
pH-Responsive N^C-Cyclometalated Iridium(III) Complexes: Synthesis, Photophysical Properties, Computational Results, and Bioimaging Application



Abstract
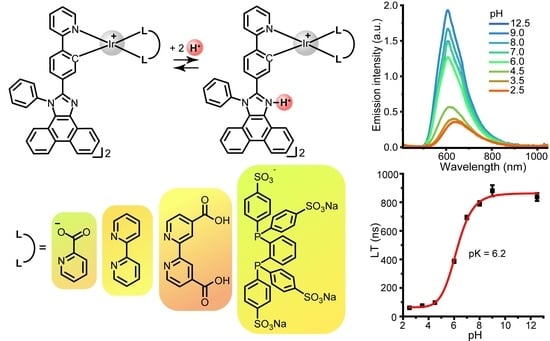
:
1. Introduction
2. Results and Discussion
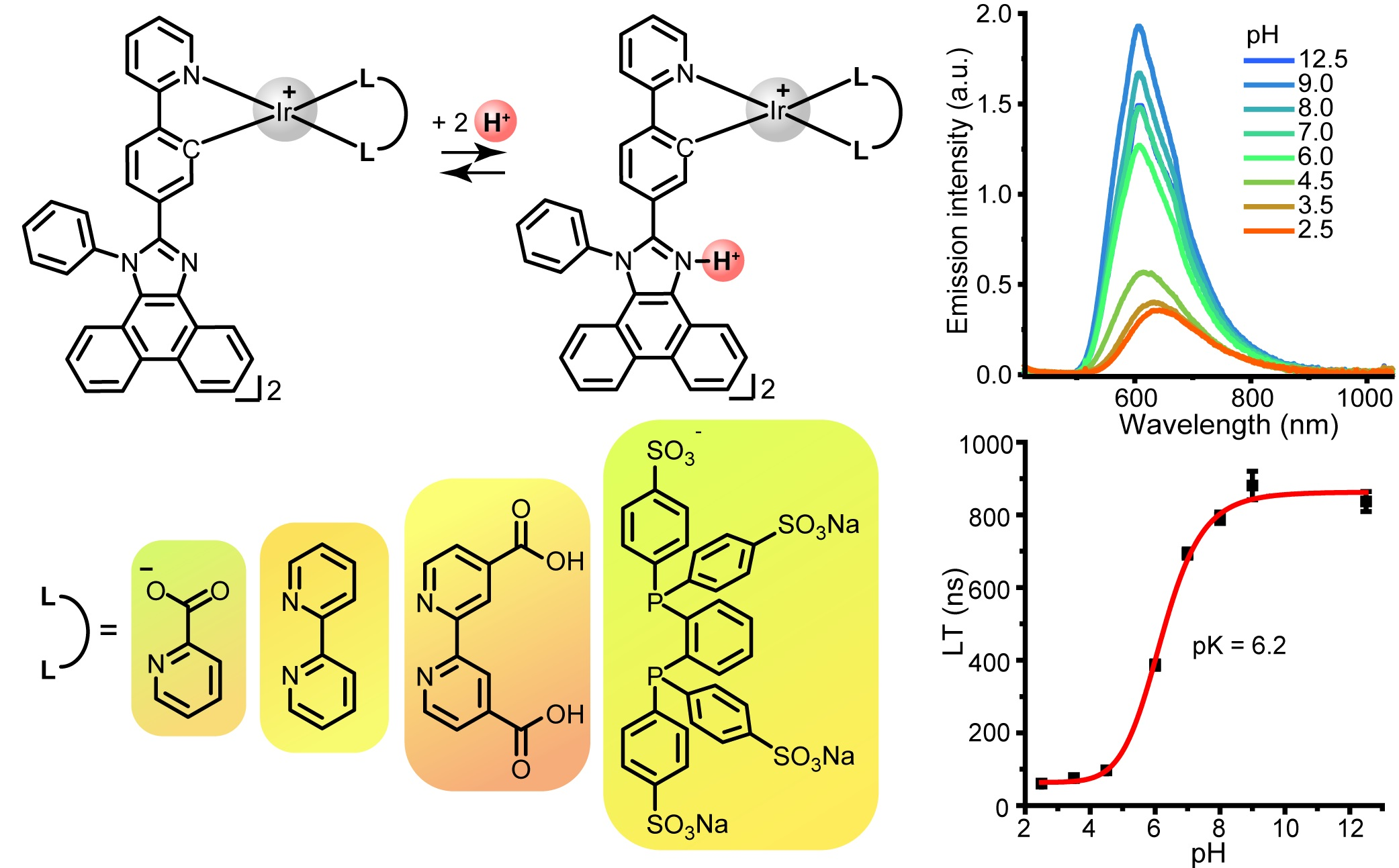
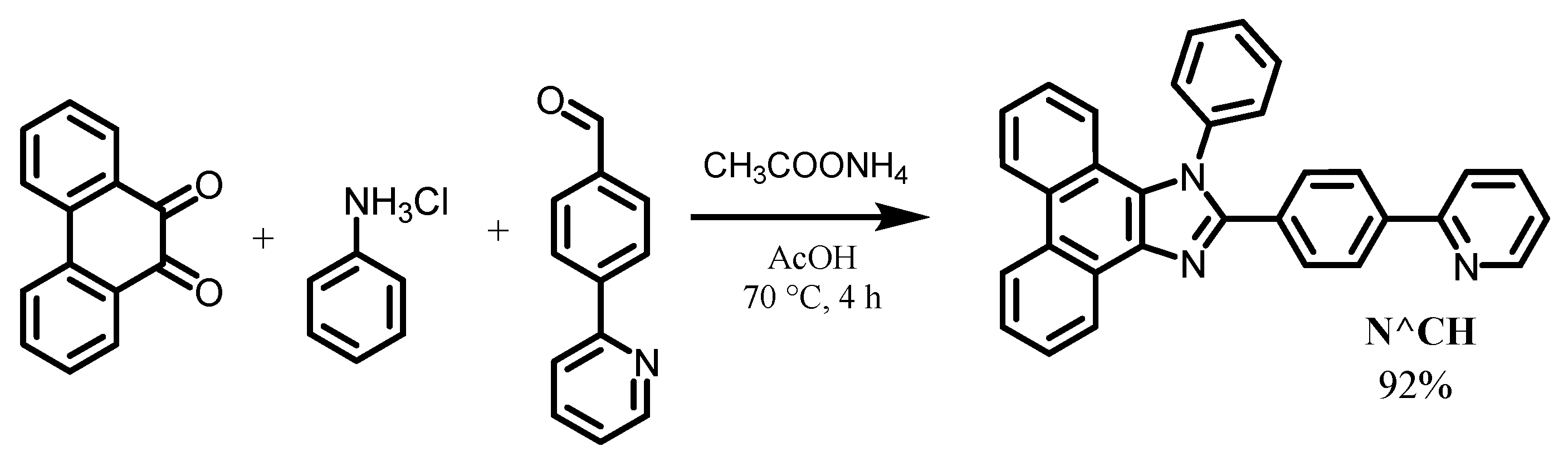
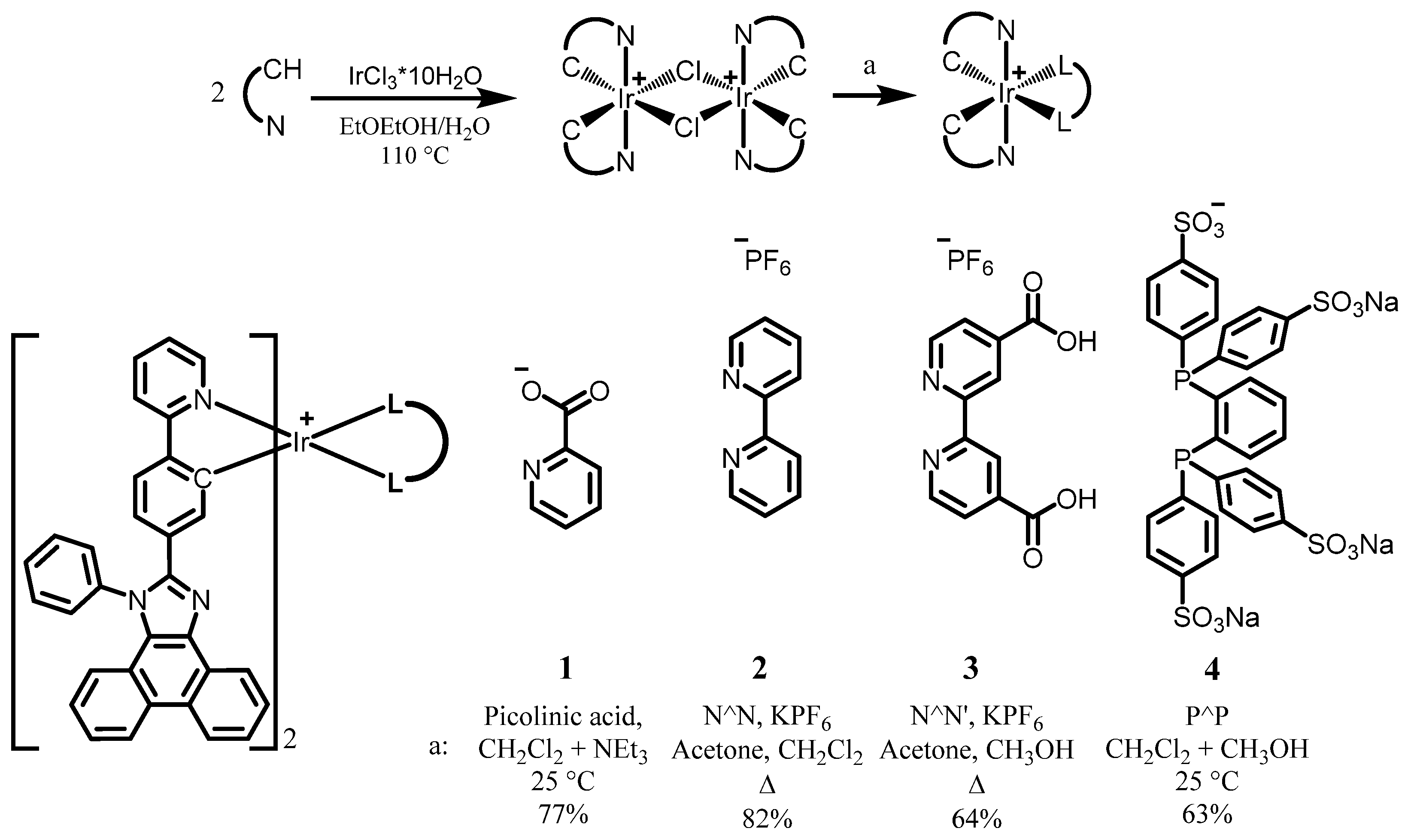
2.1. Synthesis of Ligand and Complexes
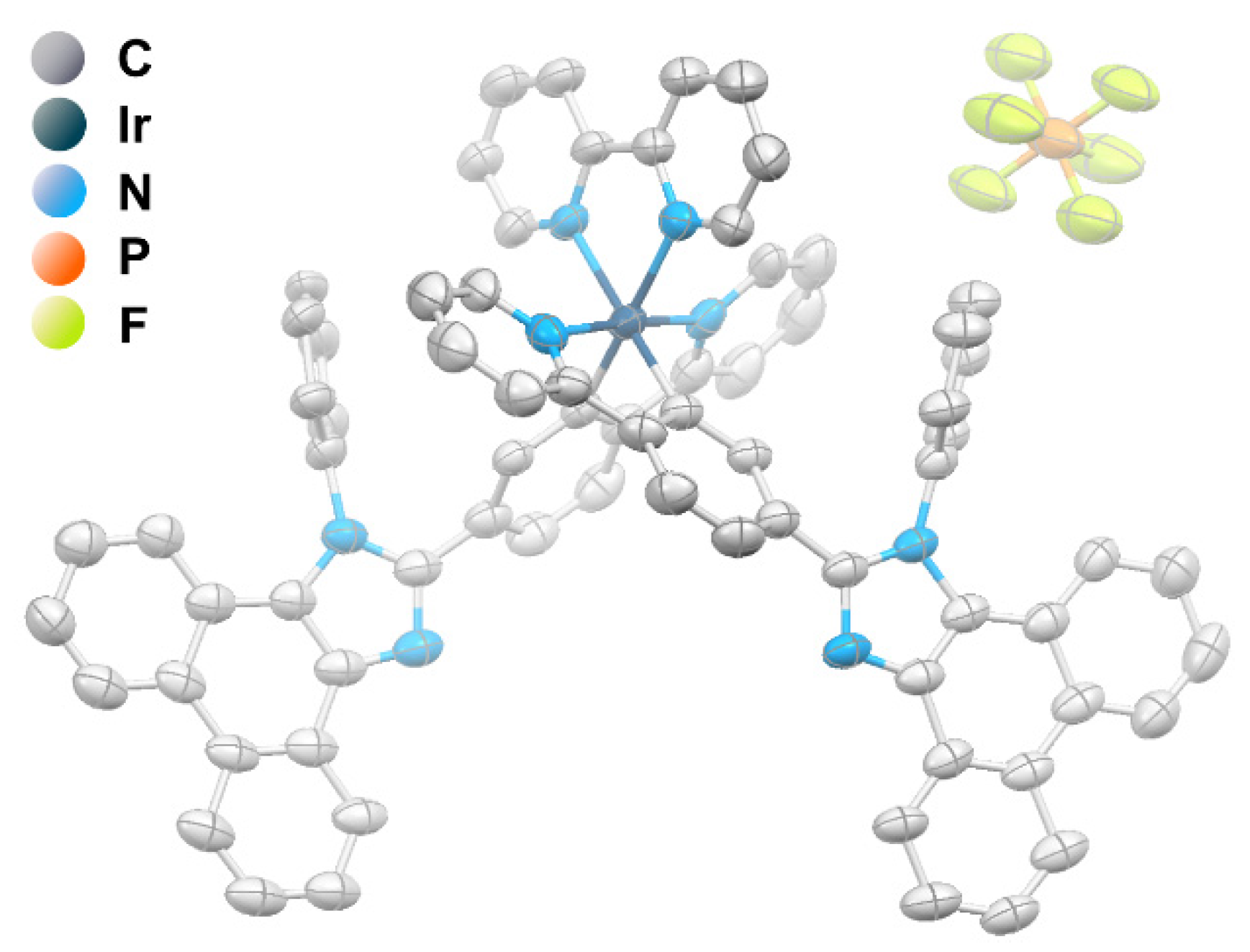
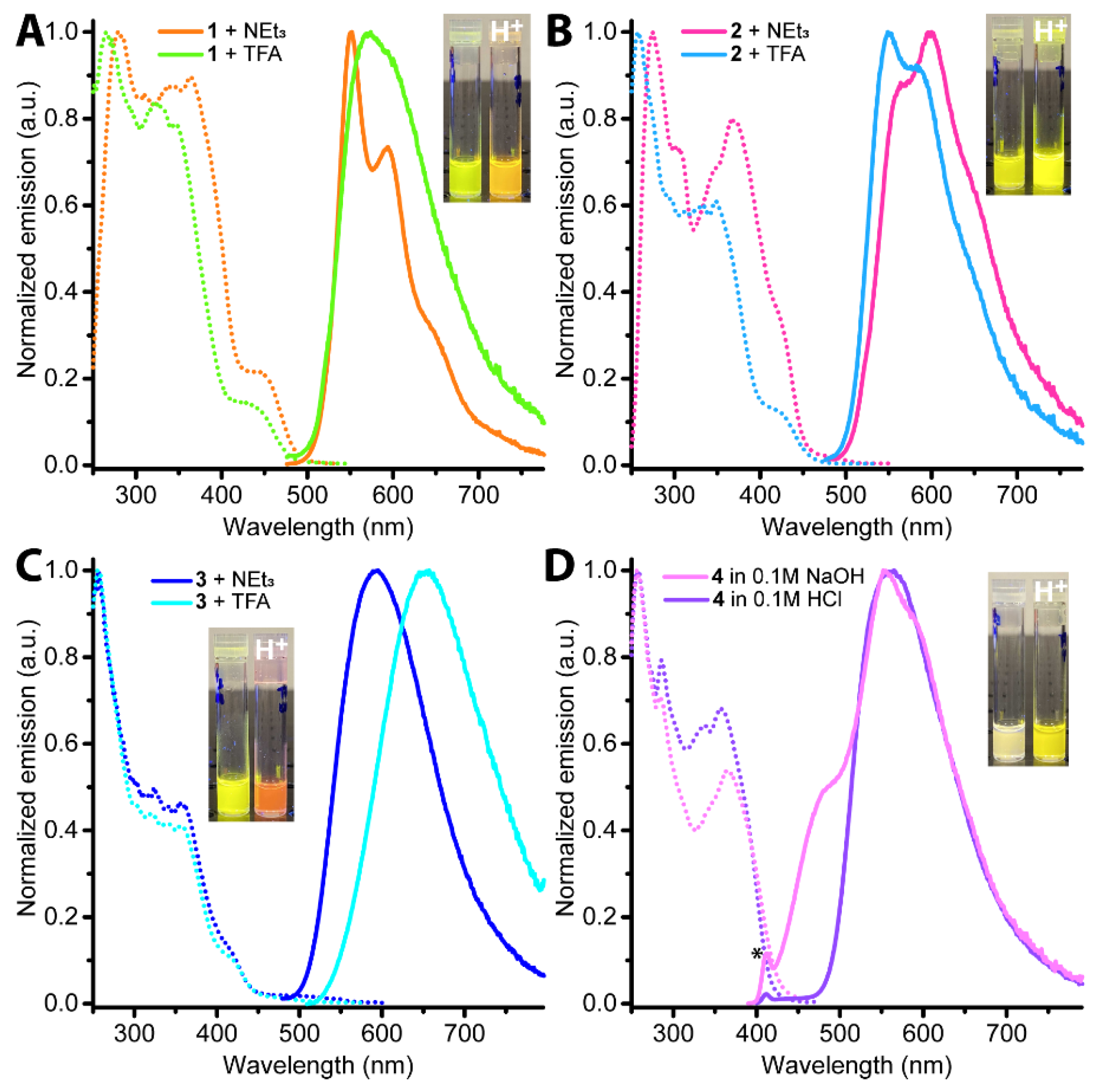
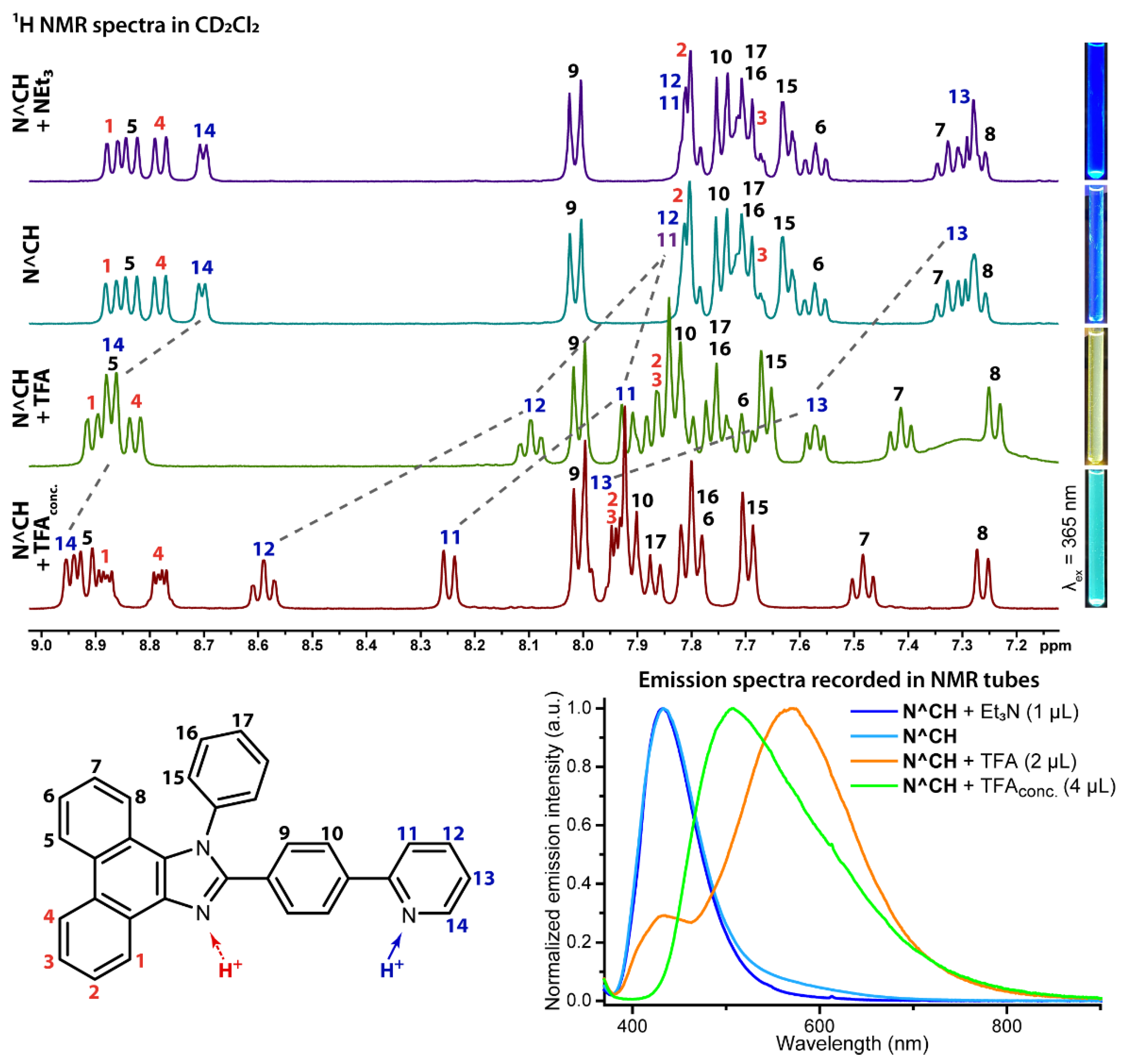
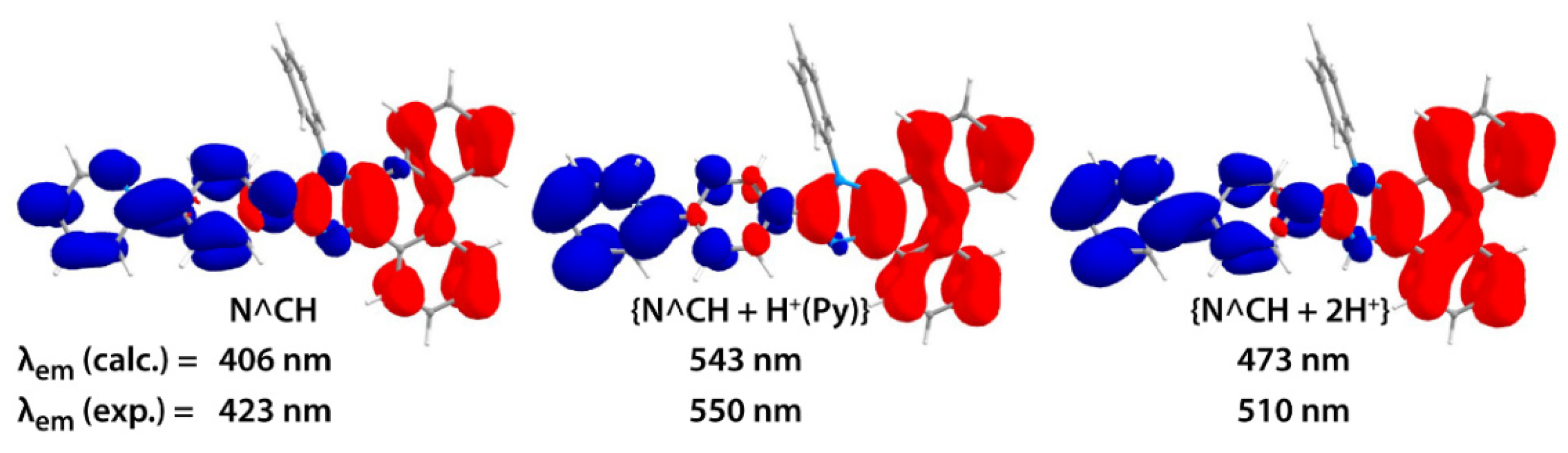
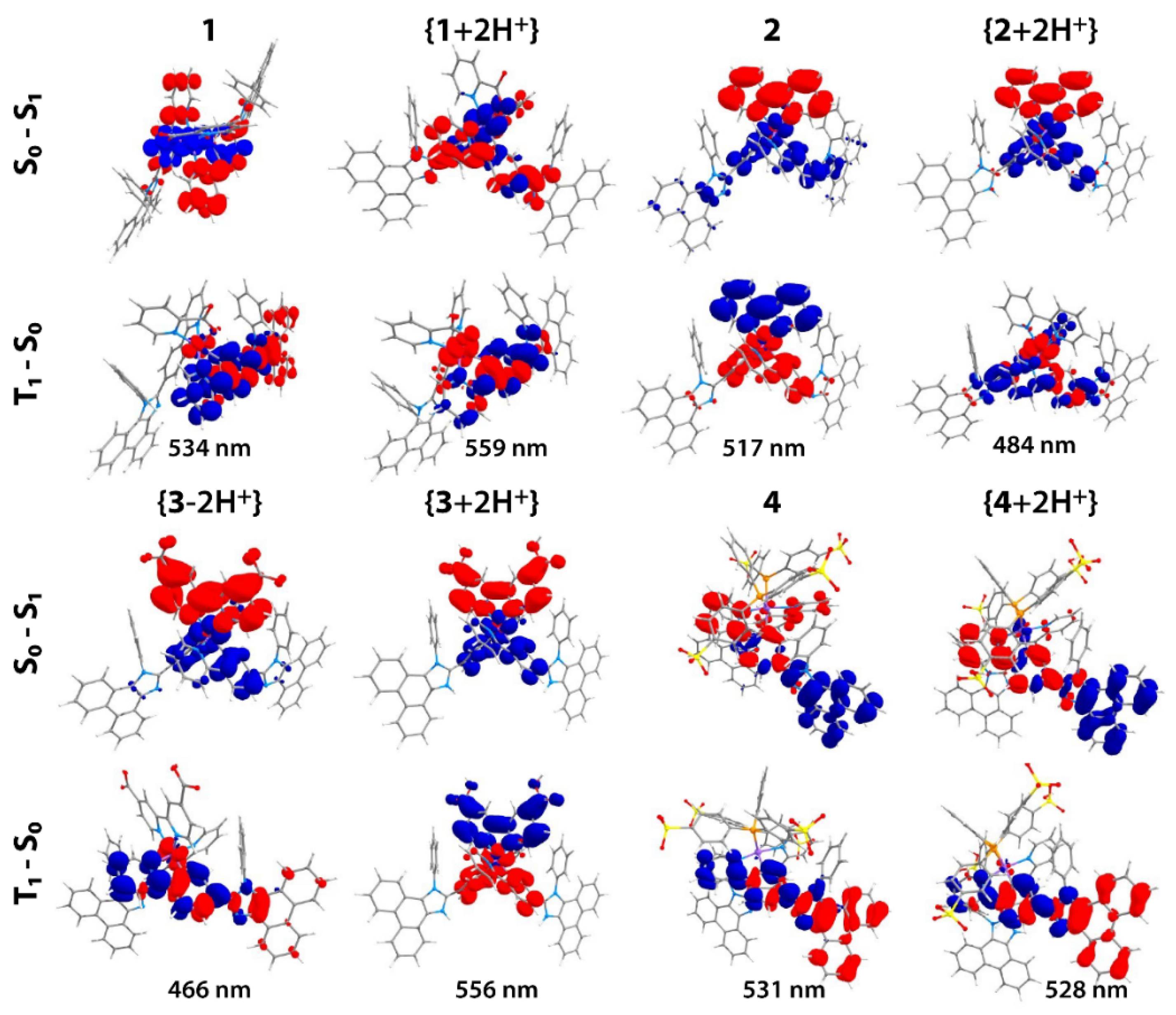
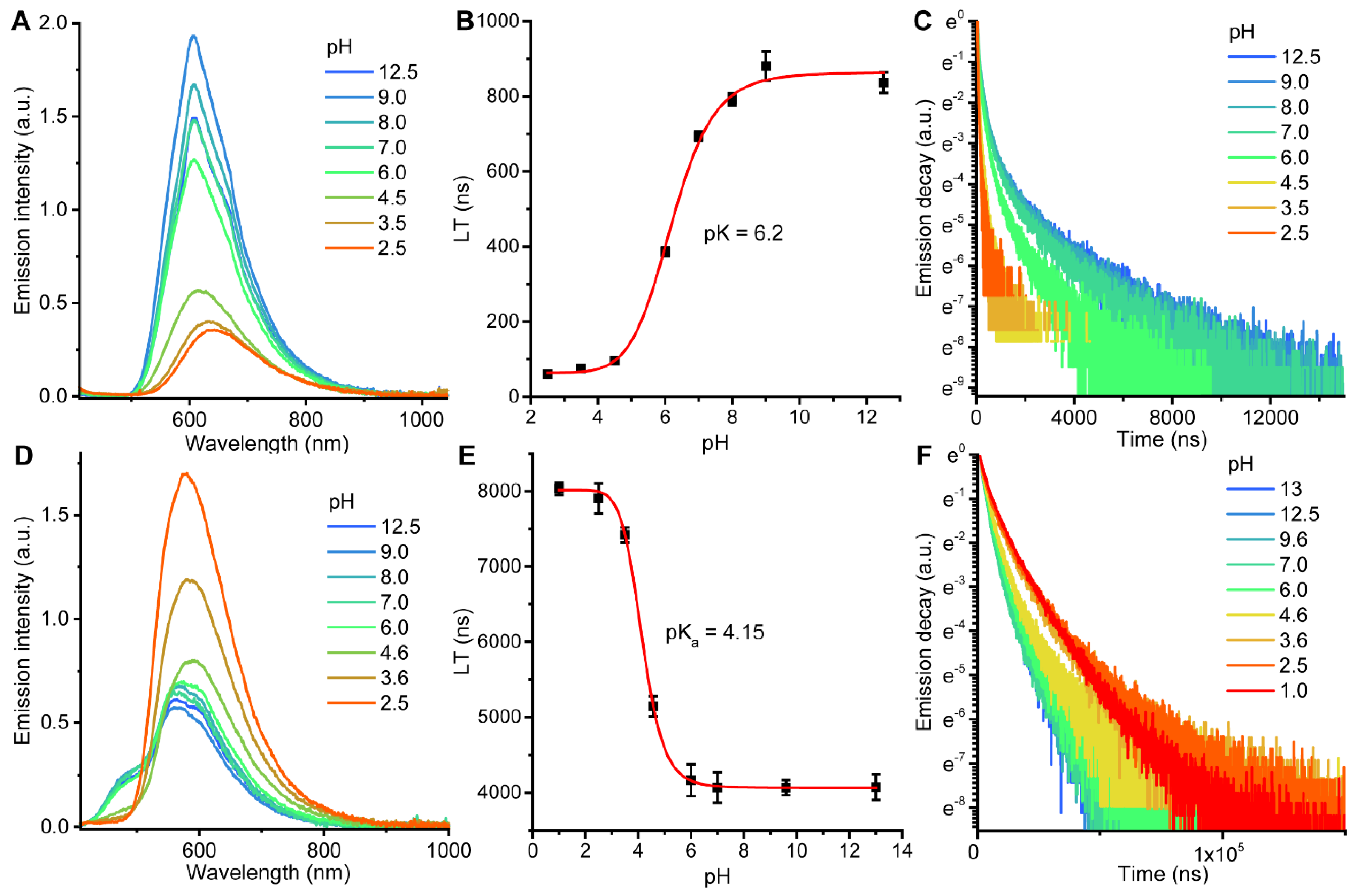
2.2. Photophysical Properties and Quantum Chemical Calculations for N^CH and Complexes 1–4
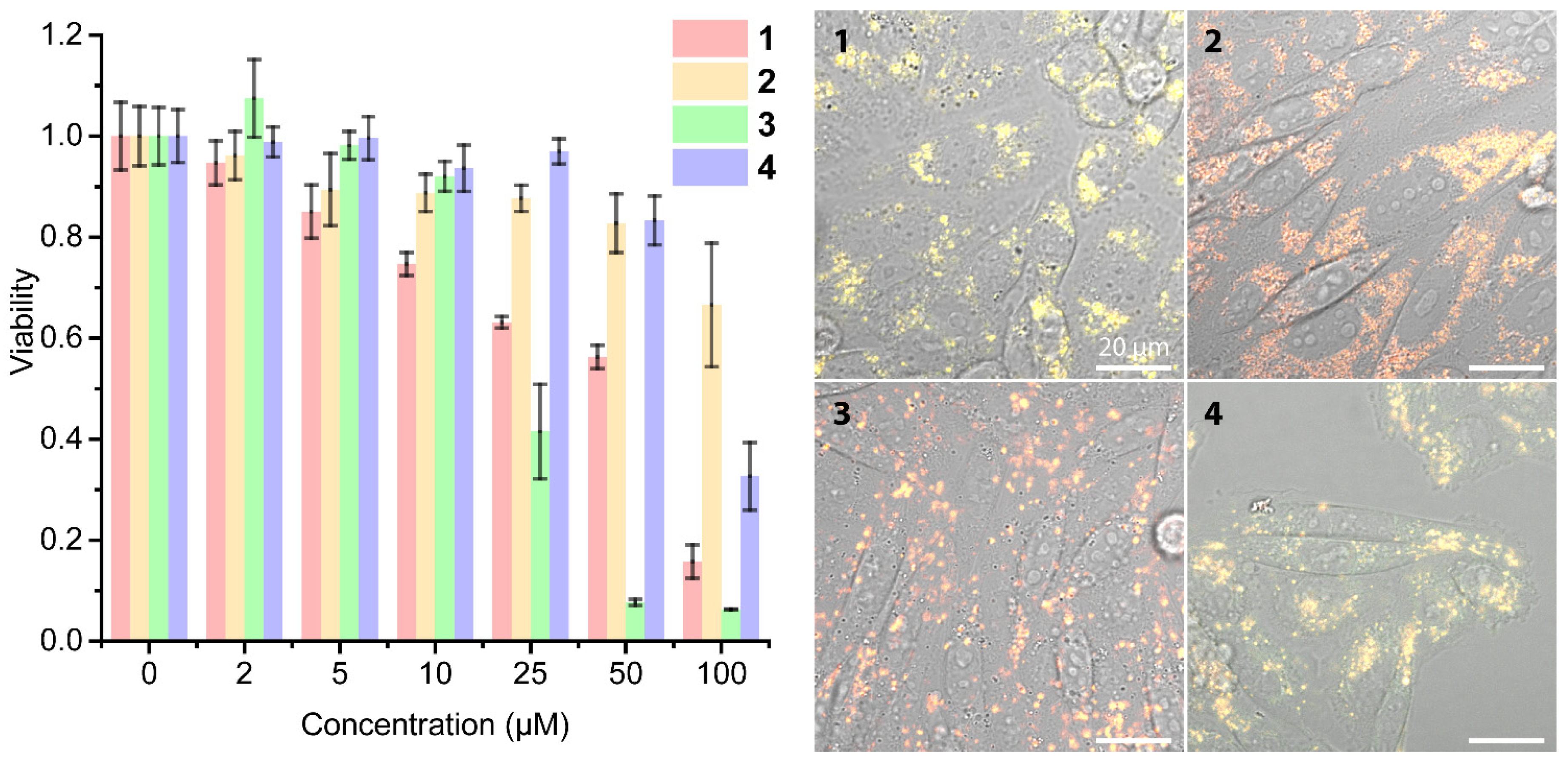
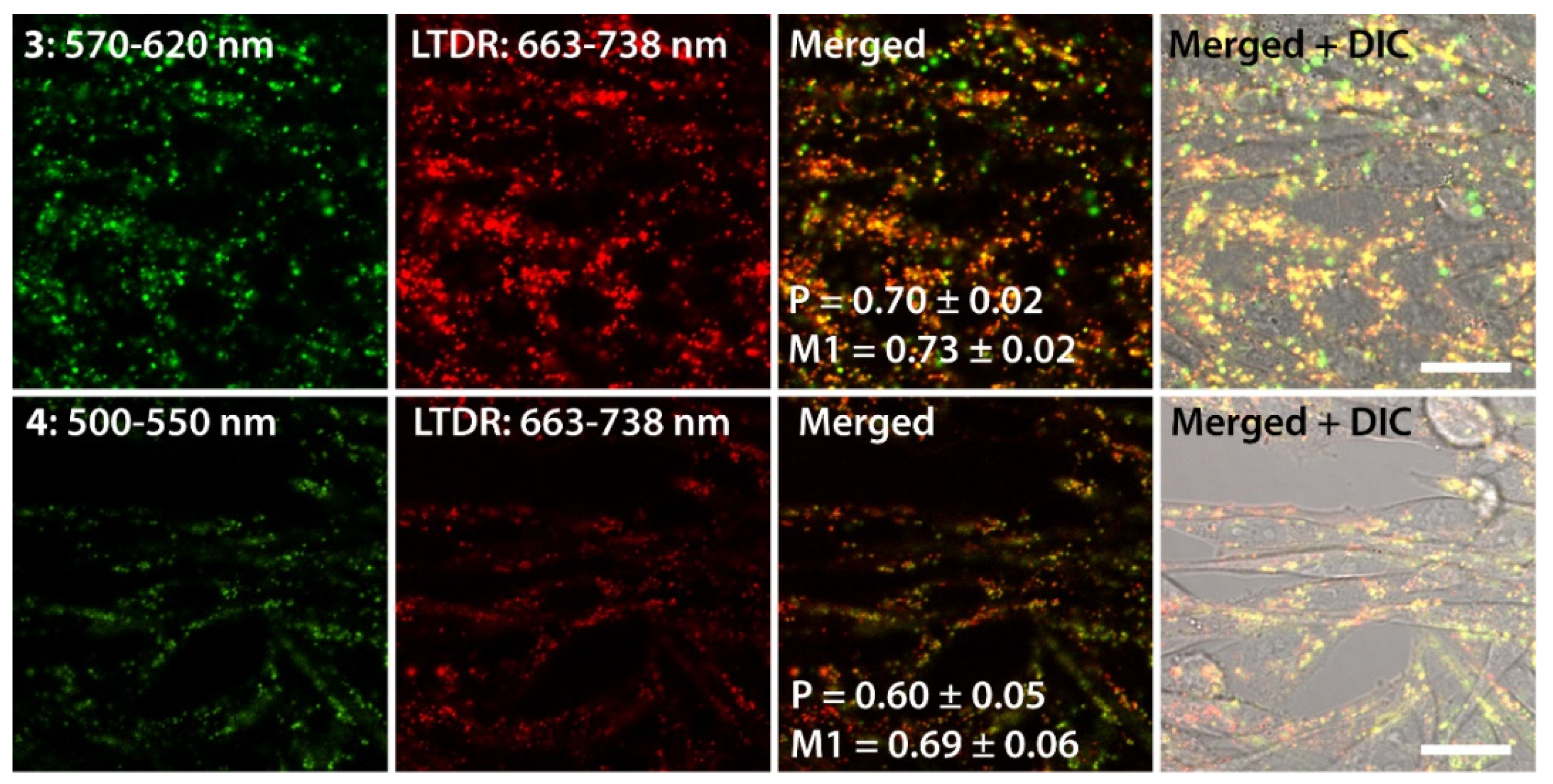
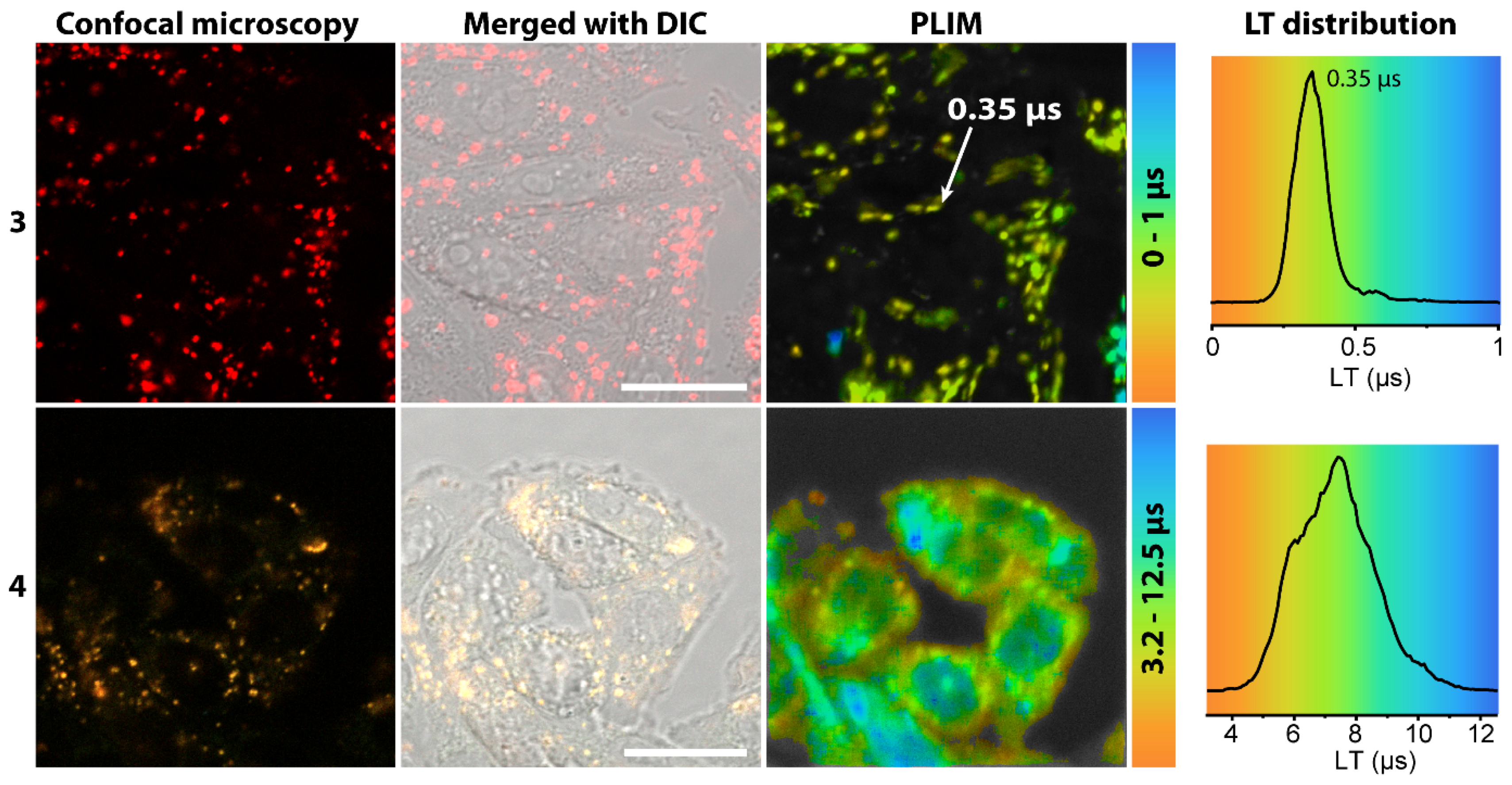
2.3. Living Cell Imaging Using 1–4
3. Materials and Methods
3.1. Synthesis of the Ligand and Complexes
3.2. X-ray Diffraction Analysis
3.3. Photophysical Measurements
3.4. Computational Details
3.5. Cell Culturing
3.6. MTT Assay
3.7. Lysosome Staining
3.8. Confocal Luminescence Microscopy and PLIM Experiment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Wencel, D.; Abel, T.; McDonagh, C. Optical Chemical pH Sensors. Anal. Chem. 2014, 86, 15–29. [Google Scholar] [CrossRef]
- Wen, Y.; Jing, N.; Huo, F.; Yin, C. Recent Progress of Organic Small Molecule-Based Fluorescent Probes for Intracellular pH Sensing. Analyst 2021, 146, 7450–7463. [Google Scholar] [CrossRef]
- Yang, Z.; Cao, J.; He, Y.; Yang, J.H.; Kim, T.; Peng, X.; Kim, J.S. Macro-/Micro-Environment-Sensitive Chemosensing and Biological Imaging. Chem. Soc. Rev. 2014, 43, 4563–4601. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Hu, Y.; Yoon, J. Fluorescent Probes and Bioimaging: Alkali Metals, Alkaline Earth Metals and pH. Chem. Soc. Rev. 2015, 44, 4619–4644. [Google Scholar] [CrossRef]
