
Triptycene Derivatives: From Their Synthesis to Their Unique Properties


Abstract
:1. Introduction
2. Progress in Triptycene Synthesis and Derivatization
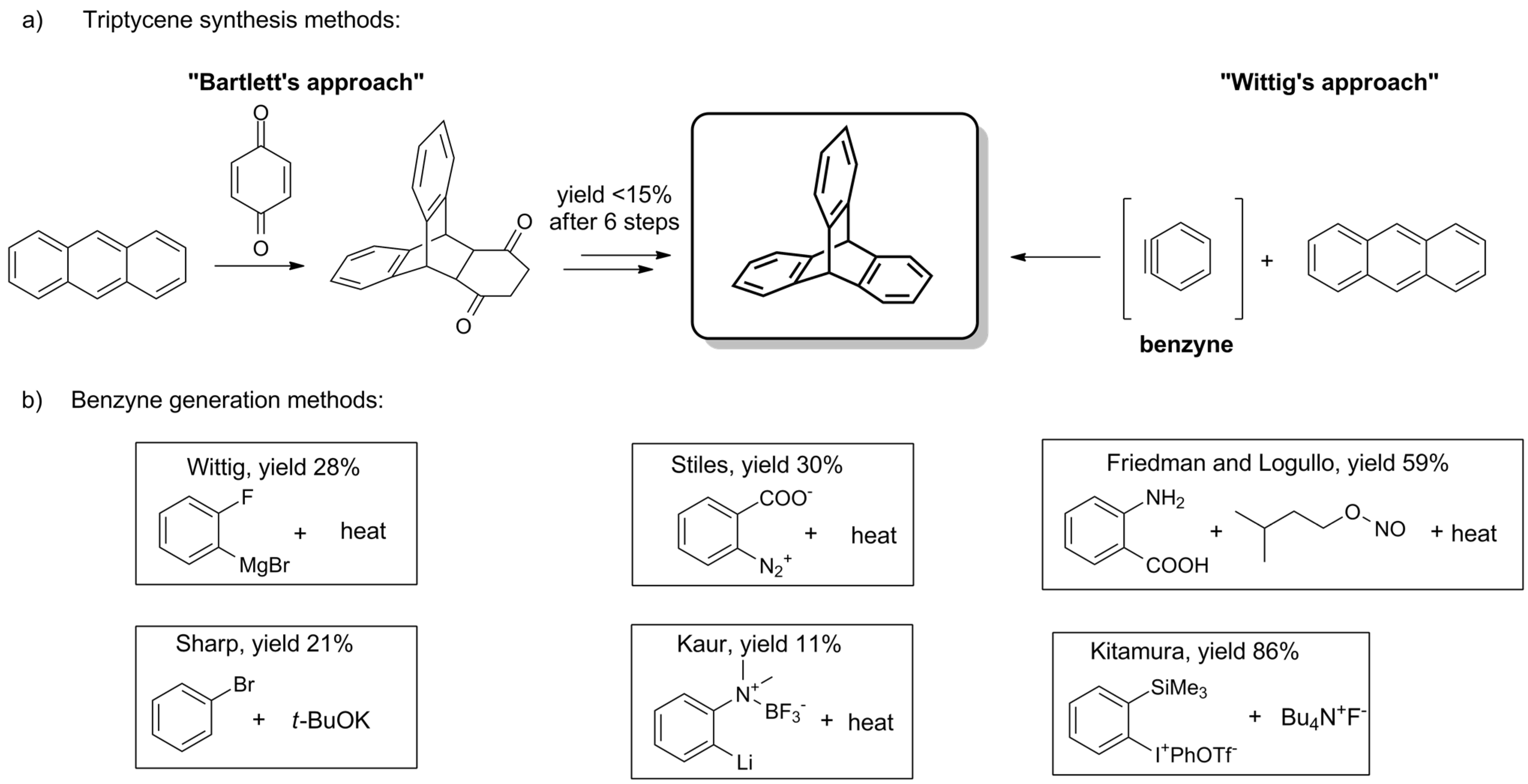
2.1. General Synthesis of the Triptycene Unit
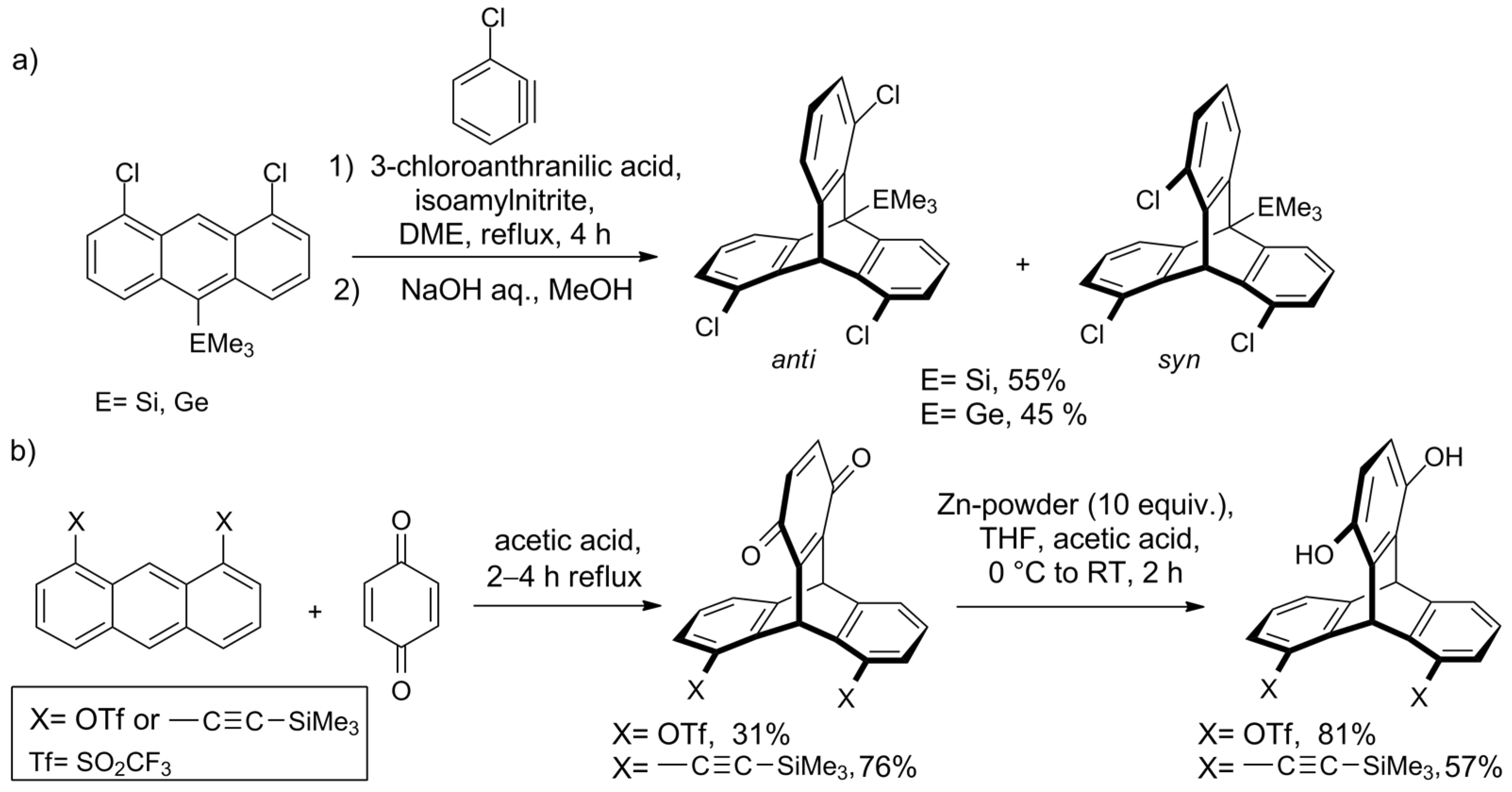
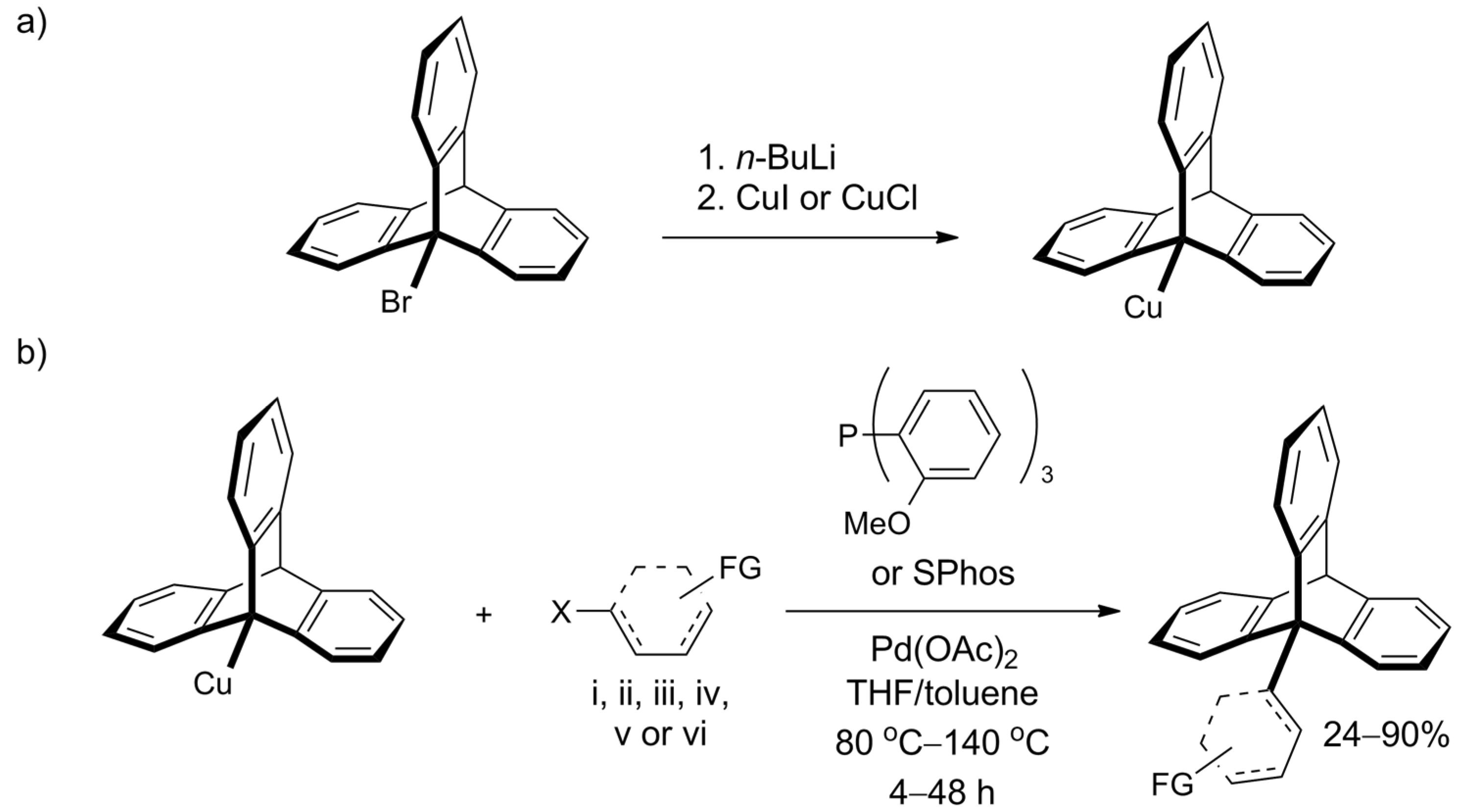
2.2. Synthesis of Ortho-Functionalized Triptycenes
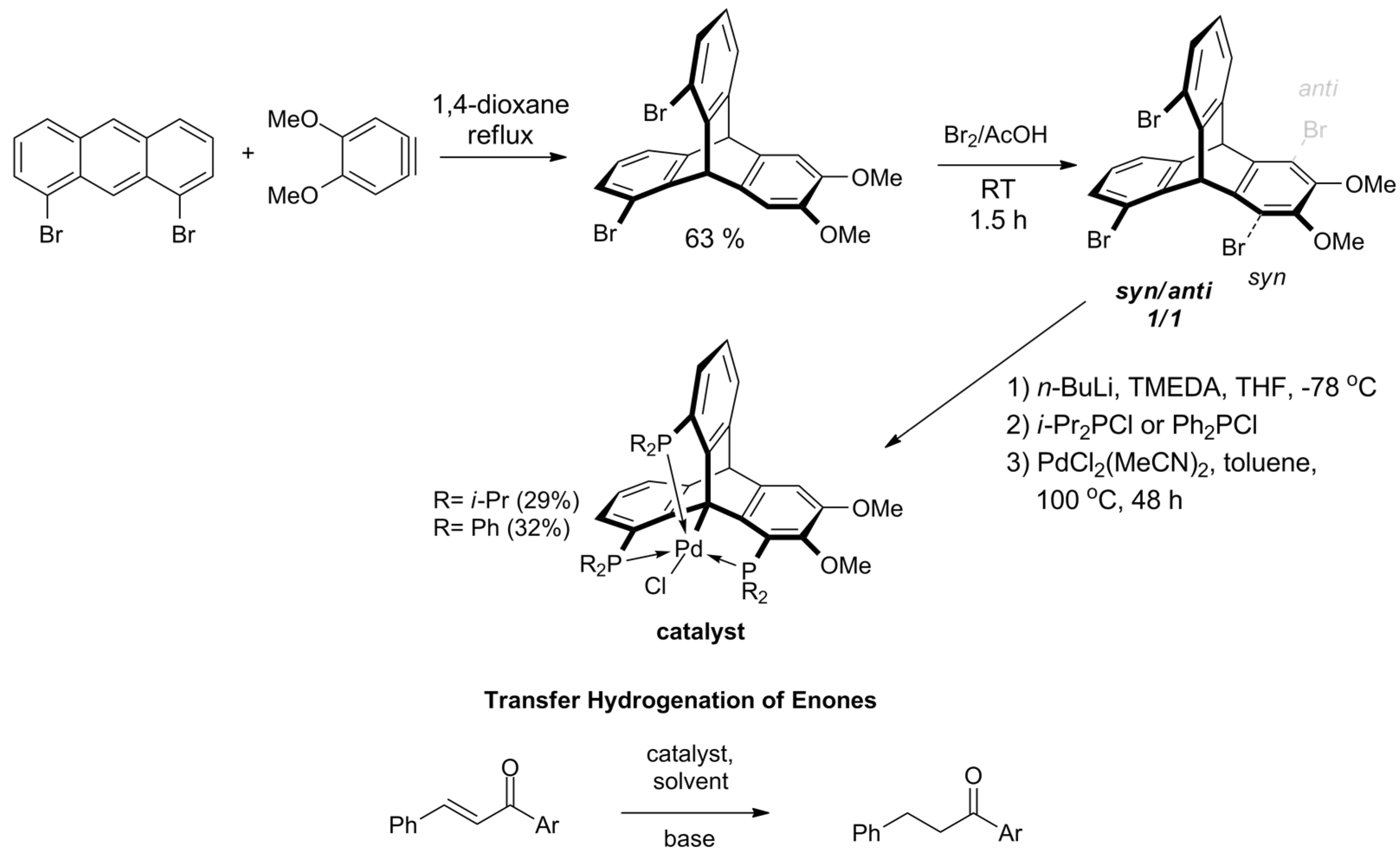
2.3. Functionalization of the Bridgehead Positions of the Triptycene Unit
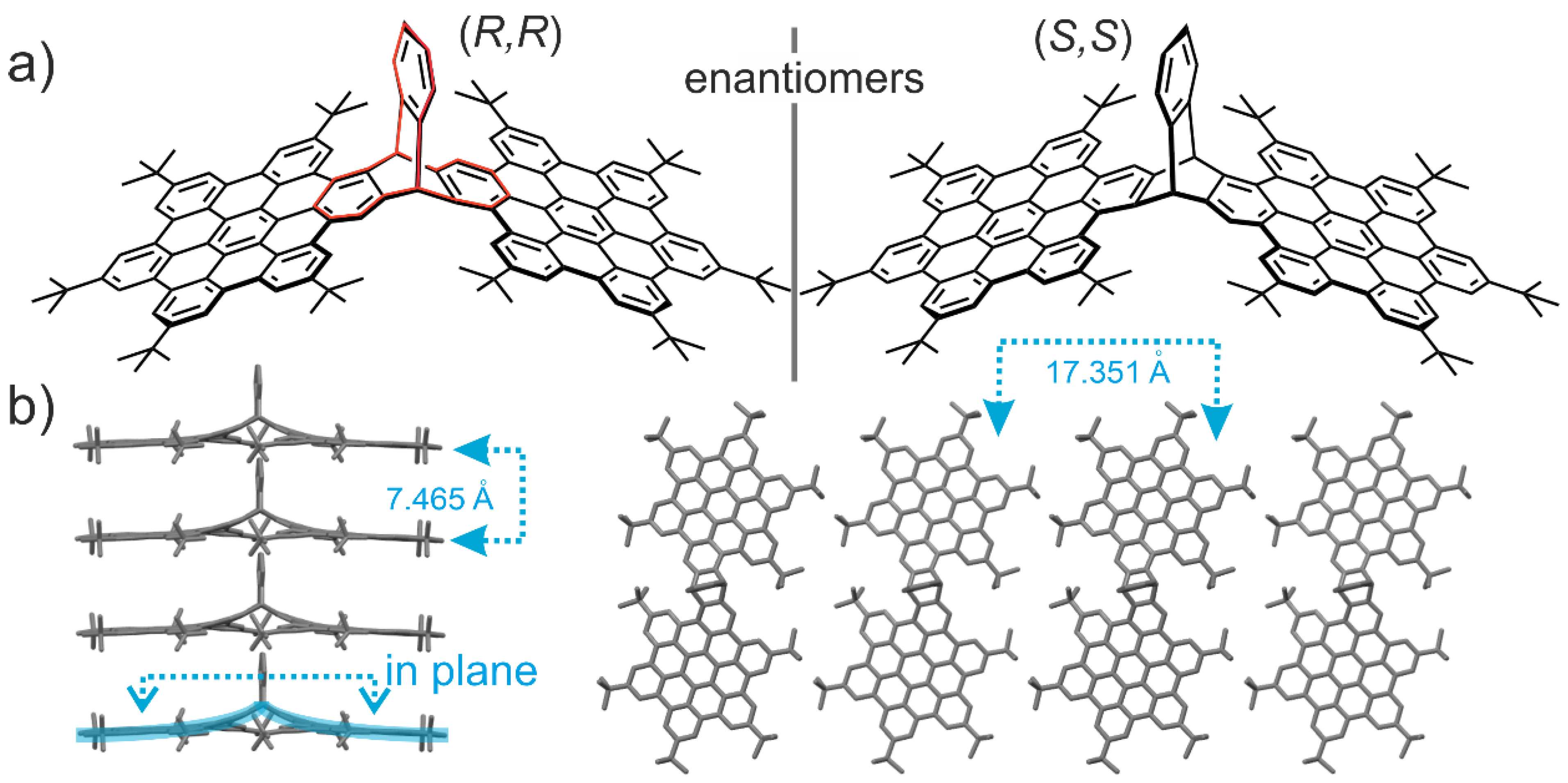
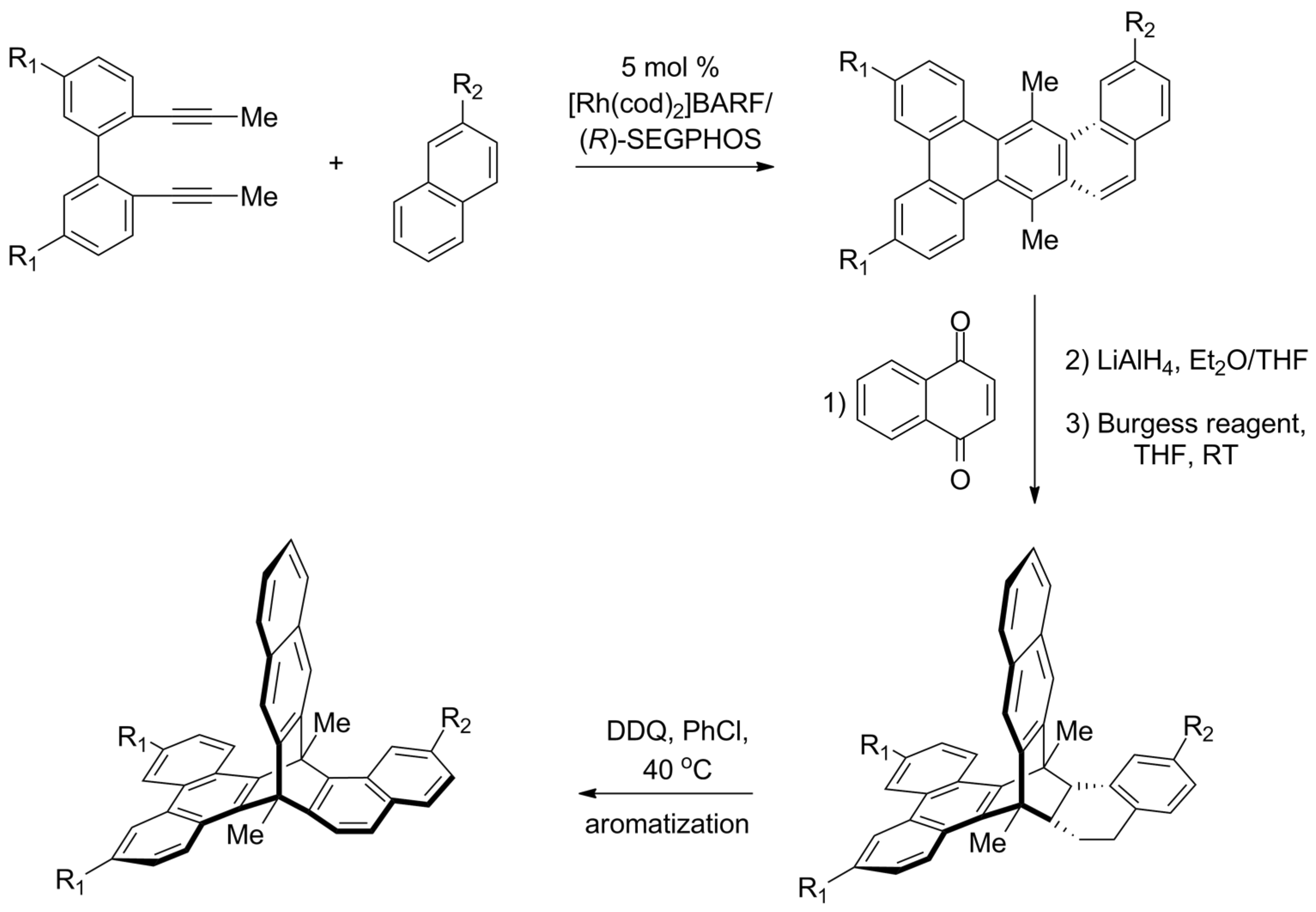
2.4. Synthesis of Chiral Triptycenes
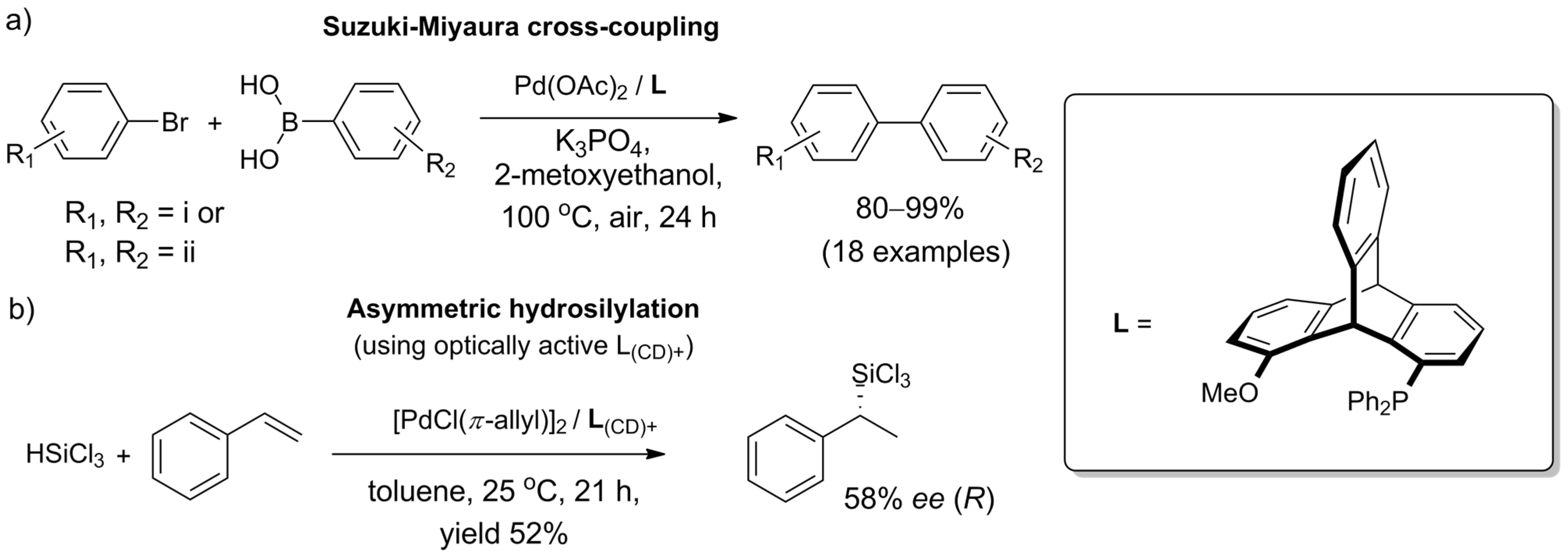
2.5. Triptycenes in Catalysis
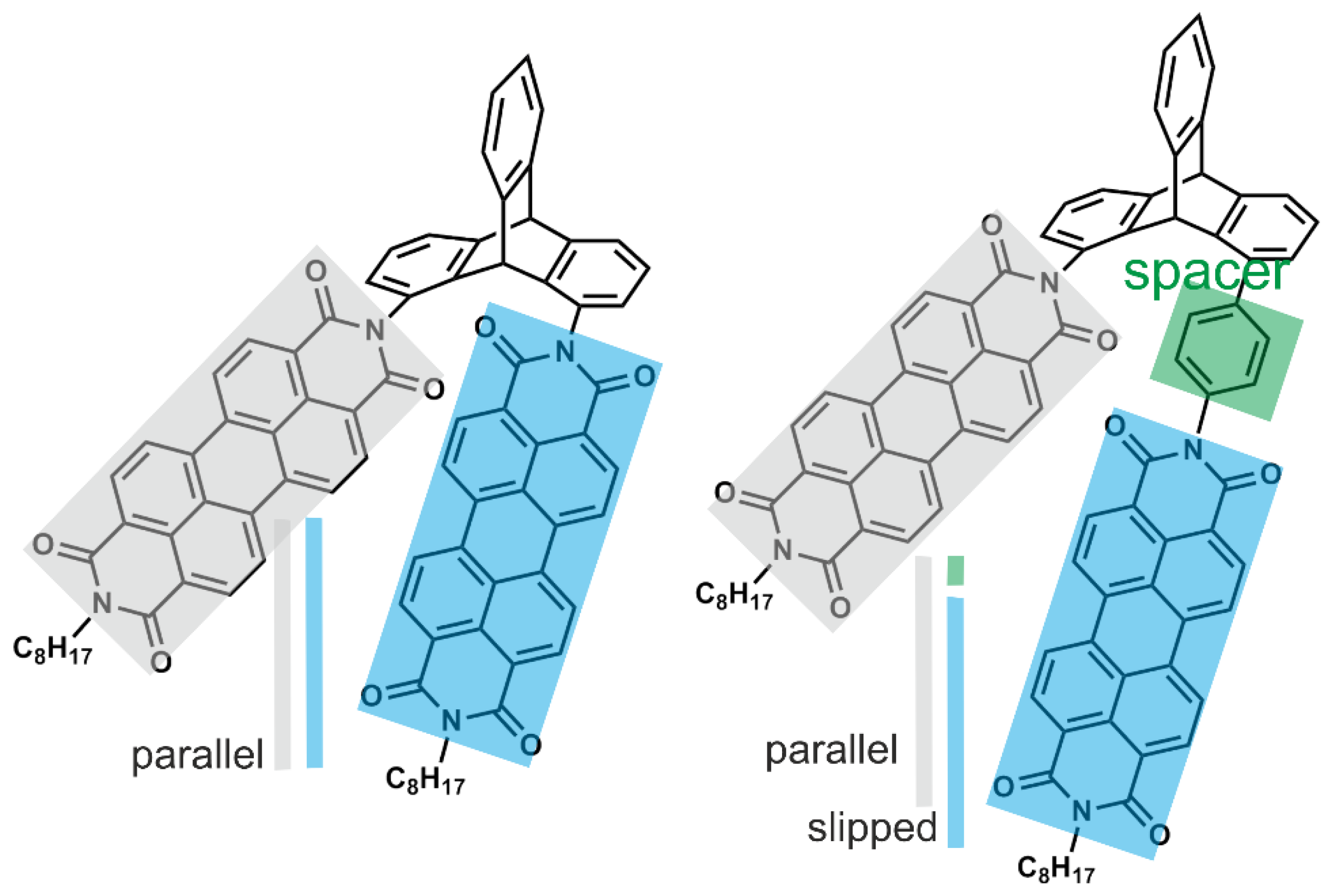
3. Triptycenes: From the Inter- and Intramolecular Interactions to the Properties
3.1. Triptycenes and Molecular Interactions
3.2. Photo-Physical Properties of Some Triptycenes
4. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bartlett, P.D.; Ryan, M.J.; Cohen, S.G. Triptycene1 (9,10-O-Benzenoanthracene). J. Am. Chem. Soc. 1942, 64, 2649–2653. [Google Scholar] [CrossRef]
- Wittig, G.; Ludwig, R. Triptycen aus Anthracen und Dehydrobenzol. Angew. Chem. 1956, 68, 40. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, C.-F. Recent Developments in Synthesis and Applications of Triptycene and Pentiptycene Derivatives. Eur. J. Org. Chem. 2011, 2011, 6377–6403. [Google Scholar] [CrossRef]
- Chen, C.F.; Ma, Y.X. Iptycenes Chemistry: From Synthesis to Applications; Springer: Berlin/Heidelberg, Germany, 2013; ISBN 978-3-642-32887-9. [Google Scholar]
- Zhao, L.; Li, Z.; Wirth, T. Triptycene Derivatives: Synthesis and Applications. Chem. Lett. 2010, 39, 658–667. [Google Scholar] [CrossRef]
- Ma, Y.-X.; Meng, Z.; Chen, C.-F. Synthesis of Substituted Iptycenes. Synlett 2015, 26, 6–30. [Google Scholar]
- Preda, G.; Nitti, A.; Pasini, D. Chiral Triptycenes in Supramolecular and Materials Chemistry. ChemistryOpen 2020, 9, 719–727. [Google Scholar] [CrossRef]
- Long, T.M.; Swager, T.M. Minimization of Free Volume: Alignment of Triptycenes in Liquid Crystals and Stretched Polymers. Adv. Mater. 2001, 13, 601–604. [Google Scholar] [CrossRef]
- Gakh, A.A. Molecular Devices: An Introduction to Technomimetics and Its Biological Applications; John Wiley & Sons: Hoboken, NJ, USA, 2018; ISBN 978-1-119-44813-6. [Google Scholar]
- Drioli, E.; Giorno, L. Comprehensive Membrane Science and Engineering; Elsevier: Amsterdam, The Netherlands, 2017; ISBN 978-0-444-53204-6. [Google Scholar]
- Chen, C.-F.; Han, Y. Triptycene-Derived Macrocyclic Arenes: From Calixarenes to Helicarenes. Acc. Chem. Res. 2018, 51, 2093–2106. [Google Scholar] [CrossRef] [PubMed]
- Ben Saida, A.; Chardon, A.; Osi, A.; Tumanov, N.; Wouters, J.; Adjieufack, A.I.; Champagne, B.; Berionni, G. Pushing the Lewis Acidity Boundaries of Boron Compounds With Non-Planar Triarylboranes Derived from Triptycenes. Angew. Chem. Int. Ed. 2019, 58, 16889–16893. [Google Scholar] [CrossRef]
- Hu, L.; Mahaut, D.; Tumanov, N.; Wouters, J.; Robiette, R.; Berionni, G. Complementary Synthetic Approaches toward 9-Phosphatriptycene and Structure–Reactivity Investigations of Its Association with Sterically Hindered Lewis Acids. J. Org. Chem. 2019, 84, 11268–11274. [Google Scholar] [CrossRef]
- Locke, G.M.; Bernhard, S.S.R.; Senge, M.O. Nonconjugated Hydrocarbons as Rigid-Linear Motifs: Isosteres for Material Sciences and Bioorganic and Medicinal Chemistry. Chem. Eur. J. 2019, 25, 4590–4647. [Google Scholar] [CrossRef]
- McGlinchey, M.J.; Nikitin, K. Palladium-Catalysed Coupling Reactions En Route to Molecular Machines: Sterically Hindered Indenyl and Ferrocenyl Anthracenes and Triptycenes, and Biindenyls. Molecules 2020, 25, 1950. [Google Scholar] [CrossRef] [PubMed]
- Usman, M.; Ahmed, A.; Yu, B.; Peng, Q.; Shen, Y.; Cong, H. A review of different synthetic approaches of amorphous intrinsic microporous polymers and their potential applications in membrane-based gases separation. Eur. Polym. J. 2019, 120, 109262. [Google Scholar] [CrossRef]
- Zhou, H.; Jin, W. Membranes with Intrinsic Micro-Porosity: Structure, Solubility, and Applications. Membranes 2018, 9, 3. [Google Scholar] [CrossRef] [Green Version]
- Mastalerz, M. Porous Shape-Persistent Organic Cage Compounds of Different Size, Geometry, and Function. Acc. Chem. Res. 2018, 51, 2411–2422. [Google Scholar] [CrossRef]
- Jacquot de Rouville, H.-P.; Kammerer, C.; Rapenne, G. From the Synthesis of Nanovehicles to Participation in the First Nanocar Race—View from the French Team. Molecules 2018, 23, 612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Rong, H.; Zou, X.; Zhu, G. Engineering microporous organic framework membranes for CO2 separations. Mol. Syst. Des. Eng. 2017, 2, 182–190. [Google Scholar] [CrossRef]
