
High Performance Liquid Chromatography versus Stacking-Micellar Electrokinetic Chromatography for the Determination of Potentially Toxic Alkenylbenzenes in Food Flavouring Ingredients

Abstract
:1. Introduction
2. Material and Methods
2.1. Standards and Reagents
2.2. Instrumentation and Software
2.3. Sample Preparation
2.4. Reversed-Phase HPLC Conditions
2.5. MEKC Analysis
3. Results and Discussion
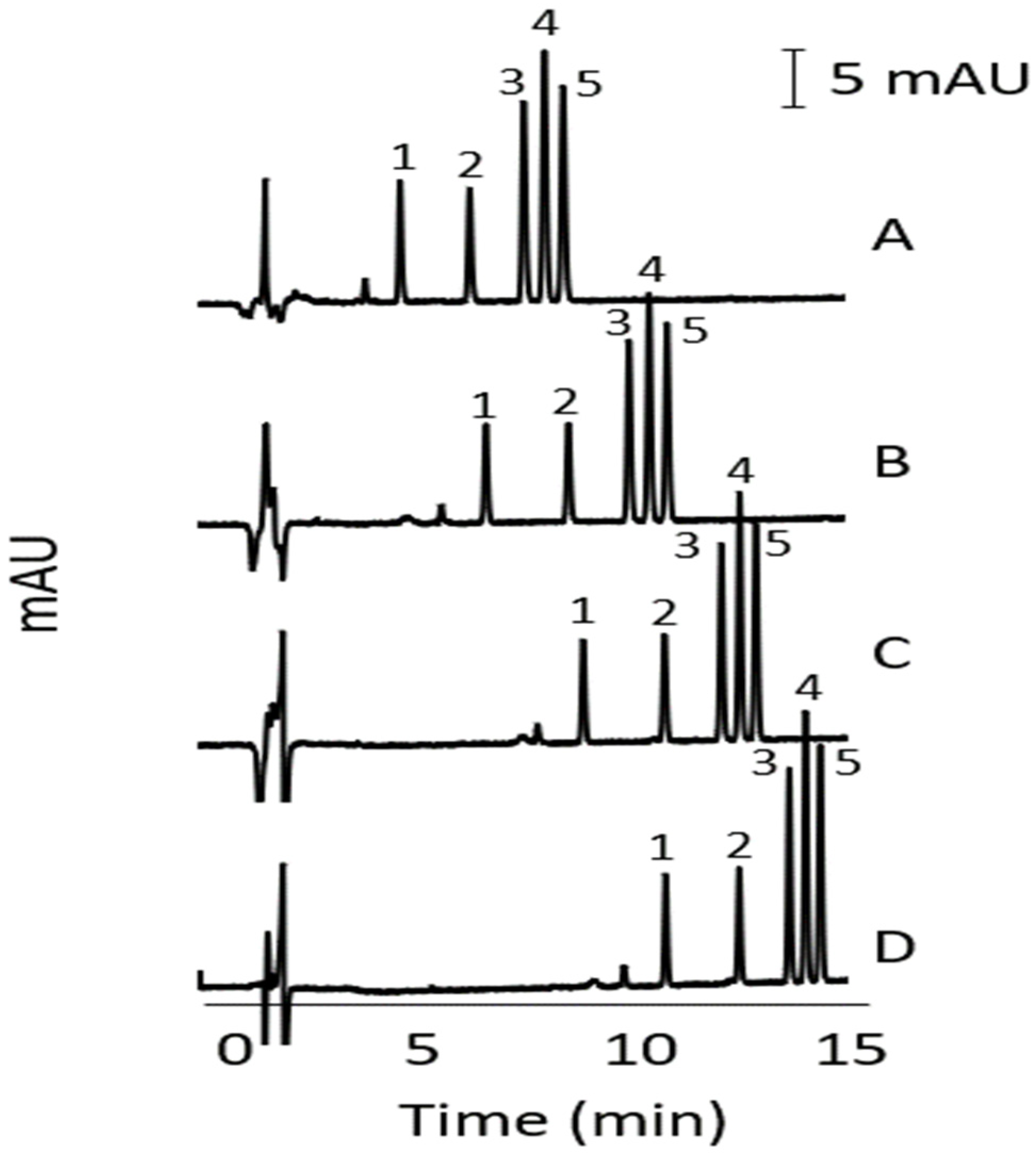
3.1. Reversed-Phase HPLC Method Development
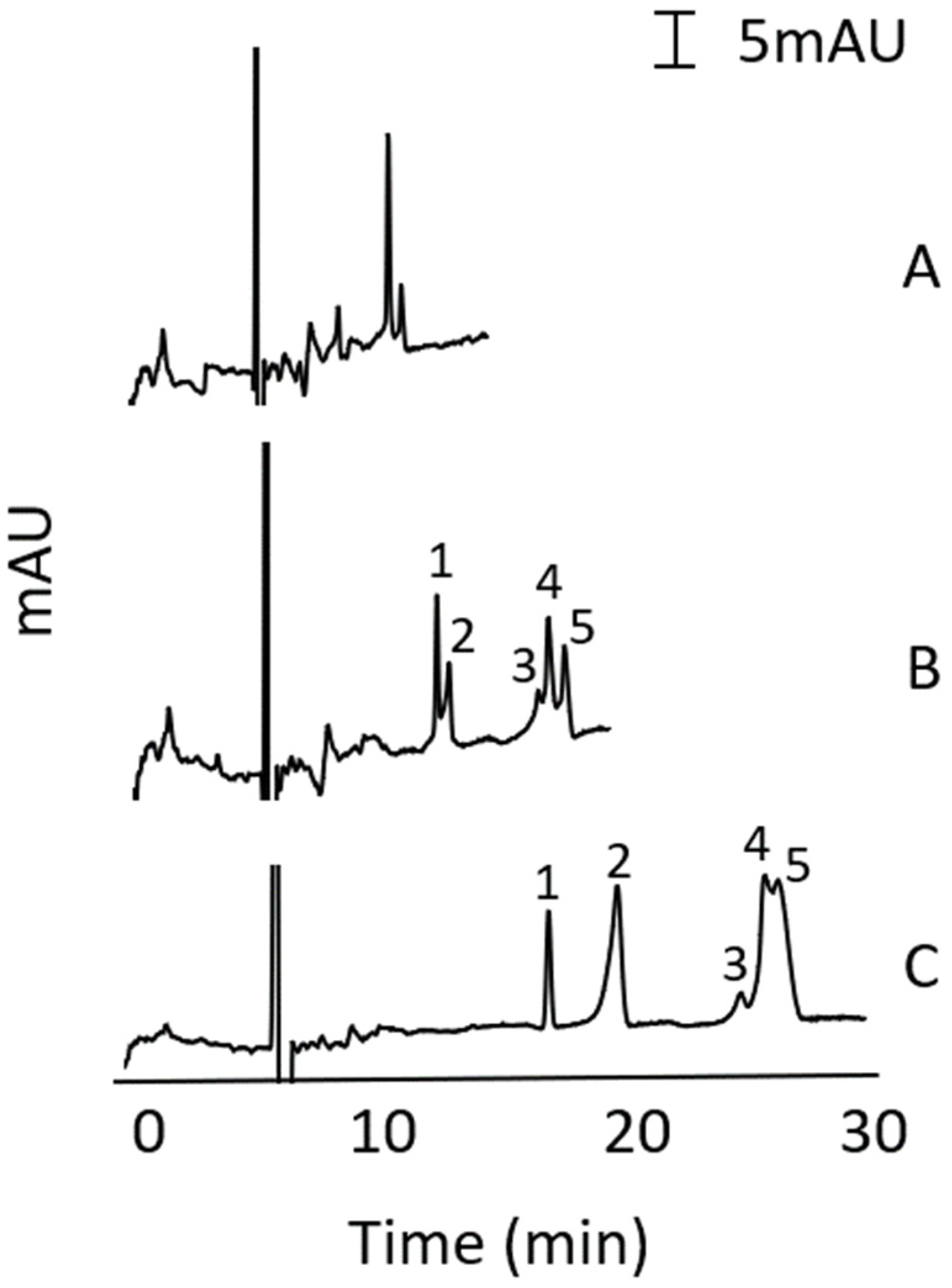
3.2. Stacking-MEKC Method Development
3.3. Stacking Method Development in MEKC
3.4. HPLC and Stacking-MEKC Comparison of Analytical Figures of Merit
3.4.1. Linearity
3.4.2. LOD and LOQ
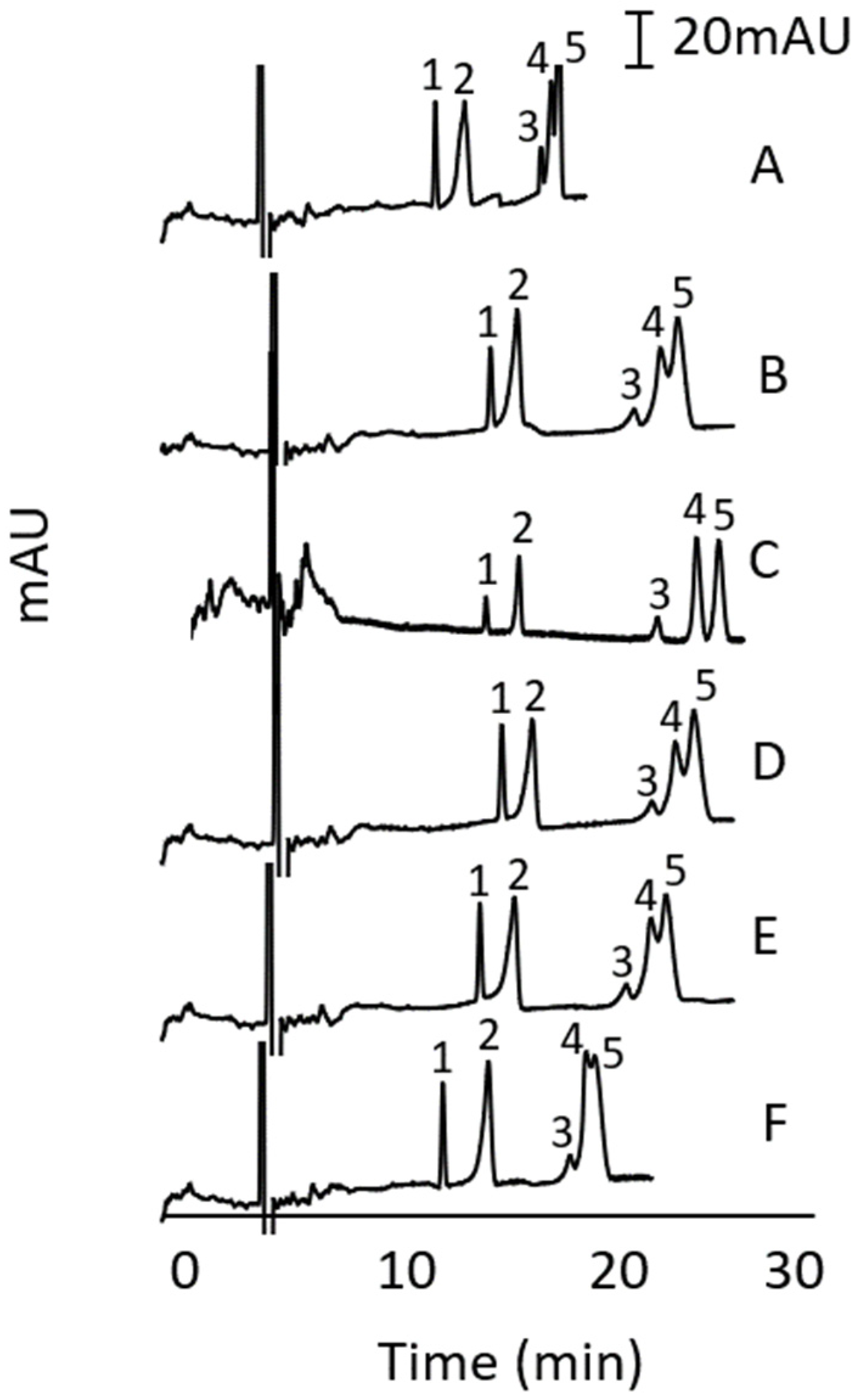
3.4.3. Intra- and Inter-Day Repeatability
3.5. Optimisation of Sample Preparation via MeOH Extraction
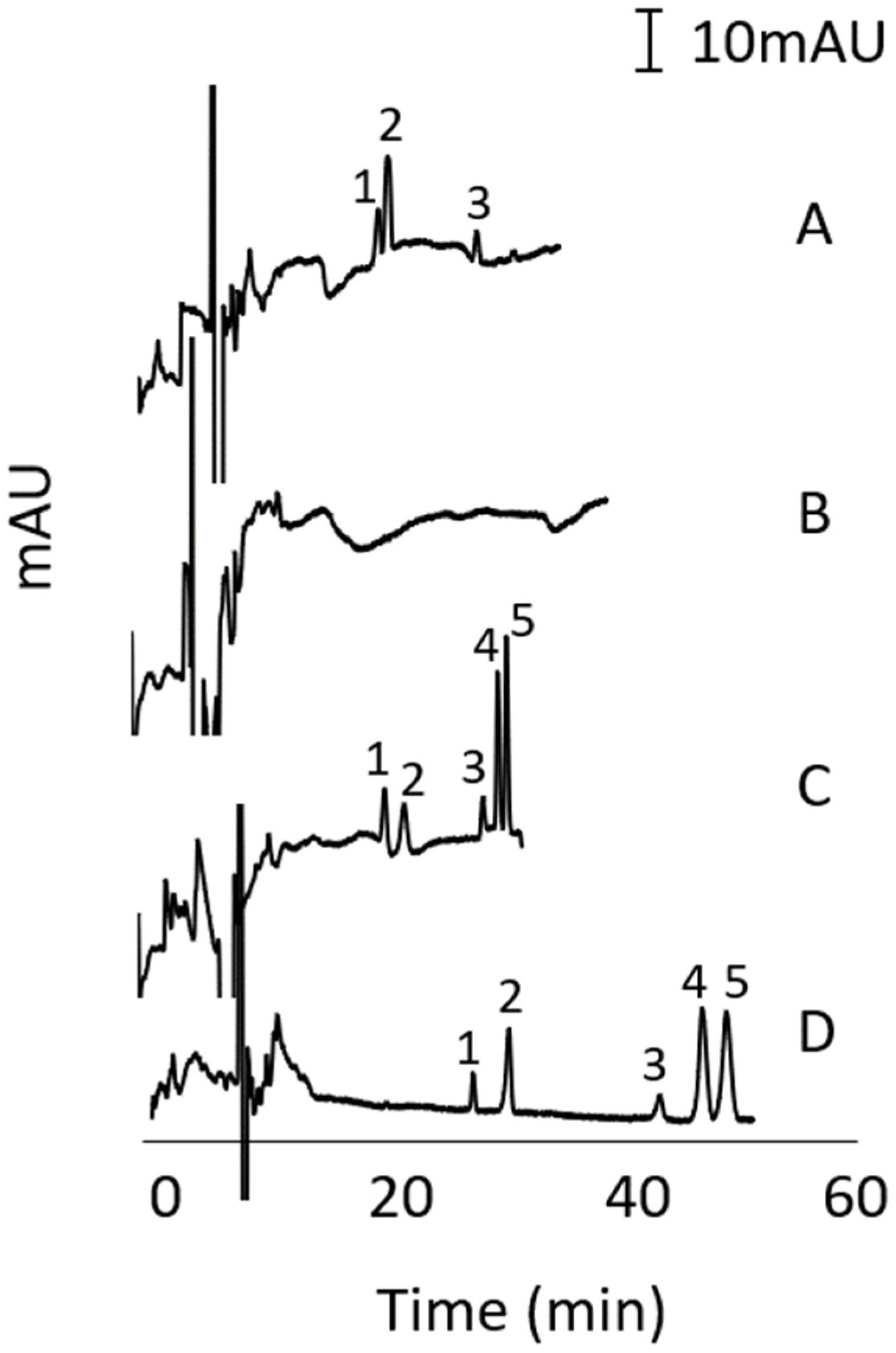
3.6. Recovery
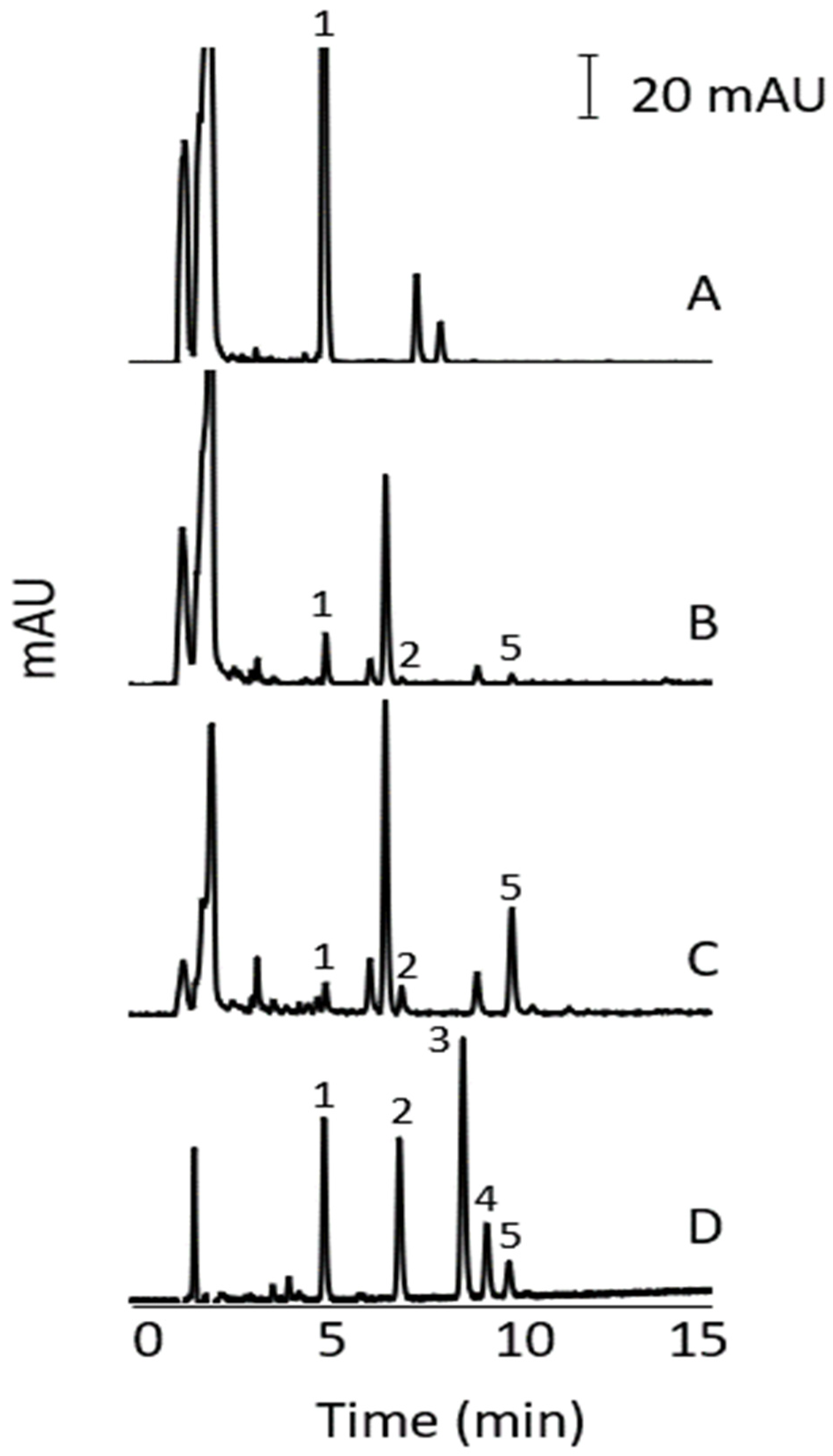
3.7. Method Application
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Sample Availability
Abbreviations
| HPLC | High performance liquid chromatography |
| MEKC | Micellar electrokinetic chromatography |
| GC | Gas chromatography |
References
- Martins, C.; Rueff, J.; Rodrigues, A.S. Genotoxic alkenylbenzene flavourings, a contribution to risk assessment. Food Chem. Toxicol. 2018, 118, 861–879. [Google Scholar] [CrossRef]