- Schwartz, L.; Peres, S.; Jolicoeur, M.; da Veiga Moreira, J. Cancer and Alzheimer’s Disease: Intracellular pH Scales the Metabolic Disorders. Biogerontology 2020, 21, 683–694. [Google Scholar] [CrossRef]
- Srivastava, J.; Barber, D.L.; Jacobson, M.P. Intracellular pH Sensors: Design Principles and Functional Significance. Physiology 2007, 22, 30–39. [Google Scholar] [CrossRef]
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and Regulators of Intracellular pH. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Griffiths, J.R. How and Why Are Cancers Acidic? Carbonic Anhydrase IX and the Homeostatic Control of Tumour Extracellular pH. Cancers 2020, 12, 1616. [Google Scholar] [CrossRef] [PubMed]
- Izumi, H.; Torigoe, T.; Ishiguchi, H.; Uramoto, H.; Yoshida, Y.; Tanabe, M.; Ise, T.; Murakami, T.; Yoshida, T.; Nomoto, M.; et al. Cellular pH Regulators: Potentially Promising Molecular Targets for Cancer Chemotherapy. Cancer Treat. Rev. 2003, 29, 541–549. [Google Scholar] [CrossRef]
- Hao, G.; Xu, Z.P.; Li, L. Manipulating Extracellular Tumour pH: An Effective Target for Cancer Therapy. RSC Adv. 2018, 8, 22182–22192. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.-T.; Ren, W.X.; Li, K.; Seo, J.; Sharma, A.; Yu, X.-Q.; Kim, J.S. Fluorescent Bioimaging of pH: From Design to Applications. Chem. Soc. Rev. 2017, 46, 2076–2090. [Google Scholar] [CrossRef]
- Di Costanzo, L.; Panunzi, B. Visual pH Sensors: From a Chemical Perspective to New Bioengineered Materials. Molecules 2021, 26, 2952. [Google Scholar] [CrossRef]
- Han, J.; Burgess, K. Fluorescent Indicators for Intracellular pH. Chem. Rev. 2010, 110, 2709–2728. [Google Scholar] [CrossRef] [PubMed]
- Steinegger, A.; Wolfbeis, O.S.; Borisov, S.M. Optical Sensing and Imaging of pH Values: Spectroscopies, Materials, and Applications. Chem. Rev. 2020, 120, 12357–12489. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Li, F.; Huang, C. Phosphorescent Chemosensors Based on Heavy-Metal Complexes. Chem. Soc. Rev. 2010, 39, 3007–3030. [Google Scholar] [CrossRef] [PubMed]
