
Synthesis of Mixed Dinucleotides by Mechanochemistry



Abstract
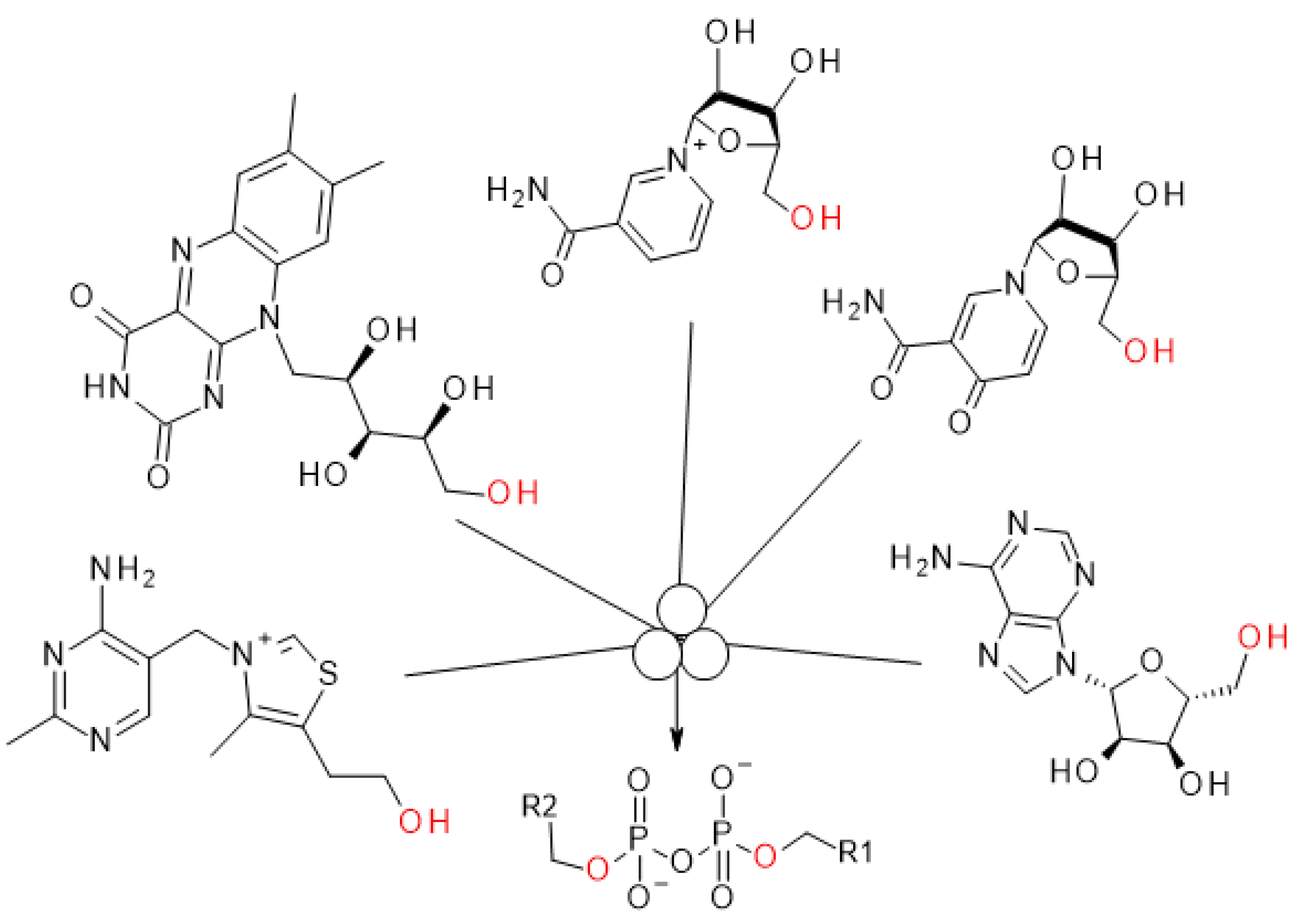
:1. Introduction
2. Results and Discussion
2.1. Synthesis of Monophosphates
2.2. Synthesis of Pyrophosphates
2.3. Synthesis of Labeled NAD (25)
3. Materials and Methods
3.1. General Synthetic Procedure for the Preparation of Monophosphates (11–15 & 28)
3.1.1. By Mechanochemical Ball-Milling
3.1.2. By Solution-Phase Synthesis
3.1.3. Synthesis of FMN (32)
3.2. General Synthetic Procedure for the Preparation of Monophospho-Imidazolates (16–19 & 29) by Mechanochemical Ball-Milling
3.3. General Synthetic Procedure for the Preparation of Pyrophosphates (20–26, 30a, 31, 33 & 37) by Mechanochemical Ball-Milling
3.4. Synthesis of M+4 NAD
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Peterson, C.T.; Rodionov, D.A.; Osterman, A.L.; Peterson, S.N. B Vitamins and Their Role in Immune Regulation and Cancer. Nutrients 2020, 12, 3380. [Google Scholar] [CrossRef]
- Luongo, T.S.; Eller, J.M.; Lu, M.-J.; Niere, M.; Raith, F.; Perry, C.; Bornstein, M.R.; Oliphint, P.; Wang, L.; McReynolds, M.R.; et al. SLC25A51 is a mammalian mitochondrial NAD(+) transporter. Nature 2020, 588, 174–179. [Google Scholar] [CrossRef]
- Makarov, M.V.; Harris, N.W.; Rodrigues, M.; Migaud, M.E. Scalable syntheses of traceable ribosylated NAD(+) precursors. Org. Biomol. Chem. 2019, 17, 8716–8720. [Google Scholar] [CrossRef]
- Hrubša, M.; Siatka, T.; Nejmanová, I.; Vopršalová, M.; Krčmová, L.K.; Matoušová, K.; Javorská, L.; Macáková, K.; Mercolini, L.; Remião, F.; et al. Biological Properties of Vitamins of the B-Complex, Part 1: Vitamins B(1), B(2), B(3), and B(5). Nutrients 2022, 14, 484. [Google Scholar] [CrossRef]
- Makarov, M.V.; Trammell, S.A.; Migaud, M.E. The chemistry of the vitamin B3 metabolome. Biochem. Soc. Trans. 2019, 47, 131–147. [Google Scholar] [CrossRef]
- Shats, I.; Williams, J.G.; Liu, J.; Makarov, M.V.; Wu, X.; Lih, F.B.; Deterding, L.J.; Lim, C.; Xu, X.; Randall, T.; et al. Bacteria Boost Mammalian Host NAD Metabolism by Engaging the Deamidated Biosynthesis Pathway. Cell Metab. 2020, 31, 564–579.e7. [Google Scholar] [CrossRef]
- Doi, H.; Mawatari, A.; Kanazawa, M.; Nozaki, S.; Nomura, Y.; Kitayoshi, T.; Akimoto, K.; Suzuki, M.; Ninomiya, S.; Watanabe, Y. Synthesis of 11C-Labeled Thiamine and Fursultiamine for in Vivo Molecular Imaging of Vitamin B1 and Its Prodrug Using Positron Emission Tomography. J. Org. Chem. 2015, 80, 6250–6258. [Google Scholar] [CrossRef]