- Mohammadi Nejad, S.; Özgüneş, H.; Başaran, N. Pharmacological and Toxicological Properties of Eugenol. Turk. J. Pharm. Sci. 2017, 14, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Scientific Committee on Food. Opinion of the Scientific Committee on Food on Estragole (1-Allyl-4-Methoxybenzene); European Commission Health and Consumer Protection Directorate-General: Brussel, Belgium, 2001; pp. 1–10. [Google Scholar]
- Scientific Committee on Food. Opinion of the Scientific Committee on Food on the Safety of the Presence of Safrole (1-Allyl-3,4-Methylene Dioxy Benzene) in Flavourings and Other Food Ingredients with Flavouring Properties; European Commission Health and Consumer Protection Directorate-General: Brussel, Belgium, 2002. [Google Scholar]
- Scientific Committee on Food; European Commission. Opinion of the Scientific Committee on Food on Methyleugenol (4-Allyl-1,2-Dimethoxybenzene); European Commission Health and Consumer Protection Directorate-General: Brussel, Belgium, 2001. [Google Scholar]
- Alajlouni, A.M.; Al-Malahmeh, A.J.; Isnaeni, F.N.; Wesseling, S.; Vervoort, J.; Rietjens, I.M.C.M. Level of Alkenylbenzenes in Parsley and Dill Based Teas and Associated Risk Assessment Using the Margin of Exposure Approach. J. Agric. Food Chem. 2016, 64, 8640–8646. [Google Scholar] [CrossRef] [PubMed]