- Weidman, J.R.; Guo, R. The Use of Iptycenes in Rational Macromolecular Design for Gas Separation Membrane Applications. Ind. Eng. Chem. Res. 2017, 56, 4220–4236. [Google Scholar] [CrossRef]
- Chérioux, F.; Galangau, O.; Palmino, F.; Rapenne, G. Controlled Directional Motions of Molecular Vehicles, Rotors, and Motors: From Metallic to Silicon Surfaces, a Strategy to Operate at Higher Temperatures. ChemPhysChem 2016, 17, 1742–1751. [Google Scholar] [CrossRef]
- Kelly, T.R.; Silva, R.A.; De Silva, H.; Jasmin, S.; Zhao, Y. A Rationally Designed Prototype of a Molecular Motor. J. Am. Chem. Soc. 2000, 122, 6935–6949. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Guo, J.-B.; Chen, C.-F. A bifunctionalized [3]rotaxane and its incorporation into a mechanically interlocked polymer. Chem. Commun. 2010, 46, 5536–5538. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Sun, W.-T.; Lu, H.-F.; Chao, I.; Huang, G.-J.; Lin, Y.-C.; Huang, S.-L.; Huang, H.-H.; Lin, Y.-D.; Yang, J.-S. A Pentiptycene-Derived Molecular Brake: Photochemical E→Z and Electrochemical Z→E Switching of an Enone Module. Chem. Eur. J. 2011, 17, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.-T.; Huang, S.-L.; Yao, H.-H.; Chen, I.-C.; Lin, Y.-C.; Yang, J.-S. An Antilock Molecular Braking System. Org. Lett. 2012, 14, 4154–4157. [Google Scholar] [CrossRef]
- Frantz, D.K.; Linden, A.; Baldridge, K.K.; Siegel, J.S. Molecular Spur Gears Comprising Triptycene Rotators and Bibenzimidazole-Based Stators. J. Am. Chem. Soc. 2012, 134, 1528–1535. [Google Scholar] [CrossRef]
- Nikitin, K.; Müller-Bunz, H.; Ortin, Y.; Risse, W.; McGlinchey, M.J. Twin Triptycyl Spinning Tops: A Simple Case of Molecular Gearing with Dynamic C2 Symmetry. Eur. J. Org. Chem. 2008, 2008, 3079–3084. [Google Scholar] [CrossRef]
- Kelly, T.R.; De Silva, H.; Silva, R.A. Unidirectional rotary motion in a molecular system. Nature 1999, 401, 150–152. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Xiang, J.-F.; Chen, C.-F. Tristable [n]rotaxanes: From molecular shuttle to molecular cable car. Chem. Sci. 2014, 5, 1520–1525. [Google Scholar] [CrossRef]
- Zhang, G.; Presly, O.; White, F.; Oppel, I.M.; Mastalerz, M. A Shape-Persistent Quadruply Interlocked Giant Cage Catenane with Two Distinct Pores in the Solid State. Angew. Chem. Int. Ed. 2014, 53, 5126–5130. [Google Scholar] [CrossRef]
- Zhu, X.-Z.; Chen, C.-F. A Highly Efficient Approach to [4]Pseudocatenanes by Threefold Metathesis Reactions of a Triptycene-Based Tris[2]pseudorotaxane. J. Am. Chem. Soc. 2005, 127, 13158–13159. [Google Scholar] [CrossRef]
- Jiang, Y.; Guo, J.-B.; Chen, C.-F. A New [3]Rotaxane Molecular Machine Based on a Dibenzylammonium Ion and a Triazolium Station. Org. Lett. 2010, 12, 4248–4251. [Google Scholar] [CrossRef]
- Chong, J.H.; MacLachlan, M.J. Iptycenes in supramolecular and materials chemistry. Chem. Soc. Rev. 2009, 38, 3301–3315. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Meng, Z.; Ma, Y.-X.; Chen, C.-F. Iptycene-Derived Crown Ether Hosts for Molecular Recognition and Self-Assembly. Acc. Chem. Res. 2014, 47, 2026–2040. [Google Scholar] [CrossRef]
- Ma, Y.-X.; Han, Y.; Chen, C.-F. Triptycene-derived calixarenes, heterocalixarenes and analogues. J. Incl. Phenom. Macrocycl. Chem. 2014, 79, 261–281. [Google Scholar] [CrossRef]
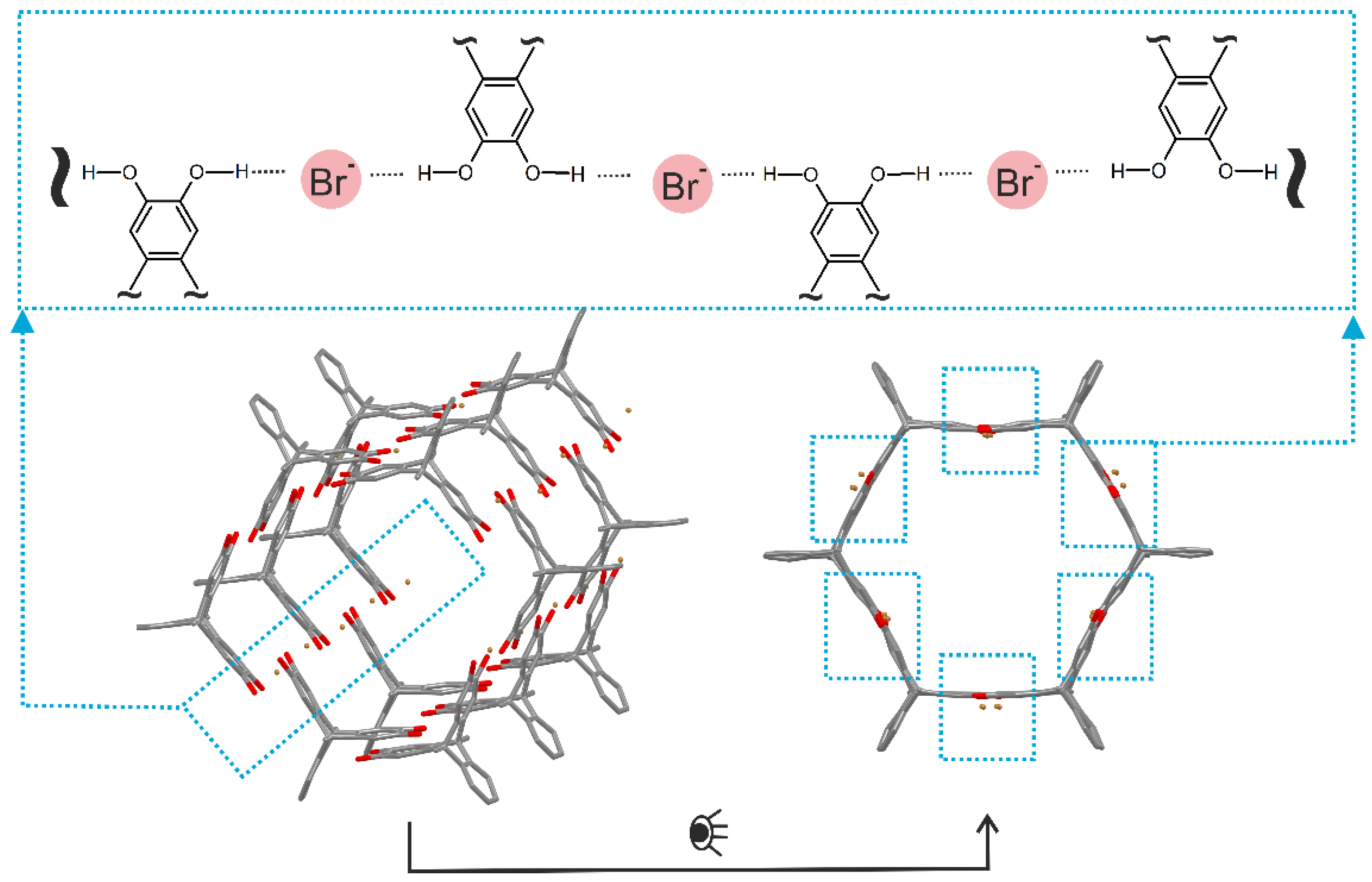
- Zhang, G.; Rominger, F.; Mastalerz, M. Hydrogen-Bonded Chains and Networks of Triptycene-Based Triboronic Acid and Tripyridinone. Cryst. Growth Des. 2016, 16, 5542–5548. [Google Scholar] [CrossRef]
- Granda, J.M.; Grabowski, J.; Jurczak, J. Synthesis, Structure, and Complexation Properties of a C3-Symmetrical Triptycene-Based Anion Receptor: Selectivity for Dihydrogen Phosphate. Org. Lett. 2015, 17, 5882–5885. [Google Scholar] [CrossRef]
- Zhang, G.-W.; Li, P.-F.; Meng, Z.; Wang, H.-X.; Han, Y.; Chen, C.-F. Triptycene-Based Chiral Macrocyclic Hosts for Highly Enantioselective Recognition of Chiral Guests Containing a Trimethylamino Group. Angew. Chem. Int. Ed. 2016, 55, 5304–5308. [Google Scholar] [CrossRef]
- Chen, C.-F. Novel triptycene-derived hosts: Synthesis and their applications in supramolecular chemistry. Chem. Commun. 2011, 47, 1674. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Jiang, Y.; Chen, C.-F. Solid state self-assembly of triptycene-based catechol derivatives by multiple OH⋯O hydrogen bonds. Chin. Chem. Lett. 2013, 24, 475–478. [Google Scholar] [CrossRef]
- Han, T.; Chen, C.-F. Formation of Ternary Complexes between a Macrotricyclic Host and Hetero-Guest Pairs: An Acid−Base Controlled Selective Complexation Process. Org. Lett. 2007, 9, 4207–4210. [Google Scholar] [CrossRef] [PubMed]
- Cohen, O.; Grossman, O.; Vaccaro, L.; Gelman, D. Synthesis of chiral nonracemic PC(sp3)P pincer ligands. J. Organomet. Chem. 2014, 750, 13–16. [Google Scholar] [CrossRef]
