.; Fuchs, R.J.; Soejarto, D.D.; Carcach, E.J.; De Blanco, S.; Swanson, M.; et al. Cytotoxic and NF-ΚB Inhibitory Constituents of the Stems of Cratoxylum cochinchinense and Their Semisynthetic Analogues. J. Nat. Prod. 2011, 74, 1117–1125. [Google Scholar] [CrossRef] [Green Version]
- Vongsak, B.; Sumet, K.; Sunan, J.; Sasipawan, M.; Chamnan, P. In Vitro Alpha Glucosidase Inhibition and Free-Radical Scavenging Activity of Propolis from Thai Stingless Bees in Mangosteen Orchard. Rev. Bras. Farmacogn. 2015, 25, 445–450. [Google Scholar] [CrossRef] [Green Version]
- Trott, A.; Olson, J.A. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger Release 2022-1: Maestro, Schrödinger, LLC, New York, NY. 2021. Available online: https://www.schrodinger.com/ (accessed on 10 December 2021).
- Whitcomb, D.C.; Lowe, M.E. Human pancreatic digestive enzymes. Dig. Dis. Sci. 2007, 52, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Brás, N.F.; Santos-Martins, D.; Fernandes, P.A.; Ramos, M.J. Mechanistic Pathway on Human α-Glucosidase Maltase-Glucoamylase Unveiled by QM/MM Calculations. J. Phys. Chem. B 2018, 122, 3889–3899. [Google Scholar] [CrossRef] [PubMed]
- Sim, L.; Quezada-Calvillo, R.; Sterchi, E.E.; Nichols, B.L.; Rose, D.R. Human Intestinal Maltase-Glucoamylase: Crystal Structure of the N-Terminal Catalytic Subunit and Basis of Inhibition and Substrate Specificity. J. Mol. Biol. 2008, 375, 782–792. [Google Scholar] [CrossRef]
- Shang, E.; Yuan, Y.; Chen, X.; Liu, Y.; Pei, J.; Lai, L. De novo design of multitarget ligands with an iterative fragment-growing strategy. J. Chem. Inf. Model. 2014, 54, 1235–1241. [Google Scholar] [CrossRef]
- Lien-Hoa, D.N.; Vo, H.T.; Pham, H.D.; Connolly, J.D.; Harrison, L.J. Xanthones from the bark of Garcinia merguensis. Phytochemistry 2003, 63, 467–470. [Google Scholar] [CrossRef]
- Anggia, I.; Bakhtiar, A.; Arbain, D. The Isolation of xanthones from trunk latex of Garcinia mangostana Linn. and their antimicrobial activities. Indones. J. Chem. 2013, 15, 187–193. [Google Scholar] [CrossRef]
- Chen, L.G.; Yang, L.G.; Wang, C.C. Anti-inflammatory activity of mangostins from Garcinia mangostana. Food Chem. Toxicol. 2008, 46, 688–693. [Google Scholar] [CrossRef]
- Ishiguro, K.; Fukumoto, H.; Nakajima, M.; Isoi, K. Xanthones in cell suspension cultures of Hypericum paturum. Phytochemistry 1993, 33, 839–840. [Google Scholar] [CrossRef]
- Chitiva-Chitiva, L.C.; Ladino-Vargas, C.; Cuca-Suárez, L.E.; Prieto-Rodriguez, J.A.; Patiño-Ladino, O.J. Antifungal Activity of Chemical Constituents from Piper pesaresanum C. DC. and Derivatives against Phytopathogen Fungi of Cocoa. Molecules 2021, 26, 3256. [Google Scholar] [CrossRef] [PubMed]
- Torres, E.; Almeida, R.M.; Vogel, C. Síntesis y evaluación de la actividad antitumoral in vitro de un éster prenilado análogo a productos naturales. Química Viva 2015, 14, 111–119. [Google Scholar]
- Kulkarni, P.P.; Kadam, A.J.; Mane, R.B.; Desai, U.V.; Wadgaonkar, P.P. Demethylation of Methyl Aryl Ethers using Pyridine Hydrochloride in Solvent-free Conditions under Microwave Irradiation. J. Chem. Res. 1999, 6, 394–395. [Google Scholar] [CrossRef]
- Sassaki, G.L.; Iacomini, M.; Gorin, P.A.J. Methylation-GC-MS Analysis of Arabinofuranose- And Galactofuranose- Containing Structures: Rapid Synthesis of Partially O-Methylated Alditol Acetate Standards. An. Acad. Bras. Cienc. 2005, 77, 223–234. [Google Scholar] [CrossRef]
- Wang, Z. Irvine-Purdie Methylation. In Comprehensive Organic Name Reactions and Reagents, 1st ed.; Wang, Z., Ed.; Wiley-Interscience: Hoboken, NJ, USA, 2009; Volume 3, pp. 1526–1529. [Google Scholar]
