
Computational Studies of Coinage Metal Anion M− + CH3X (X = F, Cl, Br, I) Reactions in Gas Phase


Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
3.1. Reaction Enthalpies
3.2. Potential Energy Profiles
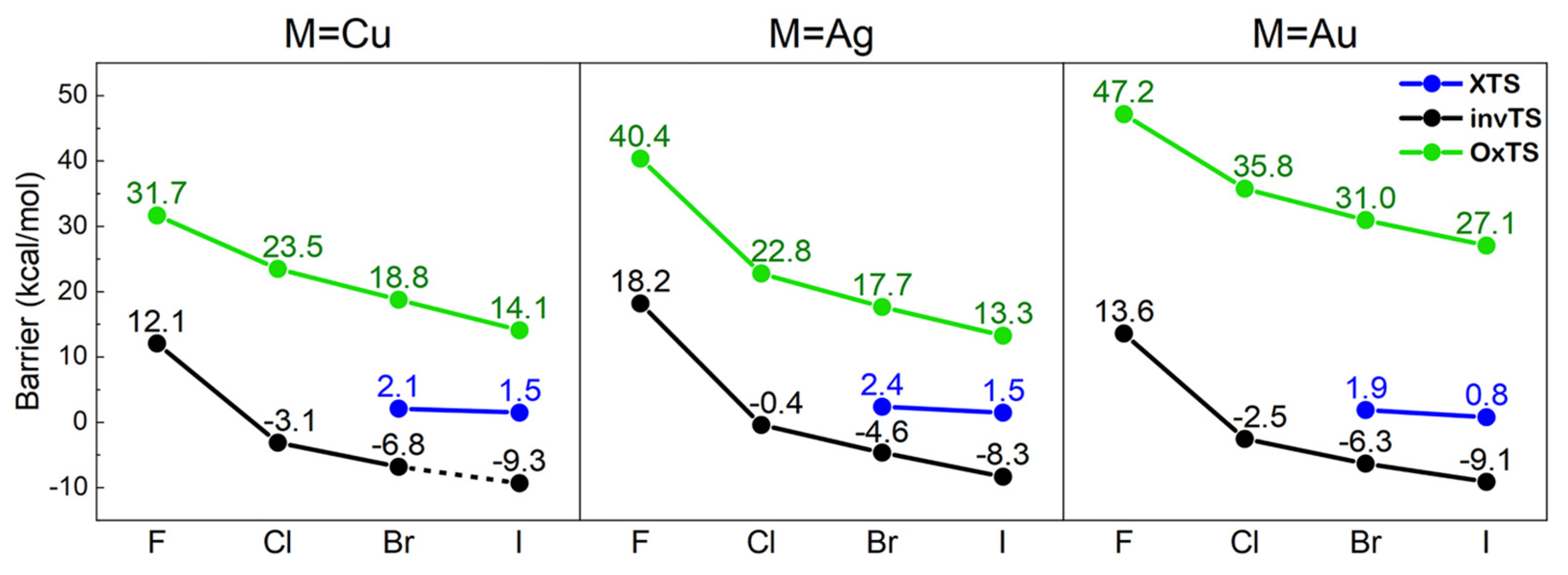
3.2.1. The Back-Side Attack Pathway
3.2.2. The Front-Side Attack at C Atom Pathway
3.2.3. The Halogen-Bonded Complex Pathway
3.2.4. Comparison between Metallic Nucleophiles M− and Main Group Nucleophiles F−
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Crabtree, R.H. Organometallic Chemistry of the Transition Metals; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Labinger, J.A.; Bercaw, J.E. Understanding and exploiting C-H bond activation. Nature 2002, 33, 507–514. [Google Scholar] [CrossRef]
- Dupont, J.; Consorti, C.S.; Spencer, J. The Potential of Palladacycles: More than Just Precatalysts. Chem. Rev. 2005, 36, 2527–2571. [Google Scholar] [CrossRef] [PubMed]
- Pud De Phatt, R.J. Platinum(IV) hydride chemistry. Coord. Chem. Rev. 2001, 219, 157–185. [Google Scholar] [CrossRef]
- Puddephatt, R.J. Coordinative Unsaturation in Platinum(IV) Chemistry: From Proposed Reaction Intermediates to the First Structurally Characterized Complexes. Angew. Chem. Int. Ed. 2002, 41, 261–263. [Google Scholar] [CrossRef]
- Rendina, L.M.; Puddephatt, R.J. Oxidative Addition Reactions of Organoplatinum(II) Complexes with Nitrogen-Donor Ligands. Chem. Rev. 1997, 28, 1735–1754. [Google Scholar] [CrossRef]
- Crespo, M.; Martinez, M.; Nabavizadeh, S.M.; Rashidi, M. Kinetico-mechanistic studies on C-X (X = H, F, Cl, Br, I) bond activation reactions on organoplatinum(II) complexes. Coord. Chem. Rev. 2014, 279, 115–140. [Google Scholar] [CrossRef]
- Diefenbach, A.; de Jong, G.T.; Bickelhaupt, F.M. Activation of H-H, C-H, C-C and C-Cl bonds by Pd and PdCl−. Understanding anion assistance in C-X bond activation. J. Chem. Theory Comput. 2005, 1, 286–298. [Google Scholar] [CrossRef] [PubMed]
- de Jong, G.T.; Bickelhaupt, F.M. Catalytic carbon−halogen bond activation: Trends in reactivity, selectivity, and solvation. J. Chem. Theory Comput. 2007, 3, 514–529. [Google Scholar] [CrossRef]
- Hartwig, J.F. Transition metal catalyzed synthesis of arylamines and aryl ethers from aryl halides and triflates: Scope and mechanism. Angew. Chem. Int. Ed. 1998, 37, 2046–2067. [Google Scholar] [CrossRef]
- Martin, R.; Buchwald, S.L. Palladium-catalyzed Suzuki-Miyaura cross-coupling reactions employing dialkylbiaryl phosphine ligands. Acc. Chem. Res. 2008, 41, 1461–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barder, T.E.; Walker, S.D.; Martinelli, J.R.; Buchwald, S.L. Catalysts for Suzuki-Miyaura coupling processes: Scope and studies of the effect of ligand structure. J. Am. Chem. Soc. 2005, 127, 4685–4696. [Google Scholar] [CrossRef]