- Dhir, S.; Tarasenko, M.; Napoli, E.; Giulivi, C. Neurological, Psychiatric, and Biochemical Aspects of Thiamine Deficiency in Children and Adults. Front. Psychiatry 2019, 10, 207. [Google Scholar] [CrossRef] [Green Version]
- Tylicki, A.; Łotowski, Z.; Siemieniuk, M.; Ratkiewicz, A. Thiamine and selected thiamine antivitamins—Biological activity and methods of synthesis. Biosci. Rep. 2018, 38, BSR20171148. [Google Scholar] [CrossRef] [Green Version]
- Barile, M.; Giancaspero, T.A.; Leone, P.; Galluccio, M.; Indiveri, C. Riboflavin transport and metabolism in humans. J. Inherit. Metab. Dis. 2016, 39, 545–557. [Google Scholar] [CrossRef]
- Kropotov, A.; Kulikova, V.; Nerinovski, K.; Yakimov, A.; Svetlova, M.; Solovjeva, L.; Sudnitsyna, J.; Migaud, M.; Khodorkovskiy, M.; Ziegler, M.; et al. Equilibrative Nucleoside Transporters Mediate the Import of Nicotinamide Riboside and Nicotinic Acid Riboside into Human Cells. Int. J. Mol. Sci. 2021, 22, 1391. [Google Scholar] [CrossRef]
- Palmieri, F.; Monné, M.; Fiermonte, G.; Palmieri, L. Mitochondrial transport and metabolism of the vitamin B-derived cofactors thiamine pyrophosphate, coenzyme A, FAD and NAD+, and related diseases: A review. IUBMB Life 2022, epub ahead of print. [CrossRef]
- Ziegler, M.; Monné, M.; Nikiforov, A.; Agrimi, G.; Heiland, I.; Palmieri, F. Welcome to the Family: Identification of the NAD(+) Transporter of Animal Mitochondria as Member of the Solute Carrier Family SLC. Biomolecules 2021, 11, 880. [Google Scholar] [CrossRef]
- Santoro, V.; Kovalenko, I.; Vriens, K.; Christen, S.; Bernthaler, A.; Haegebarth, A.; Fendt, S.-M.; Christian, S. SLC25A32 sustains cancer cell proliferation by regulating flavin adenine nucleotide (FAD) metabolism. Oncotarget 2020, 11, 801–812. [Google Scholar] [CrossRef] [Green Version]
- Grozio, A.; Mills, K.F.; Yoshino, J.; Bruzzone, S.; Sociali, G.; Tokizane, K.; Lei, H.C.; Cunningham, R.; Sasaki, Y.; Migaud, M.E.; et al. Slc12a8 is a nicotinamide mononucleotide transporter. Nat. Metab. 2019, 1, 47–57. [Google Scholar] [CrossRef]
- Dollerup, O.L.; Chubanava, S.; Agerholm, M.; Søndergård, S.D.; Altıntaş, A.; Møller, A.B.; Høyer, K.F.; Ringgaard, S.; Stødkilde-Jørgensen, H.; Lavery, G.; et al. Nicotinamide riboside does not alter mitochondrial respiration, content or morphology in skeletal muscle from obese and insulin-resistant men. J. Physiol. 2020, 598, 731–754. [Google Scholar] [CrossRef]