- Azerraf, C.; Gelman, D. Exploring the Reactivity of C(sp3)-Cyclometalated IrIII Compounds in Hydrogen Transfer Reactions. Chem. Eur. J. 2008, 14, 10364–10368. [Google Scholar] [CrossRef] [PubMed]
- Bini, L.; Müller, C.; Wilting, J.; von Chrzanowski, L.; Spek, A.L.; Vogt, D. Highly Selective Hydrocyanation of Butadiene toward 3-Pentenenitrile. J. Am. Chem. Soc. 2007, 129, 12622–12623. [Google Scholar] [CrossRef] [Green Version]
- Grossman, O.; Azerraf, C.; Gelman, D. Palladium Complexes Bearing Novel Strongly Bent Trans-Spanning Diphosphine Ligands: Synthesis, Characterization, and Catalytic Activity. Organometallics 2006, 25, 375–381. [Google Scholar] [CrossRef]
- Schmidt, S.; Abkai, G.; Rosendahl, T.; Rominger, F.; Hofmann, P. Inter- and Intramolecular Interactions in Triptycene-Derived Bisphosphite Hydroformylation Catalysts: Structures, Energies, and Caveats for DFT-Assisted Ligand Design. Organometallics 2013, 32, 1044–1052. [Google Scholar] [CrossRef]
- Kawamorita, S.; Miyazaki, T.; Iwai, T.; Ohmiya, H.; Sawamura, M. Rh-Catalyzed Borylation of N-Adjacent C(sp3)–H Bonds with a Silica-Supported Triarylphosphine Ligand. J. Am. Chem. Soc. 2012, 134, 12924–12927. [Google Scholar] [CrossRef]
- Smith, S.E.; Rosendahl, T.; Hofmann, P. Toward the Rhodium-Catalyzed Bis-Hydroformylation of 1,3-Butadiene to Adipic Aldehyde. Organometallics 2011, 30, 3643–3651. [Google Scholar] [CrossRef]
- Li, Y.; Cao, R.; Lippard, S.J. Design and Synthesis of a Novel Triptycene-Based Ligand for Modeling Carboxylate-Bridged Diiron Enzyme Active Sites. Org. Lett. 2011, 13, 5052–5055. [Google Scholar] [CrossRef] [Green Version]
- Perchellet, E.M.; Magill, M.J.; Huang, X.; Brantis, C.E.; Hua, D.H.; Perchellet, J.-P. Triptycenes: A novel synthetic class of bifunctional anticancer drugs that inhibit nucleoside transport, induce DMA cleavage and decrease the viability of leukemic cells in the nanomolar range in vitro. Anticancer. Drugs 1999, 10, 749–766. [Google Scholar] [CrossRef]
- Perchellet, E.M.; Wang, Y.; Weber, R.L.; Lou, K.; Hua, D.H.; Perchellet, J.-P.H. Antitumor triptycene bisquinones induce a caspase-independent release of mitochondrial cytochrome c and a caspase-2-mediated activation of initiator caspase-8 and -9 in HL-60 cells by a mechanism which does not involve Fas signaling. Anticancer Drugs 2004, 15, 929–946. [Google Scholar] [CrossRef]
- Wang, B.; Wu, M.; Perchellet, E.M.; McIlvain, C.J.; Sperfslage, B.J.; Huang, X.; Tamura, M.; Stephany, H.A.; Hua, D.H.; Perchellet, J.P. A synthetic triptycene bisquinone, which blocks nucleoside transport and induces DNA fragmentation, retains its cytotoxic efficacy in daunorubicin-resistant HL-60 cell lines. Int. J. Oncol. 2001, 19, 1169–1178. [Google Scholar] [CrossRef]
- Asche, C. Antitumour Quinones. Mini-Rev. Med. Chem. 2005, 5, 449–467. [Google Scholar] [CrossRef] [PubMed]
- Bernatowicz, P.; Szymański, S. Wave Properties of a Methyl Group under Ambient Conditions. Phys. Rev. Lett. 2002, 89, 23004. [Google Scholar] [CrossRef] [PubMed]
- Ratajczyk, T.; Czerski, I.; Szymański, S. Hindered rotation of the silyl group in liquid-phase NMR spectra of 9-silyltriptycene derivatives: A comparison with the methyl analogues. J. Phys. Chem. A 2008, 112, 8612–8616. [Google Scholar] [CrossRef]
- Yamamoto, G.; Mochida, H. Deamination of 1-Alkyl-9-aminomethyltriptycenes: Participation of the α-C-H Bond of the 1-Alkyl Group. Chem. Lett. 2000, 29, 454–455. [Google Scholar] [CrossRef]
- Bernatowicz, P.; Czerski, I.; Jaźwiński, J.; Szymański, S. Carr–Purcell echo spectra in the studies of lineshape effects. Nonclassical hindered rotation of methyl groups in 1,2,3,4-tetrachloro-9,10-dimethyltriptycene. J. Magn. Reson. 2004, 169, 284–292. [Google Scholar] [CrossRef]
- Czerski, I.; Bernatowicz, P.; Jaźwiński, J.; Szymański, S. Nonclassical effects in liquid-phase nuclear magnetic resonance spectra of 9-methyltriptycene derivatives. J. Chem. Phys. 2003, 118, 7157–7160. [Google Scholar] [CrossRef]
- Peurifoy, S.R.; Castro, E.; Liu, F.; Zhu, X.-Y.; Ng, F.; Jockusch, S.; Steigerwald, M.L.; Echegoyen, L.; Nuckolls, C.; Sisto, T.J. Three-Dimensional Graphene Nanostructures. J. Am. Chem. Soc. 2018, 140, 9341–9345. [Google Scholar] [CrossRef]
- Kawasumi, K.; Wu, T.; Zhu, T.; Chae, H.S.; Van Voorhis, T.; Baldo, M.A.; Swager, T.M. Thermally Activated Delayed Fluorescence Materials Based on Homoconjugation Effect of Donor–Acceptor Triptycenes. J. Am. Chem. Soc. 2015, 137, 11908–11911. [Google Scholar] [CrossRef] [Green Version]
- Ishiwari, F.; Nascimbeni, G.; Sauter, E.; Tago, H.; Shoji, Y.; Fujii, S.; Kiguchi, M.; Tada, T.; Zharnikov, M.; Zojer, E.; et al. Triptycene Tripods for the Formation of Highly Uniform and Densely Packed Self-Assembled Monolayers with Controlled Molecular Orientation. J. Am. Chem. Soc. 2019, 141, 5995–6005. [Google Scholar] [CrossRef] [Green Version]