- Cundari, T.R.; Vaddadi, S. Carbon-hydrogen and carbon-heteroatom bond activation using iridium (I) complexes. Inorg. Chim. Acta 2004, 357, 2863–2869. [Google Scholar] [CrossRef]
- Chowdhury, A.K.; Wilkins, C.L. Reactions of atomic gold ions with aliphatic and aromatic hydrocarbons and alkyl halides. J. Am. Chem. Soc. 1987, 109, 5336–5343. [Google Scholar] [CrossRef]
- Muramatsu, S.; Tsukuda, T. Reductive Activation of Small Molecules by Anionic Coinage Metal Atoms and Clusters in the Gas Phase. Chem. Asian J. 2019, 14, 3763–3772. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.C.; Wang, Y.G.; Li, J. Toward rational design of oxide-supported single-atom catalysts: Atomic dispersion of gold on ceria. J. Am. Chem. Soc. 2017, 139, 6190–6199. [Google Scholar] [CrossRef]
- Grisel, R.; Weststrate, K.J.; Gluhoi, A.; Nieuwenhuys, B.E. Catalysis by gold nanoparticles. Gold Bull. 2002, 35, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Mitsudome, T.; Kaneda, K. Gold nanoparticle catalysts for selective hydrogenations. Green Chem. 2013, 15, 2636–2654. [Google Scholar] [CrossRef]
- Muramatsu, S.; Koyasu, K.; Tsukuda, T. Oxidative Addition of CH3I to Au− in the Gas Phase. J. Phys. Chem. A 2016, 120, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhao, Y.X.; Jiang, L.X.; Chen, J.J.; He, S.G. Coupling of methane and carbon dioxide mediated by diatomic copper boride cations. Angew. Chem. Int. Ed. 2018, 57, 14134–14138. [Google Scholar] [CrossRef]
- Wester, R. Fifty years of nucleophilic substitution in the gas phase. Mass Spectrom. Rev. 2021. [Google Scholar] [CrossRef]
- Ma, J.B.; Xu, L.L.; Liu, Q.Y.; He, S.G. Activation of methane and ethane as mediated by the triatomic anion HNbN−: Electronic structure similarity with a Pt atom. Angew. Chem. Int. Ed. 2016, 55, 4947–4951. [Google Scholar] [CrossRef]
- Wang, W.; Feng, W.; Wang, W.; Li, P. Ab initio molecular dynamics simulation study on the stereo reactions between atomic oxygen anion and methane. Molecules 2018, 23, 2495. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Hase, W.L. Rethinking the SN2 reaction. Science 2016, 352, 32–33. [Google Scholar] [CrossRef]
- Szabó, I.; Czakó, G. Revealing a double-inversion mechanism for the F− + CH3Cl SN2 reaction. Nat. Commun. 2015, 6, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Rijs, N.J.; González-Navarrete, P.; Schlangen, M.; Schwarz, H. Penetrating the Elusive Mechanism of Copper-Mediated Fluoromethylation in the Presence of Oxygen through the Gas-Phase Reactivity of Well-Defined [LCuO]+ Complexes with Fluoromethanes (CH4–nFn, n= 1–3). J. Am. Chem. Soc. 2016, 138, 3125–3135. [Google Scholar] [CrossRef]
- Meyer, J.; Tajti, V.; Carrascosa, E.; Győri, T.; Stei, M.; Michaelsen, T.; Bastian, B.; Czakó, G.; Wester, R. Atomistic dynamics of elimination and nucleophilic substitution disentangled for the F− + CH3CH2Cl reaction. Nat. Chem. 2021, 13, 977–981. [Google Scholar] [CrossRef] [PubMed]