- Sonavane, M.; Hayat, F.; Makarov, M.; Migaud, M.E.; Gassman, N.R. Dihydronicotinamide riboside promotes cell-specific cytotoxicity by tipping the balance between metabolic regulation and oxidative stress. PLoS ONE 2020, 15, e0242174. [Google Scholar] [CrossRef]
- Tarantini, S.; Valcarcel-Ares, M.N.; Toth, P.; Yabluchanskiy, A.; Tucsek, Z.; Kiss, T.; Hertelendy, P.; Kinter, M.; Ballabh, P.; Süle, Z.; et al. Nicotinamide mononucleotide (NMN) supplementation rescues cerebromicrovascular endothelial function and neurovascular coupling responses and improves cognitive function in aged mice. Redox Biol. 2019, 24, 101192. [Google Scholar] [CrossRef]
- Sims, C.A.; Guan, Y.; Mukherjee, S.; Singh, K.; Botolin, P.; Davila, A.; Baur, J.A. Nicotinamide mononucleotide preserves mitochondrial function and increases survival in hemorrhagic shock. JCI Insight 2018, 3, e120182. [Google Scholar] [CrossRef]
- Wolak, N.; Zawrotniak, M.; Gogol, M.; Kozik, A.; Rapala-Kozik, M. Vitamins B1, B2, B3 and B9—Occurrence, Biosynthesis Pathways and Functions in Human Nutrition. Mini-Rev. Med. Chem. 2017, 17, 1075–1111. [Google Scholar] [CrossRef]
- Distelmaier, F.; Haack, T.B.; Wortmann, S.B.; Mayr, J.; Prokisch, H. Treatable mitochondrial diseases: Cofactor metabolism and beyond. Brain 2016, 140, e11. [Google Scholar] [CrossRef]
- Mosegaard, S.; DiPace, G.; Bross, P.; Carlsen, J.; Gregersen, N.; Olsen, R.K.J. Riboflavin Deficiency—Implications for General Human Health and Inborn Errors of Metabolism. Int. J. Mol. Sci. 2020, 21, 3847. [Google Scholar] [CrossRef]
- Ruprecht, J.; Kunji, E.R.S. The SLC25 Mitochondrial Carrier Family: Structure and Mechanism. Trends Biochem. Sci. 2020, 45, 244–258. [Google Scholar] [CrossRef] [Green Version]
- Beztsinna, N.; Tsvetkova, Y.; Bartneck, M.; Lammers, T.; Kiessling, F.; Bestel, I. Amphiphilic Phospholipid-Based Riboflavin Derivatives for Tumor Targeting Nanomedicines. Bioconjug. Chem. 2016, 27, 2048–2061. [Google Scholar] [CrossRef]
- Xu, Z. A review on the chemical synthesis of pyrophosphate bonds in bioactive nucleoside diphosphate analogs. Bioorg. Med. Chem. Lett. 2015, 25, 3777–3783. [Google Scholar] [CrossRef]