- Murray, D.J.; Patterson, D.D.; Payamyar, P.; Bhola, R.; Song, W.; Lackinger, M.; Schlüter, A.D.; King, B.T. Large Area Synthesis of a Nanoporous Two-Dimensional Polymer at the Air/Water Interface. J. Am. Chem. Soc. 2015, 137, 3450–3453. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, M.G.; Reich, T.E.; Kassab, R.M.; Jackson, K.T.; El-Kaderi, H.M. High CO2 uptake and selectivity by triptycene-derived benzimidazole-linked polymers. Chem. Commun. 2012, 48, 1141–1143. [Google Scholar] [CrossRef]
- Ghanem, B.S.; Msayib, K.J.; McKeown, N.B.; Harris, K.D.M.; Pan, Z.; Budd, P.M.; Butler, A.; Selbie, J.; Book, D.; Walton, A. A triptycene-based polymer of intrinsic microposity that displays enhanced surface area and hydrogen adsorption. Chem. Commun. 2007, 67–69. [Google Scholar] [CrossRef]
- Ghanem, B.S.; Hashem, M.; Harris, K.D.M.; Msayib, K.J.; Xu, M.; Budd, P.M.; Chaukura, N.; Book, D.; Tedds, S.; Walton, A.; et al. Triptycene-Based Polymers of Intrinsic Microporosity: Organic Materials That Can Be Tailored for Gas Adsorption. Macromolecules 2010, 43, 5287–5294. [Google Scholar] [CrossRef]
- Mastalerz, M. Permanent Porous Materials from Discrete Organic Molecules-Towards Ultra-High Surface Areas. Chem. Eur. J. 2012, 18, 10082–10091. [Google Scholar] [CrossRef]
- Mastalerz, M.; Oppel, I.M. Rational Construction of an Extrinsic Porous Molecular Crystal with an Extraordinary High Specific Surface Area. Angew. Chem. Int. Ed. 2012, 51, 5252–5255. [Google Scholar] [CrossRef]
- McKeown, N.B. Organic Molecules of Intrinsic Microporosity. Org. Mater. 2020, 2, 020–025. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Chen, C.-F. Synthesis and Structure of a Triptycene-Based Nanosized Molecular Cage. J. Org. Chem. 2007, 72, 9339–9341. [Google Scholar] [CrossRef]
- Zhang, G.; Presly, O.; White, F.; Oppel, I.M.; Mastalerz, M. A Permanent Mesoporous Organic Cage with an Exceptionally High Surface Area. Angew. Chem. Int. Ed. 2014, 53, 1516–1520. [Google Scholar] [CrossRef]
- Schneider, M.W.; Oppel, I.M.; Mastalerz, M. Exo-Functionalized Shape-Persistent [2+3] Cage Compounds: Influence of Molecular Rigidity on Formation and Permanent Porosity. Chem. Eur. J. 2012, 18, 4156–4160. [Google Scholar] [CrossRef]
- Schneider, M.W.; Oppel, I.M.; Ott, H.; Lechner, L.G.; Hauswald, H.-J.S.; Stoll, R.; Mastalerz, M. Periphery-Substituted [4 + 6] Salicylbisimine Cage Compounds with Exceptionally High Surface Areas: Influence of the Molecular Structure on Nitrogen Sorption Properties. Chem. Eur. J. 2012, 18, 836–847. [Google Scholar] [CrossRef]
- Bhat, A.S.; Elbert, S.M.; Zhang, W.-S.; Rominger, F.; Dieckmann, M.; Schröder, R.R.; Mastalerz, M. Transformation of a [4 + 6] Salicylbisimine Cage to Chemically Robust Amide Cages. Angew. Chem. Int. Ed. 2019, 58, 8819–8823. [Google Scholar] [CrossRef] [PubMed]
- Crane, A.K.; Patrick, B.O.; MacLachlan, M.J. New metal–organic frameworks from triptycene: Structural diversity from bulky bridges. Dalton Trans. 2013, 42, 8026–8033. [Google Scholar] [CrossRef] [PubMed]
- Roy, X.; Chong, J.H.; Patrick, B.O.; MacLachlan, M.J. Molecular Scaffolding of Prussian Blue Analogues Using a Phenanthroline-Extended Triptycene Ligand. Cryst. Growth Des. 2011, 11, 4551–4558. [Google Scholar] [CrossRef]
- Lee, C.-H.; Filler, R.; Lee, J.; Li, J.; Mandal, B.K. Synthesis and hydrogen adsorption properties of a new phthalocyanine-based metal–organic framework. Renew. Energy 2010, 35, 1592–1595. [Google Scholar] [CrossRef]
- Vagin, S.; Ott, A.; Weiss, H.-C.; Karbach, A.; Volkmer, D.; Rieger, B. Metal-Organic Frameworks (MOFs) Composed of (Triptycenedicarboxylato)zinc. Eur. J. Inorg. Chem. 2008, 2008, 2601–2609. [Google Scholar] [CrossRef]
- Vagin, S.I.; Ott, A.K.; Hoffmann, S.D.; Lanzinger, D.; Rieger, B. Synthesis and Properties of (Triptycenedicarboxylatio)zinc Coordination Networks. Chem. Eur. J. 2009, 15, 5845–5853. [Google Scholar] [CrossRef]
- Chong, J.H.; MacLachlan, M.J. Robust Non-Interpenetrating Coordination Frameworks from New Shape-Persistent Building Blocks. Inorg. Chem. 2006, 45, 1442–1444. [Google Scholar] [CrossRef]
- Brutschy, M.; Schneider, M.W.; Mastalerz, M.; Waldvogel, S.R. Direct gravimetric sensing of GBL by a molecular recognition process in organic cage compounds. Chem. Commun. 2013, 49, 8398–8400. [Google Scholar] [CrossRef]
- Barman, S.; Anand Garg, J.; Blacque, O.; Venkatesan, K.; Berke, H. Triptycene based luminescent metal–organic gels for chemosensing. Chem. Commun. 2012, 48, 11127–11129. [Google Scholar] [CrossRef] [Green Version]
- Cordovilla, C.; Swager, T.M. Strain Release in Organic Photonic Nanoparticles for Protease Sensing. J. Am. Chem. Soc. 2012, 134, 6932–6935. [Google Scholar] [CrossRef] [Green Version]