- Alajlouni, A.M.; Al-Malahmeh, A.J.; Wesseling, S.; Kalli, M.; Vervoort, J.; Rietjens, I.M.C.M. Risk assessment of combined exposure to alkenylbenzenes through consumption of plant food supplements containing parsley and dill. Food Addit. Contam.-Part A Chem. Anal. Control Expo. Risk Assess. 2017, 34, 2201–2211. [Google Scholar] [CrossRef] [PubMed]
- Suparmi, S.; Wahidin, D.; Rietjens, I.M.C.M. Risk characterisation of constituents present in jamu to promote its safe use. Crit. Rev. Toxicol. 2021, 51, 183–191. [Google Scholar] [CrossRef]
- Choong, Y.M.; Lin, H.J. A rapid and simple gas chromatography method for direct determination of safrole in soft drinks. J. Food Drug Anal. 2001, 9, 27–32. [Google Scholar]
- Raffo, A.; D’Aloise, A.; Magrì, A.L.; Leclercq, C. Quantitation of tr-cinnamaldehyde, safrole and myristicin in cola-flavoured soft drinks to improve the assessment of their dietary exposure. Food Chem. Toxicol. 2013, 59, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Ávila, M.; Zougagh, M.; Escarpa, A.; Ríos, Á. Determination of alkenylbenzenes and related flavour compounds in food samples by on-column preconcentration-capillary liquid chromatography. J. Chromatogr. A 2009, 1216, 7179–7185. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Park, W. Anti-inflammatory effect of myristicin on RAW 264.7 macrophages stimulated with polyinosinic-polycytidylic acid. Molecules 2011, 16, 7132–7142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woehrlin, F.; Fry, H.; Abraham, K.; Preiss-Weigert, A. Quantification of Flavoring Constituents in Cinnamon: High Variation of Coumarin in Cassia Bark from the German Retail Market and in Authentic Samples from Indonesia. J. Agric. Food Chem. 2010, 58, 10568–10575. [Google Scholar] [CrossRef]
- Khalil, A.A.; ur Rahman, U.; Khan, M.R.; Sahar, A.; Mehmood, T.; Khan, M. Essential oil eugenol: Sources, extraction techniques and nutraceutical perspectives. RSC Adv. 2017, 7, 32669–32681. [Google Scholar] [CrossRef] [Green Version]
- Environment Canada Health Canada. Benzenes, 1,2-Dimethoxy-4-(2-Propenyl)-(Methyl Eugenol); Environment Canada Health Canada: Ottawa, ON, Canada, 2010. [Google Scholar]
- Stein, U.; Greyer, H.; Hentschel, H. Nutmeg (myristicin) poisoning—report on a fatal case and a series of cases recorded by a poison information centre. Forensic Sci. Int. 2001, 118, 87–90. [Google Scholar] [CrossRef]
- Febrina, A.; Mutakin, M.; Lestari, K.; Levita, J. Determination of Safrole in Ethanol Extract of Nutmeg (Myristica fragrans Houtt) Using Reversed-Phase High Performance Liquid Chromatography. Int. J. Chem. 2014, 6. [Google Scholar] [CrossRef] [Green Version]