- Richter, C.; Schneider, C.; Quick, M.T.; Volz, P.; Mahrwald, R.; Hughes, J.; Dick, B.; Alexiev, U.; Ernsting, N.P. Dual-Fluorescence pH Probe for Bio-Labelling. Phys. Chem. Chem. Phys. 2015, 17, 30590–30597. [Google Scholar] [CrossRef] [Green Version]
- Tunik, S.P.; Chelushkin, P.S.; Shakirova, J.R.; Kritchenkov, I.; Baigildin, V.A. Phosphorescent NIR Emitters for Biomedicine: Applications, Advances and Challenges. Dalton Trans. 2021. [Google Scholar] [CrossRef]
- Lu, N.; Luo, Y.; Zhang, Q.; Zhang, P. Microenvironment-Sensitive Iridium(III) Complexes for Disease Theranostics. Dalton Trans. 2020, 49, 9182–9190. [Google Scholar] [CrossRef]
- Caporale, C.; Massi, M. Cyclometalated Iridium(III) Complexes for Life Science. Coord. Chem. Rev. 2018, 363, 71–91. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.-X.; Mak, E.C.-L.; Lo, K.K.-W. Photofunctional Transition Metal Complexes as Cellular Probes, Bioimaging Reagents and Phototherapeutics. Inorg. Chem. Front. 2021, 8, 4553–4579. [Google Scholar] [CrossRef]
- Shaikh, S.; Wang, Y.; ur Rehman, F.; Jiang, H.; Wang, X. Phosphorescent Ir (III) Complexes as Cellular Staining Agents for Biomedical Molecular Imaging. Coord. Chem. Rev. 2020, 416, 213344. [Google Scholar] [CrossRef]
- You, Y. Phosphorescence Bioimaging Using Cyclometalated Ir(III) Complexes. Curr. Opin. Chem. Biol. 2013, 17, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Meng, X.; Xie, M.; Shi, Y.; Zou, L.; Guo, S.; Jiang, J.; Liu, S.; Zhao, Q. A Self-Calibrating Phosphorescent Polymeric Probe for Measuring pH Fluctuations in Subcellular Organelles and the Zebrafish Digestive Tract. J. Mater. Chem. C 2020, 8, 2265–2271. [Google Scholar] [CrossRef]