- Stei, M.; Carrascosa, E.; Kainz, M.A.; Kelkar, A.H.; Meyer, J.; Szabó, I.; Czakó, G.; Wester, R. Influence of the leaving group on the dynamics of a gas-phase SN2 reaction. Nat. Chem. 2016, 8, 151–156. [Google Scholar] [CrossRef] [Green Version]
- Viggiano, A.A.; Ard, S.G.; Shuman, N.S. Temperature and energy dependences of ion-molecule reactions: Studies inspired by Diethard Böhme. Mass Spectrom. Rev. 2021. [Google Scholar] [CrossRef]
- Muramatsu, S.; Koyasu, K.; Tsukuda, T. Formation of Grignard Reagent-like Complex [CH3–M–I]− via Oxidative Addition of CH3I on Coinage Metal Anions M−(M= Cu, Ag, Au) in the Gas Phase. Chem. Lett. 2017, 46, 676–679. [Google Scholar] [CrossRef] [Green Version]
- Mikosch, J.; Trippel, S.; Eichhorn, C.; Otto, R.; Lourderaj, U.; Zhang, J.X.; Hase, W.L.; Weidemüller, M.; Wester, R. Imaging nucleophilic substitution dynamics. Science 2008, 319, 183–186. [Google Scholar] [CrossRef]
- Xie, J.; Sun, R.; Siebert, M.R.; Otto, R.; Wester, R.; Hase, W.L. Direct dynamics simulations of the product channels and atomistic mechanisms for the OH– + CH3I reaction. Comparison with experiment. J. Phys. Chem. A 2013, 117, 7162–7178. [Google Scholar] [CrossRef]
- Zhang, J.X.; Mikosch, J.; Trippel, S.; Otto, R.; Weidemüller, M.; Wester, R.; Hase, W.L. F− + CH3I → FCH3 + I− Reaction Dynamics. Nontraditional Atomistic Mechanisms and Formation of a Hydrogen-Bonded Complex. J. Phys. Chem. Lett. 2010, 18, 2747–2752. [Google Scholar] [CrossRef]
- Purvis III, G.D.; Bartlett, R.J. A full coupled-cluster singles and doubles model: The inclusion of disconnected triples. J. Chem. Phys. 1982, 76, 1910–1918. [Google Scholar] [CrossRef]
- Pople, J.A.; Head-Gordon, M.; Raghavachari, K. Quadratic configuration interaction. A general technique for determining electron correlation energies. J. Chem. Phys. 1987, 87, 5968–5975. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Tasi, D.A.; Fábián, Z.; Czakó, G. Benchmark ab Initio Characterization of the Inversion and Retention Pathways of the OH– + CH3Y [Y= F, Cl, Br, I] SN2 Reactions. J. Phys. Chem. A 2018, 122, 5773–5780. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. J. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical meta-generalized gradient approximation designed for molecules and solids. J. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Ji, X.Y.; Zhao, C.Y.; Xie, J. Investigating the role of halogen-bonded complexes in microsolvated Y−(H2O)n + CH3I SN2 reactions. Phys. Chem. Chem. Phys. 2021, 23, 6349–6360. [Google Scholar] [CrossRef]
- Lee, T.J.; Taylor, P.R. A diagnostic for determining the quality of single-reference electron correlation methods. Int. J. Quantum Chem. 1989, 36, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Roos, B.O.; Taylor, P.R.; Sigbahn, P.E.M. A complete active space SCF method (CASSCF) using a density matrix formulated super-CI approach. Chem. Phys. 1980, 48, 157–173. [Google Scholar] [CrossRef]