- Esser, B.; Swager, T.M. Detection of Ethylene Gas by Fluorescence Turn-On of a Conjugated Polymer. Angew. Chem. Int. Ed. 2010, 49, 8872–8875. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.-S.; Swager, T.M. Fluorescent Porous Polymer Films as TNT Chemosensors: Electronic and Structural Effects. J. Am. Chem. Soc. 1998, 120, 11864–11873. [Google Scholar] [CrossRef]
- Choi, S.-J.; Savagatrup, S.; Kim, Y.; Lang, J.H.; Swager, T.M. Precision pH Sensor Based on WO3 Nanofiber-Polymer Composites and Differential Amplification. ACS Sens. 2019, 4, 2593–2598. [Google Scholar] [CrossRef]
- Lohr, A.; Swager, T.M. Stabilization of the nematic mesophase by a homogeneously dissolved conjugated polymer. J. Mater. Chem. 2010, 20, 8107–8111. [Google Scholar] [CrossRef]
- Zhu, Z.; Swager, T.M. Conjugated Polymer Liquid Crystal Solutions: Control of Conformation and Alignment. J. Am. Chem. Soc. 2002, 124, 9670–9671. [Google Scholar] [CrossRef]
- Norvez, S. Liquid crystalline triptycene derivatives. J. Org. Chem. 1993, 58, 2414–2418. [Google Scholar] [CrossRef]
- Long, T.M.; Swager, T.M. Triptycene-containing bis(phenylethynyl)benzene nematic liquid crystals. J. Mater. Chem. 2002, 12, 3407–3412. [Google Scholar] [CrossRef]
- Long, T.M.; Swager, T.M. Using “Internal Free Volume” to Increase Chromophore Alignment. J. Am. Chem. Soc. 2002, 124, 3826–3827. [Google Scholar] [CrossRef]
- Ohira, A.; Swager, T.M. Ordering of Poly(p-phenylene ethynylene)s in Liquid Crystals. Macromolecules 2007, 40, 19–25. [Google Scholar] [CrossRef]
- Skvarchenko, V.R.; Shalaev, V.K.; Klabunovskii, E.I. Advances in the Chemistry of Triptycene. Russ. Chem. Rev. 1974, 43, 951–966. [Google Scholar] [CrossRef]
- Craig, A.; Wilcox, C., Jr. Communications: A New Synthesis of Triptycene. J. Org. Chem. 1959, 24, 1619. [Google Scholar] [CrossRef]
- Stiles, M.; Miller, R.G. Decomposition of benzenediazonium-2-carboxylate. J. Am. Chem. Soc. 1960, 82, 3802. [Google Scholar] [CrossRef]
- Friedman, L.; Logullo, F.M. Benzynes via Aprotic Diazotization of Anthranilic Acids: A Convenient Synthesis of Triptycene and Derivatives. J. Am. Chem. Soc. 1963, 85, 1549. [Google Scholar] [CrossRef]
- Cadogan, J.I.G.; Hall, J.K.A.; Sharp, J.T. The formation of arynes by reaction of potassium t-butoxide with aryl halides. J. Chem. Soc. C Org. 1967, 1860–1862. [Google Scholar] [CrossRef]
- Kessar, S.V.; Singh, P.; Singh, K.N.; Bharatam, P.V.; Sharma, A.K.; Lata, S.; Kaur, A. A Study of BF3-Promoted ortho Lithiation of Anilines and DFT Calculations on the Role of Fluorine–Lithium Interactions. Angew. Chem. Int. Ed. 2008, 47, 4703–4706. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Yamane, M.; Inoue, K.; Todaka, M.; Fukatsu, N.; Meng, Z.; Fujiwara, Y. A New and Efficient Hypervalent Iodine−Benzyne Precursor, (Phenyl)[o-(trimethylsilyl)phenyl]iodonium Triflate: Generation, Trapping Reaction, and Nature of Benzyne. J. Am. Chem. Soc. 1999, 121, 11674–11679. [Google Scholar] [CrossRef]
- Klanderman, B.H.; Perkins, W.C. Nitration of triptycene. J. Org. Chem. 1969, 34, 630–633. [Google Scholar] [CrossRef]
- Tanida, H.; Muneyuki, R. Nitration of bridged hydronaphthalenes. Fused -effect. Tetrahedron Lett. 1964, 5, 2787–2790. [Google Scholar] [CrossRef]
- Streitwieser, A.; Ziegler, G.R.; Mowery, P.C.; Lewis, A.; Lawler, R.G. Some generalizations concerning the reactivity of aryl positions adjacent to fused strained rings. J. Am. Chem. Soc. 1968, 90, 1357–1358. [Google Scholar] [CrossRef]
- Huang, W.; Einzinger, M.; Zhu, T.; Chae, H.S.; Jeon, S.; Ihn, S.-G.; Sim, M.; Kim, S.; Su, M.; Teverovskiy, G.; et al. Molecular Design of Deep Blue Thermally Activated Delayed Fluorescence Materials Employing a Homoconjugative Triptycene Scaffold and Dihedral Angle Tuning. Chem. Mater. 2018, 30, 1462–1466. [Google Scholar] [CrossRef]
- Chen, Z.; Swager, T.M. Synthesis and Characterization of Poly(2,6-triptycene). Macromolecules 2008, 41, 6880–6885. [Google Scholar] [CrossRef]
- White, N.G.; MacLachlan, M.J. Soluble Tetraaminotriptycene Precursors. J. Org. Chem. 2015, 80, 8390–8397. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, C.-F. Synthesis and Structure of 2,6,14- and 2,7,14-Trisubstituted Triptycene Derivatives. J. Org. Chem. 2006, 71, 6626–6629. [Google Scholar] [CrossRef] [PubMed]
- Paget, C.J.; Burger, A. Acetylation of Triptycene1. J. Org. Chem. 1965, 30, 1329–1331. [Google Scholar] [CrossRef]
- Li, P.-F.; Chen, C.-F. Synthesis, Structures, and Solid State Self-Assemblies of Formyl and Acetyl Substituted Triptycenes and Their Derivatives. J. Org. Chem. 2012, 77, 9250–9259. [Google Scholar] [CrossRef] [PubMed]