- Nakagawa, A.; Hisamatsu, Y.; Moromizato, S.; Kohno, M.; Aoki, S. Synthesis and Photochemical Properties of pH Responsive Tris-Cyclometalated Iridium(III) Complexes That Contain a Pyridine Ring on the 2-Phenylpyridine Ligand. Inorg. Chem. 2014, 53, 409–422. [Google Scholar] [CrossRef]
- Kando, A.; Hisamatsu, Y.; Ohwada, H.; Itoh, T.; Moromizato, S.; Kohno, M.; Aoki, S. Photochemical Properties of Red-Emitting Tris(cyclometalated) Iridium(III) Complexes Having Basic and Nitro Groups and Application to pH Sensing and Photoinduced Cell Death. Inorg. Chem. 2015, 54, 5342–5357. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Ran, G.; Zhao, Y.; Wang, C.; Song, Q. Synthesis and Application of a Water-Soluble Phosphorescent Iridium Complex as Turn-On Sensing Material for Human Serum Albumin. Dalton Trans. 2018, 47, 2330–2336. [Google Scholar] [CrossRef] [PubMed]
- Alam, P.; Kaur, G.; Sarmah, A.; Roy, R.K.; Choudhury, A.R.; Laskar, I.R. Highly Selective Detection of H+ and OH− with a Single-Emissive Iridium(III) Complex: A Mild Approach to Conversion of Non-AIEE to AIEE Complex. Organometallics 2015, 34, 4480–4490. [Google Scholar] [CrossRef]
- Ohno, K.; Sakata, T.; Shiiba, M.; Nagasawa, A.; Fujihara, T. A Water-Soluble Cyclometalated Iridium( III) Complex for pH Sensing Based on Aggregation-Induced Enhanced Phosphorescence. Dalton Trans. 2019, 48, 8068–8075. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Sheth, S.; Zhao, Y.; Song, Q. A Novel Cyclometallated Iridium(III) Complex Based Dual-Mode Phosphorescent Probe for Detection of Acidity and Bovine Serum Albumin. Anal. Methods 2019, 11, 3033–3040. [Google Scholar] [CrossRef]
- He, L.; Tan, C.P.; Ye, R.R.; Zhao, Y.Z.; Liu, Y.H.; Zhao, Q.; Ji, L.N.; Mao, Z.W. Theranostic Iridium(III) Complexes as One- and Two-Photon Phosphorescent Trackers to Monitor Autophagic Lysosomes. Angew. Chem.—Int. Ed. 2014, 53, 12137–12141. [Google Scholar] [CrossRef]