- Southwell, I.A.; Russell, M.F.; Davies, N.W. Detecting traces of methyl eugenol in essential oils: Tea tree oil, a case study. Flavour Fragr. J. 2011, 26, 336–340. [Google Scholar] [CrossRef]
- Cook, W.; Howard, A. The essential oil of Illicium anisatum Linn. Can. J. Chem. 2011, 44, 2461–2464. [Google Scholar] [CrossRef]
- National Toxicology Program. NTP Technical Report on the Toxicology Studies of Myristicin (CASRN 607-91-0) Administered by Gavage to F344/NTac Rats and B6C3F1/N Mice. In Toxicity Report 95; National Toxicology Program: Research Triangle Park, NC, USA, 2019. [Google Scholar]
- Marabini, L.; Neglia, L.; Monguzzi, E.; Galli, C.; Marinovich, M. Assessment of Toxicity of Myristicin and 1′-Hydroxymyristicin in HepG2 Cell Line. J. Pharmacol. Toxicol. 2017, 12, 170–179. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, S.; Singh, A.P. Medicinal Properties of Ginger (Zingiber Officinale Rosc.); NISCAIR-CSIR: New Delhi, India, 2003. [Google Scholar]
- Suparmi, S.; Ginting, A.J.; Mariyam, S.; Wesseling, S.; Rietjens, I.M.C.M. Levels of methyleugenol and eugenol in instant herbal beverages available on the Indonesian market and related risk assessment. Food Chem. Toxicol. 2019, 125, 467–478. [Google Scholar] [CrossRef]