- Seiki, N.; Shoji, Y.; Kajitani, T.; Ishiwari, F.; Kosaka, A.; Hikima, T.; Takata, M.; Someya, T.; Fukushima, T. Rational synthesis of organic thin films with exceptional long-range structural integrity. Science 2015, 348, 1122–1126. [Google Scholar] [CrossRef] [PubMed]
- Leung, F.K.-C.; Ishiwari, F.; Kajitani, T.; Shoji, Y.; Hikima, T.; Takata, M.; Saeki, A.; Seki, S.; Yamada, Y.M.A.; Fukushima, T. Supramolecular Scaffold for Tailoring the Two-Dimensional Assembly of Functional Molecular Units into Organic Thin Films. J. Am. Chem. Soc. 2016, 138, 11727–11733. [Google Scholar] [CrossRef]
- Kumano, M.; Ide, M.; Seiki, N.; Shoji, Y.; Fukushima, T.; Saeki, A. A ternary blend of a polymer, fullerene, and insulating self-assembling triptycene molecules for organic photovolatics. J. Mater. Chem. A 2016, 4, 18490–18498. [Google Scholar] [CrossRef]
- Tada, T.; Ishiwari, F.; Shoji, Y.; Fukushima, T. First-Principles Study of the Adsorption Behavior of Triptycene Molecular Tripods on Au(111): Site Selectivity and Unambiguous Molecular Orientation. J. Phys. Chem. C 2019, 123, 4401–4406. [Google Scholar] [CrossRef]
- Mori, I.; Kadosaka, T.; Sakata, Y.; Misumi, S. Synthesis and Spectral Properties of Chloro-substituted Triptycenes. Bull. Chem. Soc. Jpn. 1971, 44, 1649–1652. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Mague, J.T.; Pascal, R.A., Jr. An Exceptionally Close, Non-Bonded Hydrogen–Hydrogen Contact with Strong Through-Space Spin–Spin Coupling. Angew. Chem. Int. Ed. 2018, 57, 2244–2247. [Google Scholar] [CrossRef]
- Song, Q.; Ho, D.M.; Pascal, R.A. Sterically Congested in -Methylcyclophanes. J. Am. Chem. Soc. 2005, 127, 11246–11247. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.E.; Averill, B.A. Symmetrically trisubstituted triptycenes. J. Org. Chem. 1986, 51, 3308–3314. [Google Scholar] [CrossRef]
- Chmiel, J.; Heesemann, I.; Mix, A.; Neumann, B.; Stammler, H.-G.; Mitzel, N.W. The Effect of Bulky Substituents on the Formation of Symmetrically Trisubstituted Triptycenes. Eur. J. Org. Chem. 2010, 2010, 3897–3907. [Google Scholar] [CrossRef]
- Elbert, S.M.; Rominger, F.; Mastalerz, M. Synthesis of a Rigid C3v-Symmetric Tris-salicylaldehyde as a Precursor for a Highly Porous Molecular Cube. Chem. Eur. J. 2014, 20, 16707–16720. [Google Scholar] [CrossRef]
- Lamm, J.-H.; Vishnevskiy, V.Y.; Ziemann, E.; Neumann, B.; Stammler, H.-G.; Mitzel, N.W. Regiochemical Control in Triptycene Formation—An Exercise in Subtle Balancing Multiple Factors. ChemistryOpen 2018, 7, 111–114. [Google Scholar] [CrossRef] [Green Version]
- Schwartzen, A.; Rovers, M.; Neumann, B.; Stammler, H.-G.; Mitzel, N.W. Syntheses and Structures of 1,8,13,16-Substituted Triptycenes. Eur. J. Org. Chem. 2018, 2018, 5323–5333. [Google Scholar] [CrossRef]
- Himeshima, Y.; Sonoda, T.; Kobayashi, H. Fluoride-induced 1,2-elimination OF O-trimethylsilylphenyl triflate to benzyne under mild conditions. Chem. Lett. 1983, 12, 1211–1214. [Google Scholar] [CrossRef]
- Peña, D.; Cobas, A.; Pérez, D.; Guitián, E. An Efficient Procedure for the Synthesis of ortho -Trialkylsilylaryl Triflates: Easy Access to Precursors of Functionalized Arynes. Synthesis 2002, 10, 1454–1458. [Google Scholar]
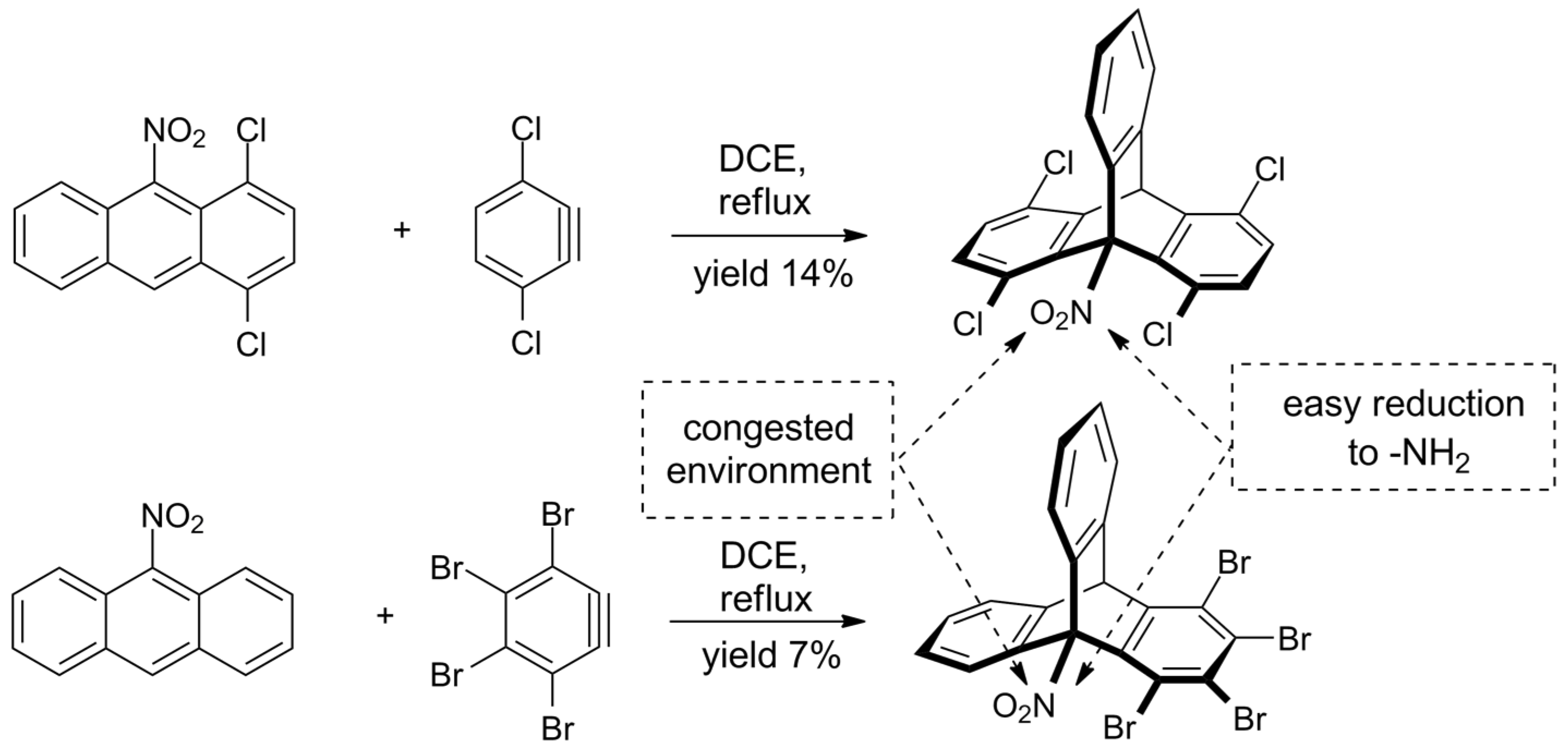
- Szupiluk, A. Synthesis of sterically crowded 9-nitrotriptycenes by the Diels–Alder cycloaddition reaction. Tetrahedron Lett. 2016, 57, 5251–5253. [Google Scholar] [CrossRef]
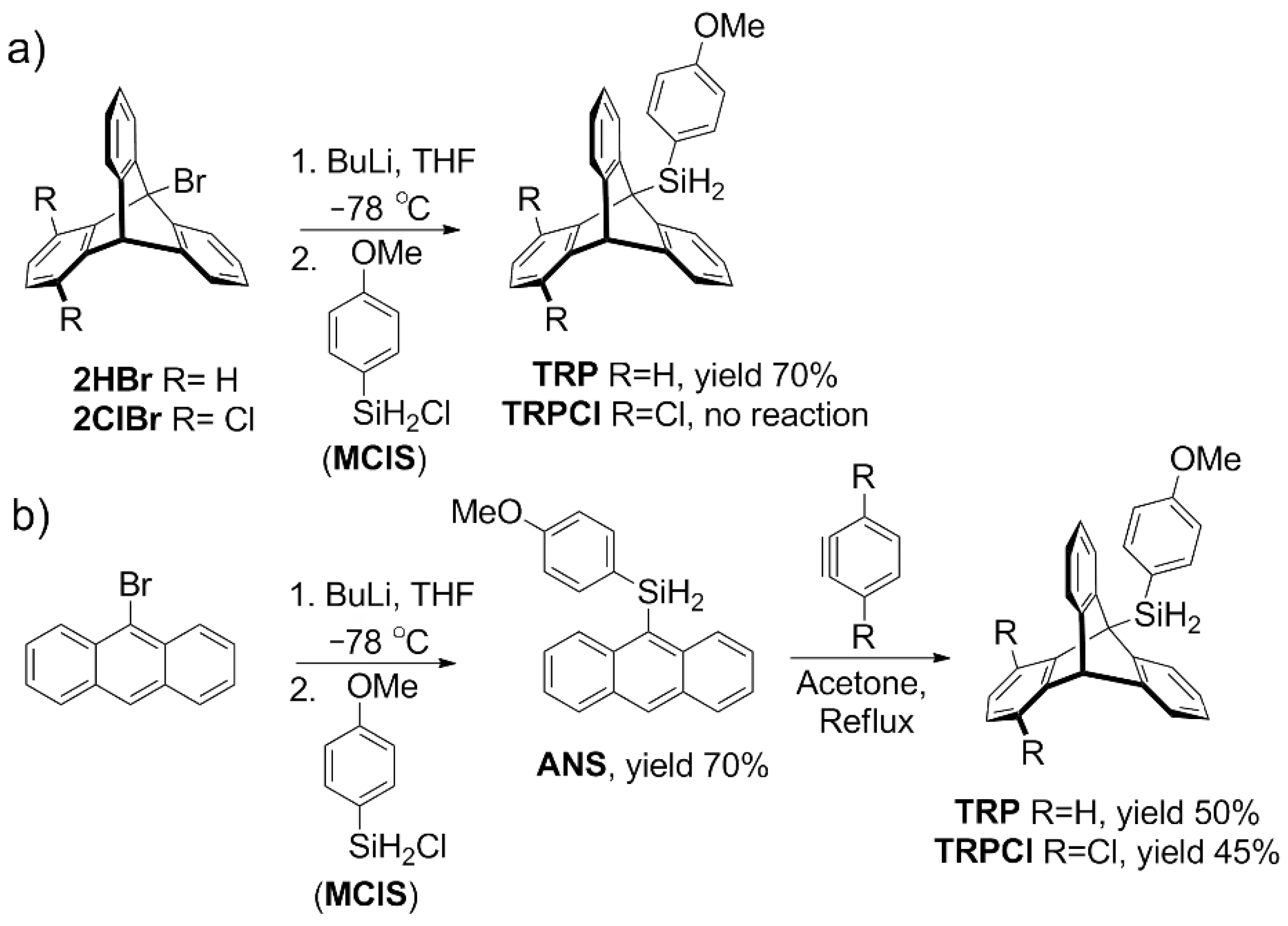
- Mames, A.; Osior, A.; Szkudlarek, P.G.; Pietrzak, M.; Szymański, S.; Ratajczyk, T. Synthesis and structural characterization of exemplary silyl triptycenes. New J. Chem. 2019, 43, 7567–7573. [Google Scholar] [CrossRef]
- Mames, A.; Gołowicz, D.; Pietrzak, M.; Kazimierczuk, K.; Szymański, S.; Ratajczyk, T. Blue-Shift Hydrogen Bonds in Silyltriptycene Derivatives: Antibonding σ* Orbitals of the Si−C Bond as Effective Acceptors of Electron Density. ChemPhysChem 2020, 21, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S.; Swager, T.M. Triptycenediols by Rhodium-Catalyzed [2 + 2 + 2] Cycloaddition. Org. Lett. 2007, 9, 3695–3697. [Google Scholar] [CrossRef] [PubMed]
- Shindo, M. Synthetic uses of ynolates. Tetrahedron 2007, 63, 10–36. [Google Scholar] [CrossRef]
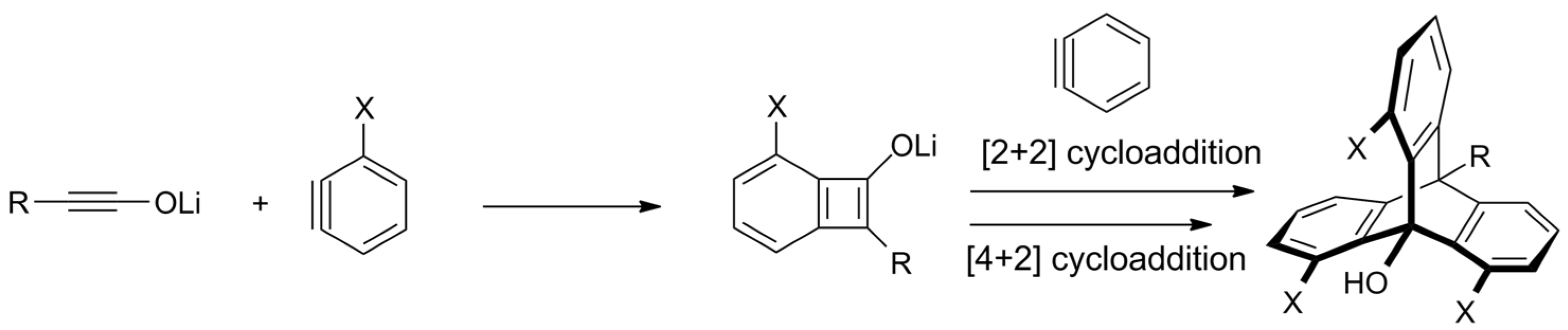
- Umezu, S.; dos Passos Gomes, G.; Yoshinaga, T.; Sakae, M.; Matsumoto, K.; Iwata, T.; Alabugin, I.; Shindo, M. Regioselective One-Pot Synthesis of Triptycenes via Triple-Cycloadditions of Arynes to Ynolates. Angew. Chem. Int. Ed. 2017, 56, 1298–1302. [Google Scholar] [CrossRef] [PubMed]
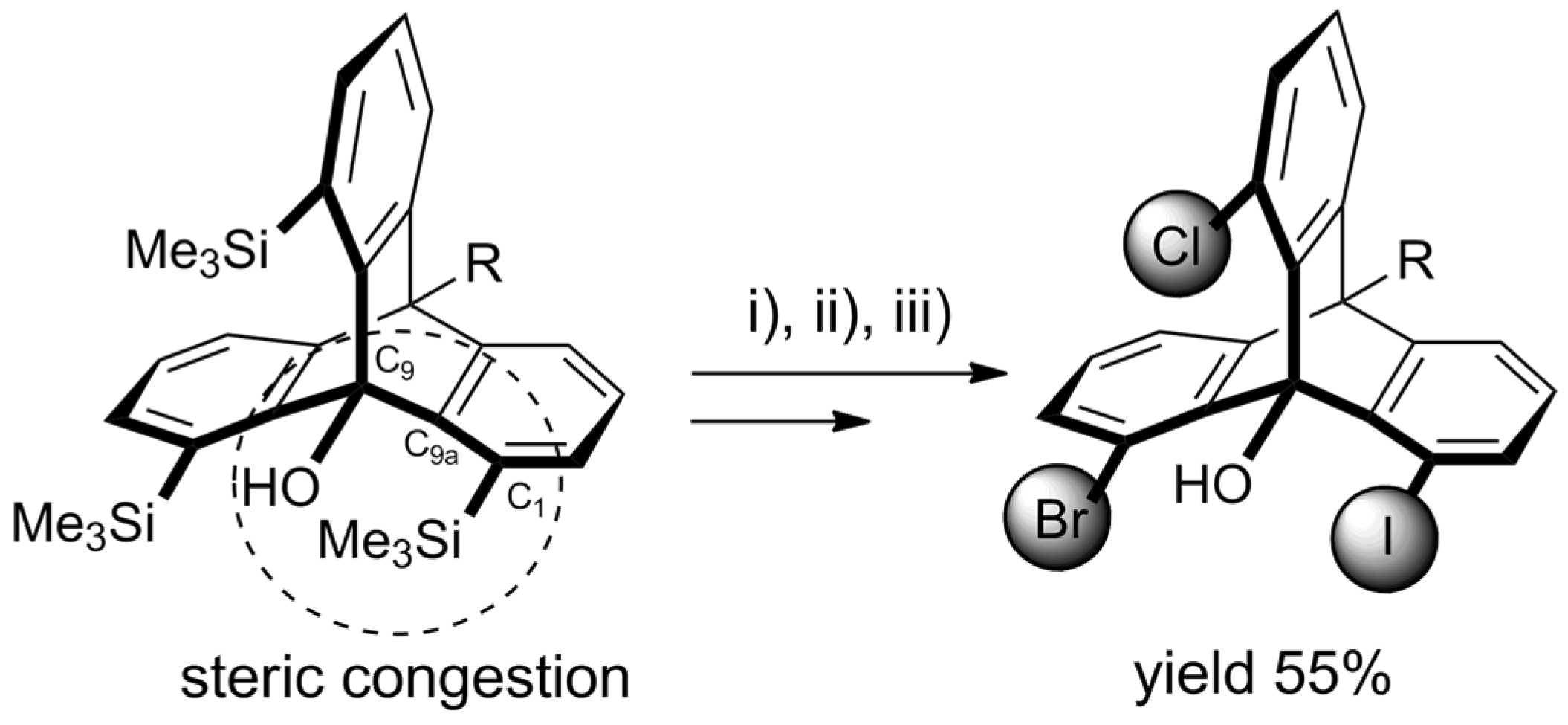
- Yoshinaga, T.; Fujiwara, T.; Iwata, T.; Shindo, M. Synthesis of Distorted 1,8,13-Trisilyl-9-hydroxytriptycenes by Triple Cycloaddition of Ynolates to 3-Silylbenzynes. Chem. Eur. J. 2019, 25, 13855–13859. [Google Scholar] [CrossRef]