- Leavens, B.B.H.; Trindle, C.O.; Sabat, M.; Altun, Z.; Demas, J.N.; DeGraff, B.A. Photophysical and Analyte Sensing Properties of Cyclometalated Ir(III) Complexes. J. Fluoresc. 2012, 22, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Liang, H.; Zeng, Y.; Yang, H.; Ho, C.L.; Xu, W.; Zhao, Q.; Huang, W.; Wong, W.Y. Phosphorescent Soft Salt for Ratiometric and Lifetime Imaging of Intracellular pH Variations. Chem. Sci. 2016, 7, 3338–3346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Z.; Wang, Y.; Chen, Y.; Fang, H.; Yuan, H.; Shi, X.; Yang, B.; Chen, Z.; He, W.; Guo, Z. A Novel Luminescent Ir(III) Complex for Dual Mode Imaging: Synergistic Response to Hypoxia and Acidity of the Tumor Microenvironment. Chem. Commun. 2020, 56, 8055–8058. [Google Scholar] [CrossRef]
- Solomatina, A.I.; Kuznetsov, K.M.; Gurzhiy, V.V.; Pavlovskiy, V.V.; Porsev, V.V.; Evarestov, R.A.; Tunik, S.P.; Solomatina, A.I.; Kuznetsov, K.M.; Gurzhiy, V.V.; et al. Luminescent Organic Dyes Containing a Phenanthro[9,10-D ]Imidazole Core and [Ir(N^C)(N^N)] + Complexes Based on the Cyclometalating and Diimine Ligands of This Type. Dalton Trans. 2020, 49, 6751–6763. [Google Scholar] [CrossRef] [PubMed]
- Nonoyama, M. Benzo[h]quinolin-10-yl-N Iridium(III) Complexes. Bull. Chem. Soc. Jpn. 1974, 47, 767–768. [Google Scholar] [CrossRef] [Green Version]
- Achelle, S.; Rodríguez-López, J.; Bureš, F.; Robin-le Guen, F. Tuning the Photophysical Properties of Push-Pull Azaheterocyclic Chromophores by Protonation: A Brief Overview of a French-Spanish-Czech Project. Chem. Rec. 2020, 20, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, T.; Gupta, S.; Milton, M.D. Smart Organic Materials with Acidochromic Properties. Curr. Org. Chem. 2020, 24, 1976–1998. [Google Scholar] [CrossRef]
- Li, K.; Tong, G.S.M.; Yuan, J.; Ma, C.; Du, L.; Yang, C.; Kwok, W.-M.; Phillips, D.L.; Che, C.-M. Excitation-Wavelength-Dependent and Auxiliary-Ligand-Tuned Intersystem-Crossing Efficiency in Cyclometalated Platinum(II) Complexes: Spectroscopic and Theoretical Studies. Inorg. Chem. 2020, 59, 14654–14665. [Google Scholar] [CrossRef] [PubMed]