- Bustanji, Y.; Al-Masri, I.M.; Mohammad, M.; Hudaib, M.; Tawaha, K.; Tarazi, H.; Alkhatib, H.S. Pancreatic lipase inhibition activity of trilactone terpenes of Ginkgo biloba. J. Enzym. Inhib. Med. Chem. 2011, 26, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Iwai, K.; Kim, M.Y.; Onodera, A.; Matsue, H. α-Glucosidase inhibitory and antihyperglycemic effects of polyphenols in the fruit of Viburnum dilatatum thunb. J. Agric. Food Chem. 2006, 54, 4588–4592. [Google Scholar] [CrossRef]
- Yung-Chi, C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Luyen, N.T.; Tram, L.H.; Hong Hanh, T.T.; Binh, P.T.; Dang, N.H.; Minh, C.V.; Dat, N.N. Inhibitors of a-glucosidase, a-amylase and lipase from Chrysanthemum morifolium. Phytochem. Lett. 2013, 6, 322–325. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Preto, J.; Gentile, F. Assessing and improving the performance of consensus docking strategies using the DockBox package. J. Comput.-Aided Mol. Des. 2019, 33, 817–829. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pancreatic Lipase | α-Amylase | α-Glucosidase | |
|---|---|---|---|
| Sample | IC50 (ppm) | IC50 (ppm) | IC50 (ppm) |
| EE | 19.6 ± 1.2 a | 718.9 ± 9.2 b | 261.3 ± 21.8 a |
| DCM | 16.5 ± 4.2 a | 141.7 ± 12.3 a | 197.6 ± 1.5 a |
| EtOAc | 59.0 ± 3.9 b | 246.1 ± 18.0 a | >1000 b |
| IPA | >1000 c >1000 c | 707.4 ± 69.9 b | >1000 b >1000 b |
| EtOH:H2O | 575.0 ± 96.2 b | ||
| Orlistat | 0.65 ± 0.1 µM | nd | nd |
| Acarbose | nd | 820.0 ± 14.0 µM | 315.2 ± 3.3 µM |
| p-value | ≤0.05 | ≤0.05 | ≤0.05 |
| Compounds | Pancreatic Lipase | α-Amylase | α-Glucosidase | ||||||
|---|---|---|---|---|---|---|---|---|---|
| IC50 (µM) | Ki (µM) | It | IC50 (µM) | Ki (µM) | It | IC50 (µM) | Ki (µM) | It | |
| 1 | 138.1 ± 6.8 e | 138.1 | NC | >1000 a | 157.1 ± 2.1 f | 154.0 | UC | ||
| 2 | 326.3 ± 7.1 d | 326.3 | NC | 165.6 ± 4.1 b | 165.6 | NC | 247.8 ± 4.0 b | 247.8 | M |
| 3 | 134.8 ± 1.5 e | 129.7 | UC | >1000 a | 123.6 ± 8.2 c | 123.6 | M | ||
| 4 | 50.6 ± 6.7 b | 50.6 | NC | 317.6 ± 4.3 c | 240.4 | C | 31.6 ± 2.6 d | 31.6 | M |
| 5 | 67.1 ± 2.8 c | 57.2 | UC | 333.5 ± 4.5 d | 138.6 | C | 84.7 ± 6.7 e | 84.7 | M |
| 6 | 69.2 ± 4.9 c | 69.2 | M | 164.4 ± 0.9 b | 28.6 | M | 33.3 ± 4.1 d | 0.3 | C |
| 7 | >1000 a | >1000 a | 37.2 ± 4.1 d | 37.2 | M | ||||
| 8 | >1000 a | >1000 a | >1000 a | ||||||
| 9 | 454.6 ± 1.9 e | 405.2 | M | 90.1 ± 6.0 e | 90.1 | C | 34.2 ± 1.8 d | 34.2 | C |
| 10 | >1000 a | >1000 a | >1000 a | ||||||
| 11 | 59.42 ± 3.8 bc | 59.4 | M | >1000 a | >1000 a | ||||
| Orlistat® | 0.65 ± 0.1 | 0.2 | II | nd | nd | ||||
| Acarbosa® | nd | 820.0 ± 14.0 | 130.0 | C | 315.2 ± 3.3 | 56.3 | C | ||
| p-value | ≤0.05 | ≤0.05 | ≤0.05 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardozo-Muñoz, J.; Cuca-Suárez, L.E.; Prieto-Rodríguez, J.A.; Lopez-Vallejo, F.; Patiño-Ladino, O.J. Multitarget Action of Xanthones from Garcinia mangostana against α-Amylase, α-Glucosidase and Pancreatic Lipase. Molecules 2022, 27, 3283. https://doi.org/10.3390/molecules27103283
Cardozo-Muñoz J, Cuca-Suárez LE, Prieto-Rodríguez JA, Lopez-Vallejo F, Patiño-Ladino OJ. Multitarget Action of Xanthones from Garcinia mangostana against α-Amylase, α-Glucosidase and Pancreatic Lipase. Molecules. 2022; 27(10):3283. https://doi.org/10.3390/molecules27103283
Chicago/Turabian StyleCardozo-Muñoz, Juan, Luis E. Cuca-Suárez, Juliet A. Prieto-Rodríguez, Fabian Lopez-Vallejo, and Oscar J. Patiño-Ladino. 2022. "Multitarget Action of Xanthones from Garcinia mangostana against α-Amylase, α-Glucosidase and Pancreatic Lipase" Molecules 27, no. 10: 3283. https://doi.org/10.3390/molecules27103283