- Feller, D. The role of databases in support of computational chemistry calculations. J. Comput. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New basis set exchange: An open, up-to-date resource for the molecular sciences community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef] [PubMed]
- Roos, B.O.; Lindh, R.; Malmqvist, P.Å.; Veryazov, V.; Widmark, P.O. New relativistic ANO basis sets for transition metal atoms. J. Phys. Chem. A 2005, 109, 6575–6579. [Google Scholar] [CrossRef] [PubMed]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis set exchange: A community database for computational sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Douglas, M.; Kroll, N.M. Quantum electrodynamical corrections to the fine structure of helium. Ann. Phys. 1974, 82, 89–155. [Google Scholar] [CrossRef]
- Hess, B.A. Applicability of the no-pair equation with free-particle projection operators to atomic and molecular structure calculations. Phys. Rev. A 1985, 32, 756. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.A. Relativistic electronic-structure calculations employing a two-component no-pair formalism with external-field projection operators. Phys. Rev. A 1986, 33, 3742. [Google Scholar] [CrossRef] [Green Version]
- Aquilante, F.; Autschbach, J.; Carlson, R.K.; Chibotaru, L.F.; Delcey, M.G.; De Vico, L.; Fdez. Galván, I.; Ferré, N.; Frutos, L.M.; Gagliardi, L.; et al. Molcas 8: New capabilities for multiconfigurational quantum chemical calculations across the periodic table. J. Comput. Chem. 2016, 506–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Bickelhaupt, F.M.; Ziegler, T. Oxidative insertion as frontside SN2 substitution: A theoretical study of the model reaction system Pd + CH3Cl. Organometallics 1995, 14, 2288–2296. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Struct. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, G.; Ciborowski, S.; Bowen, K. Stabilizing otherwise unstable anions with halogen bonding. Angew. Chem. 2017, 129, 10029–10032. [Google Scholar] [CrossRef] [Green Version]
- Mallada, B.; Gallardo, A.; Lamanec, M.; de la Torre, B.; Špirko, V.; Hobza, P.; Jelinek, P. Real-space imaging of anisotropic charge of σ-hole by means of Kelvin probe force microscopy. Science 2021, 374, 863–867. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Products | X | F | Cl | Br | I |
|---|---|---|---|---|---|
| M = Cu | |||||
| OI | [CH3–Cu–X]− | −64.3 | −79.8 | −81.2 | −82.1 |
| SN2 | CuCH3 + X− | 4.2 | −27.1 | −31.9 | −37.6 |
| XA | CuX− + CH3 | 12.3 | −5.1 | −7.2 | −9.1 |
| PT | HCu + CH2X− | 62.5 | 49.8 | 46.3 | 41.4 |
| M = Ag | |||||
| OI | [CH3–Ag–X]− | −37.5 | −57.8 | −61.0 | −63.9 |
| SN2 | AgCH3 + X− | 18.1 | −13.1 | −18.0 | −23.6 |
| XA | AgX− + CH3 | 26.4 | 4.3 | 0.6 | −3.0 |
| PT | HAg + CH2X− | 73.1 | 60.4 | 56.8 | 52.0 |
| M = Au | |||||