- Ravalico, F.; Messina, I.; Berberian, M.V.; James, S.L.; Migaud, M.E.; Vyle, J.S. Rapid synthesis of nucleotide pyrophosphate linkages in a ball mill. Org. Biomol. Chem. 2011, 9, 6496–6497. [Google Scholar] [CrossRef]
- Migaud, M.E.; Redpath, P.; Crossey, K.; Cunningham, R.; Rhonemus, T.; Venkataraman, S. Selective Solvent Free Phosphorylation, C. Queen’s University Belfast Application. U.S. Patent 20160355539A1, 2016. [Google Scholar]
- Normington, K.D.S.; David, A.; Livingston, D.; McKearin, J.M.; Szczepankiewicz, B.; Kremsky, J.N. Nicotinamide Mononucleotide Derivatives and Their Uses. WIPO Patent WO/2017/024255, 9 February 2017. [Google Scholar]
- Gomollón-Bel, F. Ten Chemical Innovations That Will Change Our World: IUPAC identifies emerging technologies in Chemistry with potential to make our planet more sustainable. Chem. Int. 2019, 41, 12–17. [Google Scholar] [CrossRef]
- Wang, G.-W. Mechanochemical organic synthesis. Chem. Soc. Rev. 2013, 42, 7668–7700. [Google Scholar] [CrossRef]
- Wortmann, S.B.; Meunier, B.; Mestek-Boukhibar, L.; Broek, F.V.D.; Maldonado, E.M.; Clement, E.; Weghuber, D.; Spenger, J.; Jaros, Z.; Taha, F.; et al. Bi-allelic Variants in TKFC Encoding Triokinase/FMN Cyclase Are Associated with Cataracts and Multisystem Disease. Am. J. Hum. Genet. 2020, 106, 256–263. [Google Scholar] [CrossRef]
- Johnson, D.C., 2nd; Widlanski, T.S. Overview of the synthesis of nucleoside phosphates and polyphosphates. Curr. Protoc. Nucleic Acid Chem. 2004, 15, 13.1.1–13.1.31. [Google Scholar] [CrossRef] [Green Version]
- Franchetti, P.; Pasqualini, M.; Petrelli, R.; Ricciutelli, M.; Vita, P.; Cappellacci, L. Stereoselective synthesis of nicotinamide β-riboside and nucleoside analogs. Bioorg. Med. Chem. Lett. 2004, 14, 4655–4658. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, M.; Kato, T.; Takenishi, T. A novel method for phosphorylation of nucleosides to 5′-nucleotides. Tetrahedron Lett. 1967, 8, 5065–5068. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Kato, T.; Takenishi, T. Studies of Phosphorylation. III. Selective Phosphorylation of Unprotected Nucleosides. Bull. Chem. Soc. Jpn. 1969, 42, 3505–3508. [Google Scholar] [CrossRef] [Green Version]
- Eguaogie, O.; Vyle, J.S. Vibration Ball Milling for the Synthesis of 5′-Thioadenosine 5′-Pyrophosphate (P′-->5′) Adenosine (dASppA). Curr. Protoc. Nucleic Acid Chem. 2017, 70, 1.41.1–1.41.12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appy, L.; Depaix, A.; Bantreil, X.; Lamaty, F.; Peyrottes, S.; Roy, B. Straightforward Ball-Milling Access to Dinucleoside 5′,5′-Polyphosphates via Phosphorimidazolide Intermediates. Chemistry 2019, 25, 2477–2481. [Google Scholar] [CrossRef] [Green Version]