- Farag, S.E.A.; Abo-Zeid, M. Degradation of the natural mutagenic compound safrole in spices by cooking and irradiation. Food/Nahr. 1997, 41, 359–361. [Google Scholar] [CrossRef]
- Zeller, A.; Rychlik, M. Impact of estragole and other odorants on the flavour of anise and tarragon. Flavour Fragr. J. 2007, 22, 105–113. [Google Scholar] [CrossRef]
- Eisenreich, A.; Götz, M.E.; Sachse, B.; Monien, B.H.; Herrmann, K.; Schäfer, B. Alkenylbenzenes in Foods: Aspects Impeding the Evaluation of Adverse Health Effects. Foods 2021, 10, 2139. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.; Crystal, K.; Coats, J. Three molecules found in rosemary or nutmeg essential oils repel ticks (Dermacentor variabilis) more effectively than DEET in a no-human assay. Pest Manag. Sci. 2021, 77, 1348–1354. [Google Scholar] [CrossRef]
- Carlson, M.; Thompson, R. Liquid chromatographic determination of safrole in sassafras-derived herbal products. J. AOAC Int. 1997, 80, 1023–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batool, S.; Khera, R.A.; Hanif, M.A.; Ayub, M.A. Bay Leaf. Med. Plants South Asia 2020, 63–74. [Google Scholar] [CrossRef]