- Streitwieser, A.; Ziegler, G.R. Acidity of hydrocarbons. XXXIII. Kinetics acidities of the hydrogens in triptycene toward cesium cyclohexylamide. J. Am. Chem. Soc. 1969, 91, 5081–5084. [Google Scholar] [CrossRef]
- Oi, M.; Takita, R.; Kanazawa, J.; Muranaka, A.; Wang, C.; Uchiyama, M. Organocopper cross-coupling reaction for C–C bond formation on highly sterically hindered structures. Chem. Sci. 2019, 10, 6107–6112. [Google Scholar] [CrossRef] [Green Version]
- Mislow, K.; Siegel, J. Stereoisomerism and local chirality. J. Am. Chem. Soc. 1984, 106, 3319–3328. [Google Scholar] [CrossRef]
- Gao, Y.; Ren, C.; Lin, X.; He, T. The Progress and Perspective of Organic Molecules with Switchable Circularly Polarized Luminescence. Front. Chem. 2020, 8, 458. [Google Scholar] [CrossRef]
- Brandt, J.R.; Salerno, F.; Fuchter, M.J. The added value of small-molecule chirality in technological applications. Nat. Rev. Chem. 2017, 1, 45. [Google Scholar] [CrossRef]
- Ikai, T.; Wada, Y.; Awata, S.; Yun, C.; Maeda, K.; Mizuno, M.; Swager, T.M. Chiral triptycene-pyrene π-conjugated chromophores with circularly polarized luminescence. Org. Biomol. Chem. 2017, 15, 8440–8447. [Google Scholar] [CrossRef] [PubMed]
- Ikai, T.; Yoshida, T.; Awata, S.; Wada, Y.; Maeda, K.; Mizuno, M.; Swager, T.M. Circularly Polarized Luminescent Triptycene-Based Polymers. ACS Macro. Lett. 2018, 7, 364–369. [Google Scholar] [CrossRef]
- Ikai, T.; Yoshida, T.; Shinohara, K.; Taniguchi, T.; Wada, Y.; Swager, T.M. Triptycene-Based Ladder Polymers with One-Handed Helical Geometry. J. Am. Chem. Soc. 2019, 141, 4696–4703. [Google Scholar] [CrossRef] [PubMed]
- Wada, Y.; Shinohara, K.; Ikai, T. Optically active triptycenes containing hexa-peri-hexabenzocoronene units. Chem. Commun. 2019, 55, 11386–11389. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, A.; Ogura, F.; Nakagawa, M. Synthesis of Trisubstituted Triptycenes and the Optical Resolution of 7-Carboxy-2,5-diacetoxytriptycene. Bull. Chem. Soc. Jpn. 1962, 35, 853–857. [Google Scholar] [CrossRef] [Green Version]
- Ikai, T.; Nagata, N.; Awata, S.; Wada, Y.; Maeda, K.; Mizuno, M.; Swager, T.M. Optically active distorted cyclic triptycenes: Chiral stationary phases for HPLC. RSC Adv. 2018, 8, 20483–20487. [Google Scholar] [CrossRef] [Green Version]
- Shibata, T.; Kamimura, Y. Asymmetric synthesis of multi-substituted triptycenes via enantioselective alkynylation of 1,5-dibromoanthracene-9,10-dione. Tetrahedron Asymmetry 2015, 26, 41–45. [Google Scholar] [CrossRef] [Green Version]
- Aida, Y.; Shibata, Y.; Tanaka, K. Enantioselective Synthesis of Distorted π-Extended Chiral Triptycenes Consisting of Three Distinct Aromatic Rings by Rhodium-Catalyzed [2+2+2] Cycloaddition. Chem. Eur. J. 2019, 26, 3004–3009. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, G.; Jouaiti, A.; Geoffroy, M.; Bernardinelli, G. 9-Substituted Triptycene as a Probe for the Study of Internal Rotation around a C−PH Bond in the Solid State: A Single Crystal EPR Study at Variable Temperature. J. Phys. Chem. 1996, 100, 10861–10868. [Google Scholar] [CrossRef]
- Grossman, O.; Gelman, D. Novel Trans-Spanned Palladium Complexes as Efficient Catalysts in Mild and Amine-Free Cyanation of Aryl Bromides under Air. Org. Lett. 2006, 8, 1189–1191. [Google Scholar] [CrossRef]
- Azerraf, C.; Grossman, O.; Gelman, D. Rigid trans-spanning triptycene-based ligands: How flexible they can be? J. Organomet. Chem. 2007, 692, 761–767. [Google Scholar] [CrossRef]
- Grossman, O.; Rueck-Braun, K.; Gelman, D. Trans-Spanned Palladium Catalyst for Mild and Efficient Amination of Aryl Halides with Benzophenone Imine. Synthesis 2008, 2008, 537–542. [Google Scholar] [CrossRef]
- Azerraf, C.; Gelman, D. New Shapes of PC(sp3)P Pincer Complexes. Organometallics 2009, 28, 6578–6584. [Google Scholar] [CrossRef]
- Levy, R.; Azerraf, C.; Gelman, D.; Rueck-Braun, K.; Kapoor, P.N. Cyclometalated phosphine-based Ir(III) pincer complex as a catalyst for Oppenauer-type oxidation of alcohols. Catal. Commun. 2009, 11, 298–301. [Google Scholar] [CrossRef]
- Kisets, I.; Gelman, D. Carbometalated Complexes Possessing Tripodal Pseudo-C3-Symmetric Triptycene-Based Ligands. Organometallics 2018, 37, 526–529. [Google Scholar] [CrossRef]
- Leung, F.K.-C.; Ishiwari, F.; Shoji, Y.; Nishikawa, T.; Takeda, R.; Nagata, Y.; Suginome, M.; Uozumi, Y.; Yamada, Y.M.A.; Fukushima, T. Synthesis and Catalytic Applications of a Triptycene-Based Monophosphine Ligand for Palladium-Mediated Organic Transformations. ACS Omega 2017, 2, 1930–1937. [Google Scholar] [CrossRef] [Green Version]
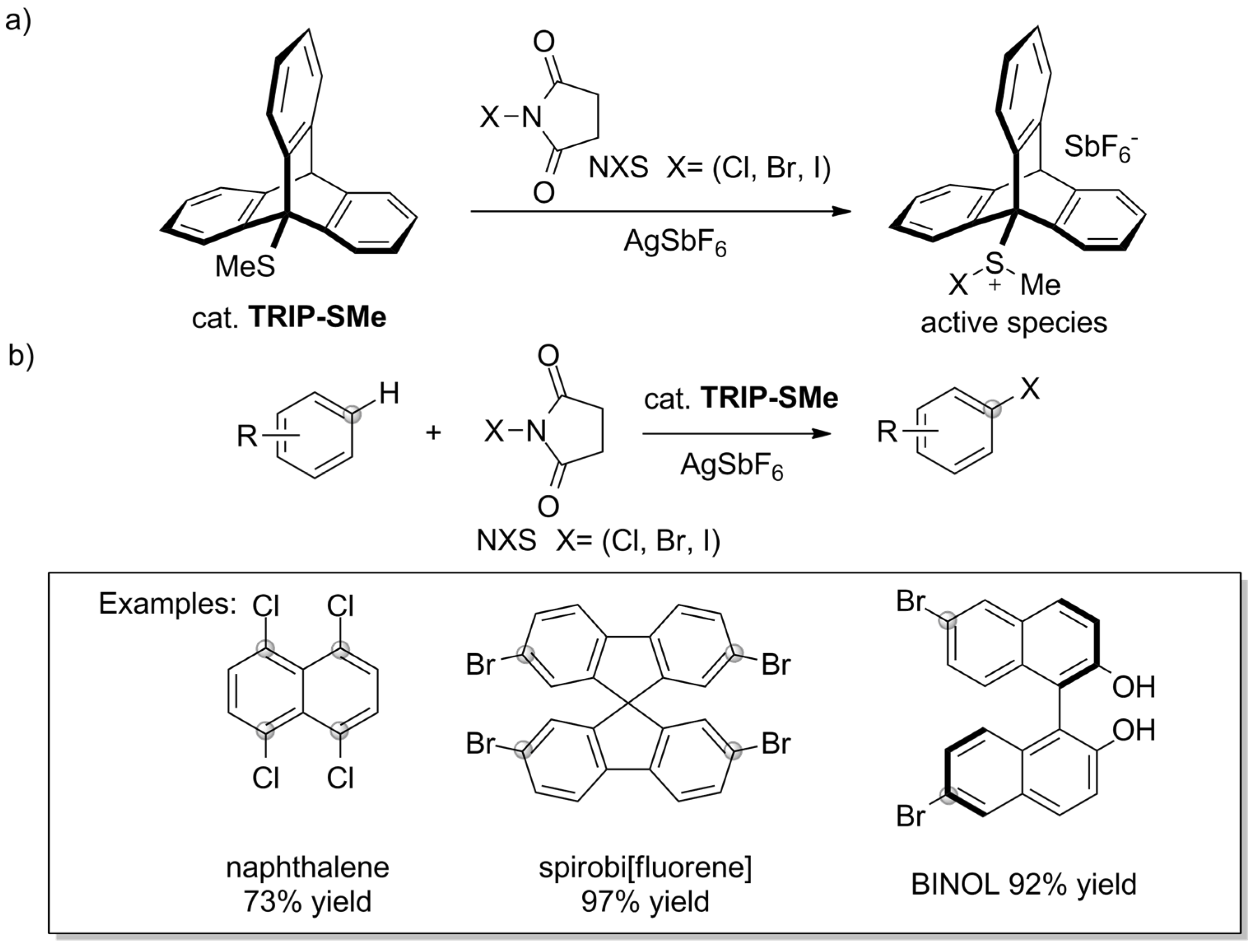
- Nishii, Y.; Ikeda, M.; Hayashi, Y.; Kawauchi, S.; Miura, M. Triptycenyl Sulfide: A Practical and Active Catalyst for Electrophilic Aromatic Halogenation Using N-Halosuccinimides. J. Am. Chem. Soc. 2020, 142, 1621–1629. [Google Scholar] [CrossRef]