- Eguaogie, O.; Vyle, J.S.; Conlon, P.F.; Gîlea, M.A.; Liang, Y. Mechanochemistry of nucleosides, nucleotides and related materials. Beilstein J. Org. Chem. 2018, 14, 955–970. [Google Scholar] [CrossRef] [Green Version]
- Appy, L.; Chardet, C.; Peyrottes, S.; Roy, B. Synthetic Strategies for Dinucleotides Synthesis. Molecules 2019, 24, 4334. [Google Scholar] [CrossRef] [Green Version]
- Canales, J.; Cabezas, A.; Pinto, R.M.; Cameselle, J.C. Fluorimetric HPLC detection of endogenous riboflavin 4′,5′-cyclic phosphate in rat liver at nanomolar concentrations. Anal. Biochem. 2005, 341, 214–219. [Google Scholar] [CrossRef]
- Pinto, R.M.; Fraiz, F.J.; Cabezas, A.; Ávalos, M.; Canales, J.; Costas, M.J.; Cameselle, J.C. Preparation of riboflavin 4′,5′-cyclic phosphate by incubation of flavin-adenine dinucleotide with Mn2+ in the absence of riboflavin 5′-phosphate cyclase. Anal. Biochem. 1999, 268, 409–411. [Google Scholar] [CrossRef]
- Johnston, C.; Hardacre, C.; Migaud, M.E. Investigations into the synthesis of a nucleotide dimer via mechanochemical phosphoramidite chemistry. R. Soc. Open Sci. 2021, 8, 201703. [Google Scholar] [CrossRef]
- Johnston, C.; Hardacre, C.; Migaud, M.E.C. Applications of Mechanochemistry for the Synthesis of DNA on Ionic Liquid Supports. Chem.-Methods 2021, 1, 382–388. [Google Scholar] [CrossRef]
- Davila, A.; Liu, L.; Chellappa, K.; Redpath, P.; Nakamaru-Ogiso, E.; Paolella, L.M.; Zhang, Z.; Migaud, M.E.; Rabinowitz, J.D.; Baur, J.A. Nicotinamide adenine dinucleotide is transported into mammalian mitochondria. eLife 2018, 7, e33246. [Google Scholar] [CrossRef] [PubMed]
- Arsenis, C.; McCormick, D.B. Purificationof Flavin Mononucleotide-dependent Enzymes by Column Chromatography on Flavin Phosphate Cellulose Compounds. J. Biol. Chem. 1966, 241, 330–334. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent | BM (30 Hz) or SP (−20 °C) | Solvent | Compound | Reaction time | Conversion (%) a | By-Product | Yields ^ | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Abbreviation | No. | >60 min | 30 min | 60 min | >60 min | |||||
| (a) Ade (1 eq.) + POCl3(4 eq.) | BM | No solvent | AMP d | 11 | n.d. | 21 a | 36 a | n.d. | Ade 64% a | 36 |