- Yahyaa, M.; Berim, A.; Nawade, B.; Ibdah, M.; Dudareva, N.; Ibdah, M. Biosynthesis of methyleugenol and methylisoeugenol in Daucus carota leaves: Characterization of eugenol/isoeugenol synthase and O-Methyltransferase. Phytochemistry 2019, 159, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Carman, A.S.; Kuan, S.S.; Francis, O.J.; Ware, G.M.; Luedtke, A.E. Rapid Screening Method for Determining Myristicin in Fresh and Frozen Carrots by Gas Chromatography. Anal. Lett. 1985, 18, 1167–1175. [Google Scholar] [CrossRef]
- Rivera-Pérez, A.; López-Ruiz, R.; Romero-González, R.; Garrido Frenich, A. A new strategy based on gas chromatography–high resolution mass spectrometry (GC–HRMS-Q-Orbitrap) for the determination of alkenylbenzenes in pepper and its varieties. Food Chem. 2020, 321, 126727. [Google Scholar] [CrossRef] [PubMed]
- Dang, H.N.P.; Quirino, J.P. Analytical separation of carcinogenic and genotoxic alkenylbenzenes in foods and related products (2010–2020). Toxins 2021, 13, 387. [Google Scholar] [CrossRef] [PubMed]
- Terabe, S.; Otsuka, K.; Ichikawa, K.; Tsuchiya, A.; Ando, T. Electrokinetic Separations with Micellar Solutions and Open-Tubular Capillaries. Anal. Chem. 1984, 56, 111–113. [Google Scholar] [CrossRef]
- Yu, R.B.; Quirino, J.P. Ionic liquids in electrokinetic chromatography. J. Chromatogr. A 2021, 1637. [Google Scholar] [CrossRef] [PubMed]