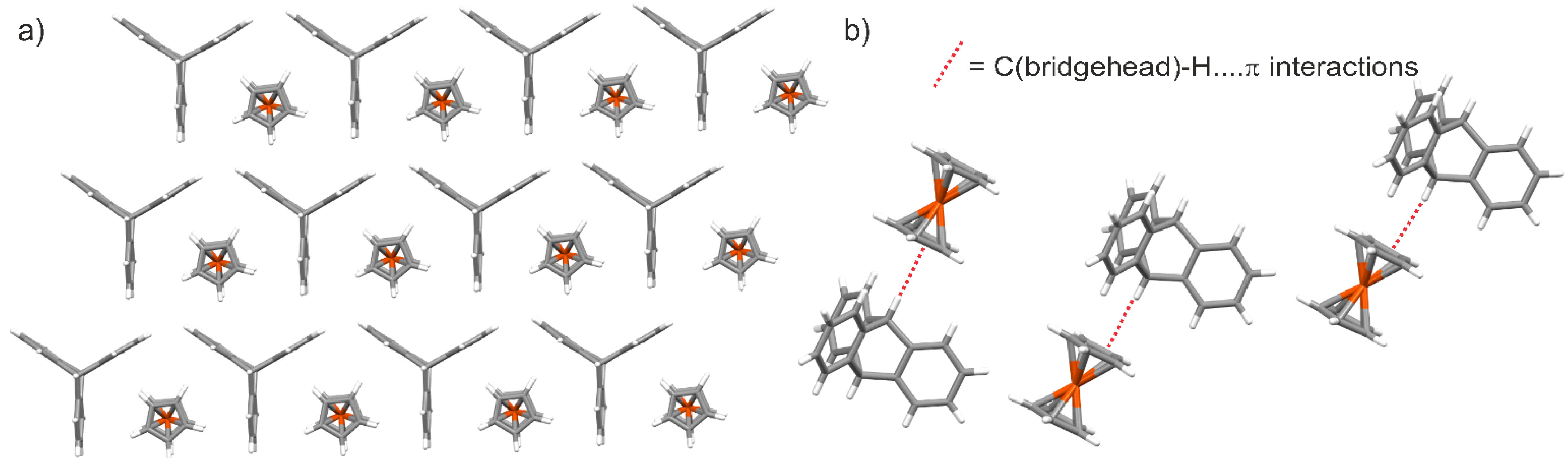
- Lari, A.; Pitak, M.B.; Coles, S.J.; Bresco, E.; Belser, P.; Beyeler, A.; Pilkington, M.; Wallis, J.D. The use of the triptycene framework for observing O⋯C O molecular interactions. CrystEngComm 2011, 13, 6978–6984. [Google Scholar] [CrossRef]
- Aliev, A.E.; Motherwell, W.B. Some Recent Advances in the Design and Use of Molecular Balances for the Experimental Quantification of Intramolecular Noncovalent Interactions of π Systems. Chem. Eur. J. 2019, 25, 10516–10530. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Yan, Q.; Liu, S.; Zhao, D. Probing the intermolecular interactions of aromatic amides containing N-heterocycles and triptycene. CrystEngComm 2014, 16, 4265–4273. [Google Scholar] [CrossRef]
- Gu, X.; Lai, Y.-H. Triptycene: No Homoconjugation Effect for Extending Optical Properties of π-Conjugated Oligomers. Org. Lett. 2010, 12, 5200–5203. [Google Scholar] [CrossRef]
- Ratajczyk, T.; Szymanski, S. Are the silyl group hydrogens in peri-substituted-9-silyltriptycenes engaged in blue-shifting hydrogen bonds? Phys. Chem. Chem. Phys. 2009, 11, 2335–2338. [Google Scholar] [CrossRef]
- Szymański, S.; Bernatowicz, P. The Symmetrization Postulate of Quantum Mechanics in NMR Spectra. Annu. Rep. NMR Spectrosc. 2004, 54, 1–39. [Google Scholar] [CrossRef]
- Chen, C.-H.; Gabbaï, F.P. Fluoride Anion Complexation by a Triptycene-Based Distiborane: Taking Advantage of a Weak but Observable C−H⋅⋅⋅F Interaction. Angew. Chem. Int. Ed. 2017, 56, 1799–1804. [Google Scholar] [CrossRef]
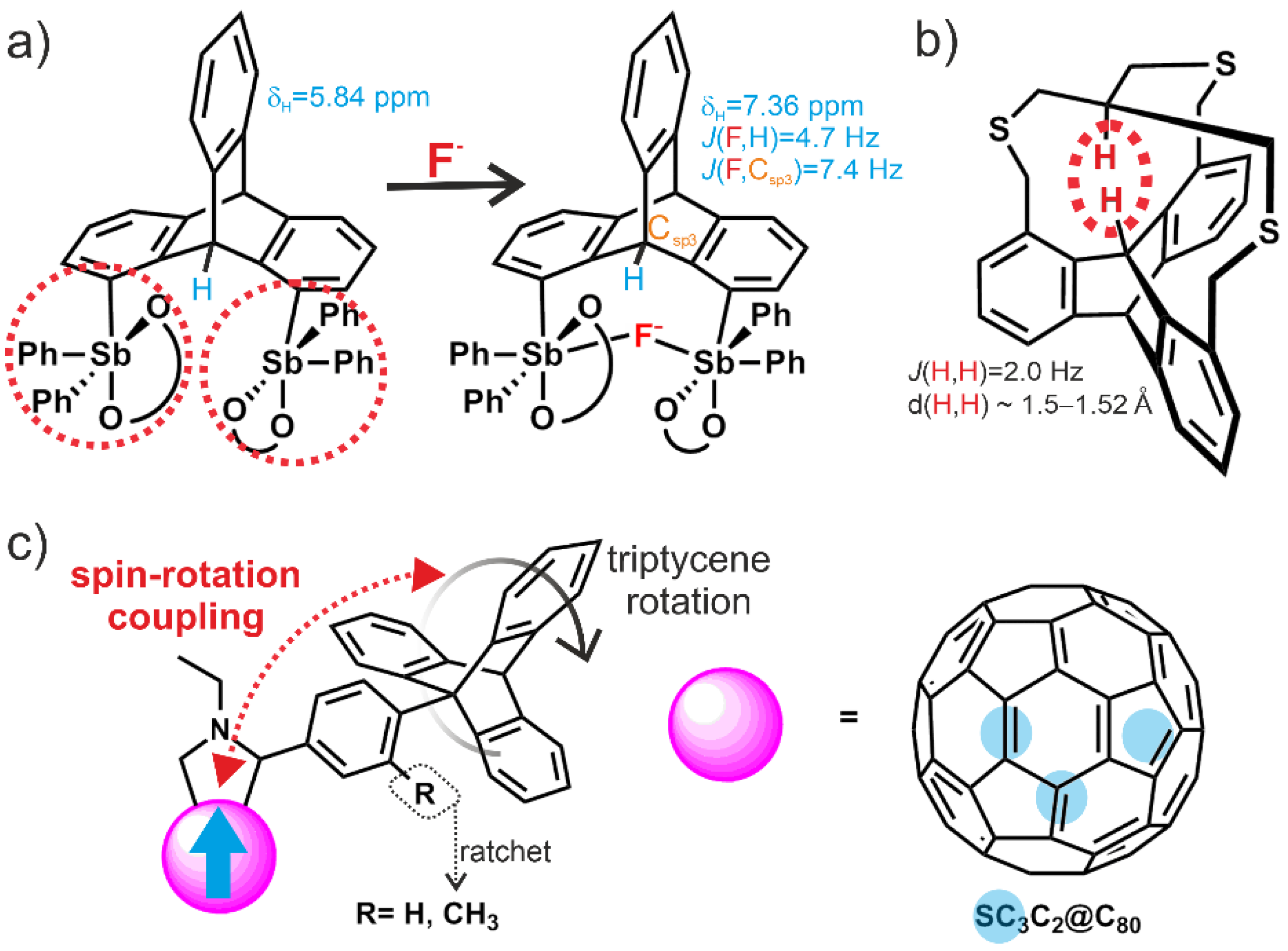
- Meng, H.; Zhao, C.; Nie, M.; Wang, C.; Wang, T. Triptycene molecular rotors mounted on metallofullerene Sc3C2@C80 and their spin–rotation couplings. Nanoscale 2018, 10, 18119–18123. [Google Scholar] [CrossRef]
- Sanmartín-Matalobos, J.; Fondo, M.; García-Deibe, A.M.; Amoza, M.; Bermejo, P.; Domínguez, M.R.; Mota, A.J.; Pérez-Lustres, J.L.; Bhowmick, S.; Das, N. Zinc-mediated diastereoselective assembly of a trinuclear circular helicate. RSC Adv. 2016, 6, 21228–21234. [Google Scholar] [CrossRef]
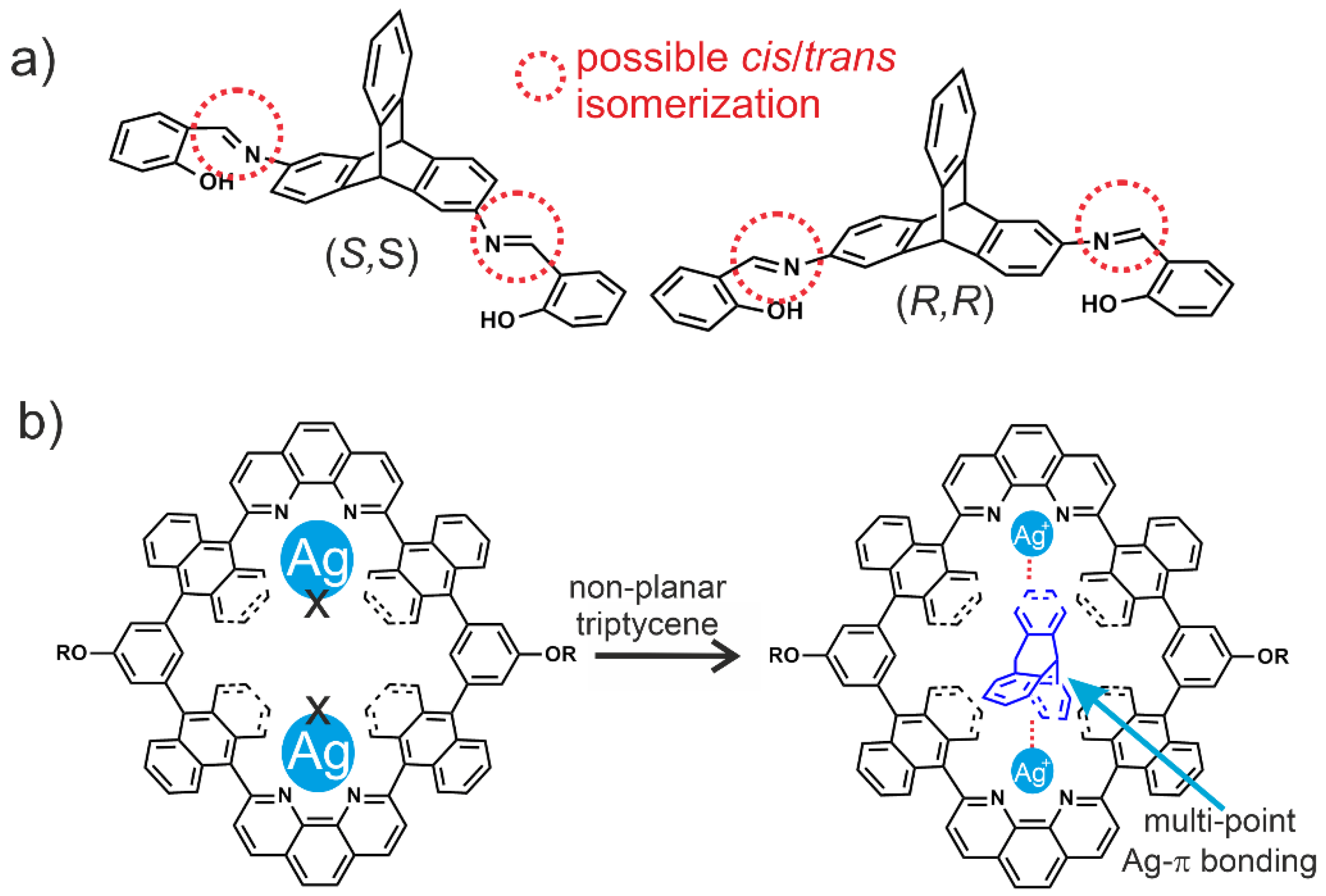
- Omoto, K.; Tashiro, S.; Shionoya, M. Molecular recognition of planar and non-planar aromatic hydrocarbons through multipoint Ag–π bonding in a dinuclear metallo-macrocycle. Chem. Sci. 2019, 10, 7172–7176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, N.G.; MacLachlan, M.J. Anion-templated hexagonal nanotubes. Chem. Sci. 2015, 6, 6245–6249. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Torubaev, Y.; Ansari, S.N.; Singh, S.K.; Mobin, S.M.; Mathur, P. The borderline: Exploring the structural landscape of triptycene in cocrystallization with ferrocene. CrystEngComm 2020, 22, 1314–1320. [Google Scholar] [CrossRef]
- Margulies, E.A.; Shoer, L.E.; Eaton, S.W.; Wasielewski, M.R. Excimer formation in cofacial and slip-stacked perylene-3,4:9,10-bis(dicarboximide) dimers on a redox-inactive triptycene scaffold. Phys. Chem. Chem. Phys. 2014, 16, 23735–23742. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-H.; Lin, Z.-Q.; Shi, N.-E.; Jin, L.-Z.; Yu, M.-N.; Xie, L.-H.; Yi, M.-D.; Huang, W. A polyhedral supramolecular system of endocyclic crystalline organic nanostructures: The case of triptycenes. CrystEngComm 2015, 17, 1448–1452. [Google Scholar] [CrossRef]
- Shuku, Y.; Mizuno, A.; Ushiroguchi, R.; Hyun, C.S.; Ryu, Y.J.; An, B.-K.; Kwon, J.E.; Park, S.Y.; Tsuchiizu, M.; Awaga, K. An exotic band structure of a supramolecular honeycomb lattice formed by a pancake π–π interaction between triradical trianions of triptycene tribenzoquinone. Chem. Commun. 2018, 54, 3815–3818. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, T.; Maruyama, M.; Okada, S.; Hatsugai, Y. Flat bands and higher-order topology in polymerized triptycene: Tight-binding analysis on decorated star lattices. Phys. Rev. Mater. 2019, 3, 114201. [Google Scholar] [CrossRef] [Green Version]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TRPCl-R1 | TRPCl-R2 | |
|---|---|---|
| 1J(Si,CPh) | 68 | - |
| 1J(Si,CPh) | - | 77 |
| 1J(H1,Si) | 214 | 215 |
| 1J(H2,Si) | 214 | 190 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woźny, M.; Mames, A.; Ratajczyk, T. Triptycene Derivatives: From Their Synthesis to Their Unique Properties. Molecules 2022, 27, 250. https://doi.org/10.3390/molecules27010250
Woźny M, Mames A, Ratajczyk T. Triptycene Derivatives: From Their Synthesis to Their Unique Properties. Molecules. 2022; 27(1):250. https://doi.org/10.3390/molecules27010250
Chicago/Turabian StyleWoźny, Mateusz, Adam Mames, and Tomasz Ratajczyk. 2022. "Triptycene Derivatives: From Their Synthesis to Their Unique Properties" Molecules 27, no. 1: 250. https://doi.org/10.3390/molecules27010250