| (b) Ade (1.eq.) + POCl3(3 eq.) | SP | (MeO)3PO | 24 h | n.d. | n.d. | 79 | n.d. | 68 | ||
| (a) NR (1 eq.) + POCl3(4 eq.) | BM | No solvent | NMN d | 12 | n.d. | 44 a | 76 a | n.d. | NR 24% a | 76 |
| (b) NR (1 eq.) + POCl3(3 eq.) | SP | (MeO)3PO | 48 h | n.d. | n.d. | 75 | n.d. | 61 | ||
| (a) PYR (1 eq.) + POCl3 (4 eq.) | BM | No solvent | PYRMP d | 13 | n.d. | 55 a | 80 a | n.d. | PYR 20% a | 80 |
| (b) PYR (1 eq.) + POCl3(3 eq.) | SP | (MeO)3PO | 48 h | 0 | 0 | 0 | mixture | 0 | ||
| C5 deuterated-Adenosine (1 eq.) + POCl3(4 eq.) | SP | (MeO)3PO | C5 deuterated-AMP † | 14 | 48 h | n.d. | n.d. | 80 (a) | C5 deuterated-Adenosine 20% a | 80 |
| 18O-Nicotinamide riboside (1 eq.) + POCl3(4 eq.) | SP | (MeO)3PO | 18O-NMN d | 15 | 120 h | n.d. | n.d. | n.d. | n.d. | 54 |
| Th.HCl (1 eq.) + POCl3(4 eq.) | BM | No solvent | ThMP d | 28 | n.d. | 87 a | 88 a | n.d. | Th 12% a | 88 |
| RF(1 eq.) + POCl3(3 eq.) | BM | No Solvent | FMN d | 32 | x | 0 | 0 | 0 | RF 100% a | 0 |
| SP | Methanol/POCl3 | 16 h | n.d. | n.d. | n.d. | n.d. | 67 | |||
| Reagent and Method (A/B) | Solvent | Compound | Reaction Time | Conversion (%) | By-Product | Yields | |
|---|---|---|---|---|---|---|---|
| Abbreviation | No. | >60 min | >60 min | ||||
| AMP-Imidazolate (1 eq.) + NMN d (1 eq.) (A) | H2O (6 eq.) | NAD+ | 20 | 90 min | [NAD+ (69%) & AP2A (10%)] a&c | (19%) a NMN | 23 * |
| AP2A | 21 | 18 * | |||||
| AMP-Imidazolate (1 eq.) + NMN d (1 eq.) (B) | CAN e | NAD+ | 20 | 2 h | [NAD+ & AP2A (12%)] b&c | [NMN (61%) & AMP-Imidazolate (26%)] b&c | n.d. |
| AP2A | 21 | ||||||
| NMN-Imidazolate + AMP d (A) | H2O (6 eq.) | NAD+ | 20 | 90 min | [NAD+ & NP2N (58%)] b&c | [NMN (22%) & AMP (17%)] b&c | 13 * |
| Double NMN | 22 | 13 * | |||||
| NMN-Imidazolate (1 eq.) + AMP d (1eq.) (B) | CAN e | NAD+ | 20 | 2 h | [NAD+ & AP2A (8%)] b&c | [AMP (51%) & NMN-Imidazolate (36%)] b&c | n.d. |
| Double NMN | 22 | ||||||
| 4PYRMP-Imidazolate (1 eq.) + AMP d (1 eq.) (A) | H2O (6 eq.) | 4-ox-NAD | 23 | 90 min | [4-ox-NAD (68%) & Double 4PYRMP (32%)] a&c | n.d. | |
| Double 4PYRMP | 24 | ||||||
| AMP-Imidazolate (1 eq.) + 4PYRMP d (1 eq.) (A) | H2O (6 eq.) | 4-ox-NAD | 23 | 90 min | [4-ox-NAD & AP2A (82%)] b&c | Monophosphates Mixture (12%) b | n.d. |
| AP2A | 21 | ||||||
| AMP-Imidazolate (1 eq.) + 4PYRMP d (1 eq.) (B) | ACN e | 4-ox-NAD | 23 | 2 h | [4-ox NAD & AP2A (81%)] b&c | [4PYRMP (12%) & AMP-Imidazolate (6%] b&c | 14 * |