| OI | [CH3−Au−X]− | −38.0 | −57.3 | −60.4 | −63.6 |
| SN2 | AuCH3 + X− | 21.8 | −9.5 | −14.3 | −20.0 |
| XA | AuX− + CH3 | 43.7 | 20.7 | 15.9 | 10.5 |
| PT | HAu + CH2X− | 77.8 | 65.1 | 61.5 | 56.6 |
| X | F | Cl | Br | I | ||||
|---|---|---|---|---|---|---|---|---|
| E | H | E | H | E | H | E | H | |
| M = Cu | ||||||||
| Cu− + CH3X | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| RC | −6.6 | −5.0 | −8.0 | −6.5 | −8.7 | −7.2 | −9.8 | −8.7 |
| invTS | 13.4 | 12.1 | −2.9 | −3.1 | −6.9 | −6.8 | −9.8 | −9.3 |
| PC | 4.7 | 3.4 | −25.6 | −25.9 | −31.1 | −31.0 | −36.4 | −35.9 |
| XC | - | - | - | - | −1.8 | −0.5 | −11.8 | −11.3 |
| XTS | - | - | - | - | 1.3 | 2.1 | 0.4 | 1.5 |
| OxTS | 33.3 | 31.7 | 24.3 | 23.5 | 19.3 | 18.8 | 14.3 | 14.1 |
| MTS | 4.6 | 2.7 | −25.7 | −26.5 | - | - | - | - |
| HPC | −0.7 | −2.5 | - | - | - | - | - | - |
| HPTS | 0.0 | −2.1 | - | - | −32.6 | −33.0 | −38.2 | −38.3 |
| M = Ag | ||||||||
| Ag− + CH3X | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| RC | −6.4 | −4.8 | −7.7 | −6.1 | −8.3 | −6.8 | −9.3 | −7.8 |
| invTS | 19.6 | 18.2 | −0.1 | −0.4 | −4.6 | −4.6 | −8.5 | −8.3 |
| PC | 17.9 | 16.8 | −11.5 | −11.6 | −17.0 | −16.7 | −22.9 | −22.4 |
| XC | - | - | - | - | -1.4 | 0.0 | −7.9 | −6.9 |
| XTS | - | - | - | - | 1.1 | 2.4 | 0.3 | 1.5 |
| OxTS | 42.0 | 40.4 | 23.7 | 22.8 | 18.2 | 17.7 | 13.6 | 13.3 |
| MTS | 17.9 | 16.3 | - | - | - | - | - | - |
| HPC | 13.3 | 13.2 | - | - | - | - | - | - |
| HPTS | 16.1 | 14.1 | −13.8 | −14.5 | −17.7 | −18.1 | −24.6 | −24.6 |
| M = Au | ||||||||
| Au− + CH3X | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| RC | −8.6 | −7.0 | −10.0 | −8.4 | −10.6 | −9.0 | −11.1 | −9.6 |
| invTS | 14.2 | 13.6 | −2.7 | −2.5 | −6.7 | −6.3 | −9.6 | −9.1 |
| PC | 12.5 | 12.0 | −15.1 | −14.5 | −20.2 | −19.3 | −25.6 | −24.5 |
| XC | - | - | - | - | −3.3 | −1.8 | −11.0 | −10.0 |
| XTS | - | - | - | - | 0.7 | 1.9 | −0.3 | 0.8 |
| OxTS | 48.2 | 47.2 | 36.2 | 35.8 | 31.1 | 31.0 | 27.2 | 27.1 |
| MTS | 12.9 | 11.7 | −14.7 | −14.8 | - | - | - | - |
| HPC | 10.3 | 9.0 | −14.7 | −14.2 | - | - | - | - |
| HPTS | 13.1 | 11.7 | −14.0 | −14.1 | −19.2 | −18.9 | −24.4 | −23.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.; Ji, X.; Ying, F.; Zhang, J.; Zhao, C.; Xie, J. Computational Studies of Coinage Metal Anion M− + CH3X (X = F, Cl, Br, I) Reactions in Gas Phase. Molecules 2022, 27, 307. https://doi.org/10.3390/molecules27010307
Wang F, Ji X, Ying F, Zhang J, Zhao C, Xie J. Computational Studies of Coinage Metal Anion M− + CH3X (X = F, Cl, Br, I) Reactions in Gas Phase. Molecules. 2022; 27(1):307. https://doi.org/10.3390/molecules27010307
Chicago/Turabian StyleWang, Fan, Xiaoyan Ji, Fei Ying, Jiatao Zhang, Chongyang Zhao, and Jing Xie. 2022. "Computational Studies of Coinage Metal Anion M− + CH3X (X = F, Cl, Br, I) Reactions in Gas Phase" Molecules 27, no. 1: 307. https://doi.org/10.3390/molecules27010307