- Sprouse, S.; King, K.A.; Spellane, P.J.; Watts, R.J. Photophysical Effects of Metal-Carbon .Sigma. Bonds in Ortho-Metalated Complexes of Iridium(III) and Rhodium(III). J. Am. Chem. Soc. 1984, 106, 6647–6653. [Google Scholar] [CrossRef]
- Bruce, M. Cyclometalation Reactions. Angew. Chem.—Int. Ed. 1977, 16, 73–86. [Google Scholar] [CrossRef]
- CrysAlisPro, Version: 1.171.39.35a; Rigaku Oxford Diffraction; Rigaku Corporation: Tokyo, Japan, 2017.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. Platon Squeeze: A Tool for the Calculation of the Disordered Solvent Contribution to the Calculated Structure Factors. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Brouwer, A.M. Standards for Photoluminescence Quantum Yield Measurements in Solution (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 2213–2228. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Kobayashi, A.; Kaneko, S.; Takehira, K.; Yoshihara, T.; Ishida, H.; Shiina, Y.; Oishi, S.; Tobita, S. Reevaluation of Absolute Luminescence Quantum Yields of Standard Solutions Using a Spectrometer with an Integrating Sphere and a Back-Thinned CCD Detector. Phys. Chem. Chem. Phys. 2009, 11, 9850–9860. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Austin, A.; Petersson, G.A.; Frisch, M.J.; Dobek, F.J.; Scalmani, G.; Throssell, K. A Density Functional with Spherical Atom Dispersion Terms. J. Chem. Theory Comput. 2012, 8, 4989–5007. [Google Scholar] [CrossRef]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-Adjustedab Initio Pseudopotentials for the Second and Third Row Transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Peverati, R.; Truhlar, D.G. Screened-Exchange Density Functionals with Broad Accuracy for Chemistry and Solid-State Physics. Phys. Chem. Chem. Phys. 2012, 14, 16187–16191. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- O’boyle, N.M.; Tenderholt, A.L.; Langner, K.M. CCLIB: A Library for Package-Independent Computational Chemistry Algorithms. J. Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.L. Natural Transition Orbitals. J. Chem. Phys. 2003, 118, 4775–4777. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N° | Absorbance, nm (ε × 10−3, M−1cm−1) | λex, nm | λem, nm | Stokes Shift *, cm−1 | τav (aer/deg **), μs | Φ (aer/deg **), % |
|---|---|---|---|---|---|---|
| N^CH in CH2Cl2 | 262 (53), 280 (25), 335 (23), 363 (19) | 315sh, 356sh, 372 | 423 | 3910 | 1.7 × 10−3 | 18 |
| N^CH in CH2Cl2 + TFA (1:1) | 250sh (49), 257 (63), 275 (26), 295 (19), 323 (19), 334 (19), 351 (20), 410sh (2.4) | 317, 355sh (at 426 nm); 343, 377 (at 600 nm) | 423, 550 | 750 | 3.1 × 10−3 (at 550 nm) | 14 |
| N^CH in CH2Cl2 + TFA conc | 255 (86), 276 (31), 292sh (23), 322 (14), 334 (16), 358 (21) | 335, 368 | 510 | 8355 | 1.1 × 10−3 | 11 |
| 1 in CH2Cl2 | 261 (124), 308sh (36), 350sh (38), 364 (40), 450sh (5) | 280, 308sh, 342, 364, 445 | 553, 596, 650sh | 4140 | 0.53/ 4.32 | 2.0/25 |
| 1 in CH2Cl2 + TFA | 256, 277sh, 298sh, 320sh, 333sh, 348, 450sh | 265, 325, 348, 430sh | 590 | 5270 | 0.65/ 1.62 | 4.3/14 |
| 1 in CH2Cl2 + Et3N | 261 (124), 308sh (36), 350sh (38), 364 (40), 450sh (5) | 280, 308sh, 342, 364, 445 | 554, 597, 650sh | 4170 | 0.46/ 6.56 | 1.8/30 |
| 2 in CH2Cl2 | 261 (149), 309sh (40), 350sh (34), 370 (41), 425sh (15) | 275, 305sh, 350sh, 370, 425sh | 570, 595 | 5990 | 0.69/ 6.14 | 2.0/17 |
| 2 in CH2Cl2 + TFA | 257, 275sh, 300sh, 310sh, 335sh, 350sh, 425sh | 258, 290sh, 335sh, 350sh, 420sh | 550, 585, 640sh | 5350 | 1.02/ 6.68 | 3.0/21 |
| 2 in CH2Cl2 + Et3N | 260 (149), 309sh (40), 350sh (34), 370 (41), 425sh (15) | 275, 305sh, 350sh, 370, 425sh | 570, 600, 650sh | 5990 | 0.54/ 7.37 | 1.9/31 |