| AP2A | 21 | 22 * | |||||
| ThP-Imidazolate (1 eq.) + AMP d (1eq.) (A) | H2O (6 eq.) | AThDP | 30a | 90 min | [AThDP (90%) & ThP2Th (10%)] a&c | (19%)a NMN | 63 * |
| ThP2Th | 30b | ^ | |||||
| ThP-Imidazolate (1 eq.) + AMP d (1 eq.) (B) | ACN e | AThDP | 30a | 2 h | [AThDP & ThP2Th (12%)] b&c | [AMP (43%) & ThP-Imidazolate (44%)] b&c | n.d. |
| ThP2Th | 30b | ||||||
| ThP-Imidazolate (1 eq.) + NMN d (1 eq.) (A) | H2O (6 eq.) | NRThDP | 31 | 90 min | [NRThDP (85%) & ThP2Th (8%)] b&c | NMN (7%) b | 52 * |
| ThP2Th | 30b | ^ | |||||
| 4PYRMP-Imidazolate (1 eq.) + Na2H2P2O7 (1 eq.) (A) | H2O (6 eq.) | Double 4PYRMP | 24 | 90 min | Double 4PYRMP (100%) b | Na2H2P2O7 (100%) b | 43 * |
| NMN-Imidazolate (1 eq.) + FMN d (1 eq.) (A) | H2O (6 eq.) | FMN-NMN | 33 | 90 min | [FMN-NMN, Double NMN (51%) & Cyclic FMN (5%)] b&c | [NMN and FMN (35%)] b&c | 32 * |
| Double NMN | 22 | 14 * | |||||
| Cyclic FMN | 34 | ^ | |||||
| AMP-Imidazolate (1 eq.) + FMN d (1 eq.) (A) | H2O (6 eq.) | FAD | 35 | 90 min | [FAD, AP2A (67%) b & Cyclic FMN (10%)] b&c | [AMP and FMN (23%)] b&c | ^ |
| AP2A | 21 | ^ | |||||
| Cyclic FMN | 34 | ^ | |||||
| AMP-Imidazolate (1 eq.) + FMN d (1 eq.) (B) | ACN e | FAD | 35 | 2 h | FAD &AP2A (14%) b&c | [FMN (36%) & AMP- Imidazolate (48%)] b | ^ |
| AP2A | 21 | ^ | |||||
| 18O-NMN-Imidazolate + C5 deuterated-AMP (A) | H2O (6 eq.) | 18O (M+4) NAD+ | 25 | 90 min | [18O (M + 4) NAD+ & 18O (M + 4) double NMN (29%) b&c | [18O (M + 2) NMN+ & (M + 2) C5-deuterated adenosine (71%)] b&c | 8 * |
| 18O (M + 4) double NMN | 26 | 11* | |||||
| AMP-Imidazolate + Thiamine diphosphate | H2O (6 eq.) | AThTP | 37 | 90 min | [AThTP (25%)& AP2A(48%)] b&c | [AMP (4%) & ThDp-imidazolate (22%)] b&c | 18* |
| AP2A | 21 | n.d. | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayat, F.; Makarov, M.V.; Belfleur, L.; Migaud, M.E. Synthesis of Mixed Dinucleotides by Mechanochemistry. Molecules 2022, 27, 3229. https://doi.org/10.3390/molecules27103229
Hayat F, Makarov MV, Belfleur L, Migaud ME. Synthesis of Mixed Dinucleotides by Mechanochemistry. Molecules. 2022; 27(10):3229. https://doi.org/10.3390/molecules27103229
Chicago/Turabian StyleHayat, Faisal, Mikhail V. Makarov, Luxene Belfleur, and Marie E. Migaud. 2022. "Synthesis of Mixed Dinucleotides by Mechanochemistry" Molecules 27, no. 10: 3229. https://doi.org/10.3390/molecules27103229