| 3 in CH3OH | 258 (155), 304 (45), 322 (42), 342 (39), 357 (40), 420sh (8), 480sh (2) | 256, 301sh, 321, 340, 357, 420sh, 480sh | 595 | 4030 | 0.235/ 0.820 | 3.6/14 |
| 3 in CH3OH +TFA | 255 (148), 301sh (47), 325 (42), 357 (40), 420sh (9), 480sh (2) | 256, 301sh, 326, 340, 357, 420sh, 480sh | 650 | 5450 | 0.035/ 0.040 | 0.60/0.64 |
| 3 in CH3OH +NEt3 | 258 (155), 304 (45), 322 (42), 342 (39), 357 (40), 420sh (8), 480sh (2) | 256, 301sh, 321, 340, 357, 420sh, 480sh | 595 | 4030 | 0.226/ 0.647 | 4.4/13 |
| 4 in PBS pH 7.0 | 233 (617), 263sh (128), 271 (117), 279 (96), 308sh (24), 347sh (24), 367 (27) | 285, 350sh, 367 | 482sh, 555, 587sh | 6500 | 4.07/ 41.87 (at 560 nm); 1.0 × 10−3 (at 480 nm) | 0.46/4.35 |
| 4 in 0.1 M HCl | 236sh (589), 264 (117), 271 (109), 280 (91), 337 (29), 357 (31) | 274, 286, 302sh, 338, 360 | 563 | 10250 | 8.03/ 12.47 | 1.64/2.24 |
| 4 in 0.1 M NaOH | 233 (593), 263sh (122), 271 (112), 279 (91), 347sh (24), 367 (26) | 285, 350sh, 367 | 482sh, 555, 587sh | 6500 | 4.08/ 51.98 | 0.42/4.96 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solomatina, A.I.; Kozina, D.O.; Porsev, V.V.; Tunik, S.P. pH-Responsive N^C-Cyclometalated Iridium(III) Complexes: Synthesis, Photophysical Properties, Computational Results, and Bioimaging Application. Molecules 2022, 27, 232. https://doi.org/10.3390/molecules27010232
Solomatina AI, Kozina DO, Porsev VV, Tunik SP. pH-Responsive N^C-Cyclometalated Iridium(III) Complexes: Synthesis, Photophysical Properties, Computational Results, and Bioimaging Application. Molecules. 2022; 27(1):232. https://doi.org/10.3390/molecules27010232
Chicago/Turabian StyleSolomatina, Anastasia I., Daria O. Kozina, Vitaly V. Porsev, and Sergey P. Tunik. 2022. "pH-Responsive N^C-Cyclometalated Iridium(III) Complexes: Synthesis, Photophysical Properties, Computational Results, and Bioimaging Application" Molecules 27, no. 1: 232. https://doi.org/10.3390/molecules27010232