- Huhn, C.; Pütz, M.; Pyell, U. Separation of very hydrophobic analytes by micellar electrokinetic chromatography. III. Characterization and optimization of the composition of the separation electrolyte using carbon number equivalents. Electrophoresis 2008, 29, 783–795. [Google Scholar] [CrossRef]
- Ranasinghe, M.; Quirino, J.P. Can we replace liquid chromatography with the greener capillary electrophoresis? Curr. Opin. Green Sustain. Chem. 2021, 31. [Google Scholar] [CrossRef]
- Laing, T.D.; Marenco, A.J.; Moore, D.M.; Moore, G.J.; Mah, D.C.W.; Lee, W.E. Capillary electrophoresis laser-induced fluorescence for screening combinatorial peptide libraries in assays of botulinum neurotoxin A. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2006, 843, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, M.; Karunaratne, H.; Quirino, J.P. Sweeping-micellar electrokinetic chromatography versus reversed phase liquid chromatography for the determination of coumarin in curry. J. Chromatogr. A 2021, 1657. [Google Scholar] [CrossRef]
- Simó, C.; Moreno-Arribas, M.V.; Cifuentes, A. Ion-trap versus time-of-flight mass spectrometry coupled to capillary electrophoresis to analyze biogenic amines in wine. J. Chromatogr. A 2008, 1195, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Quirino, J.P.; Terabe, S. Exceeding 5000-fold concentration of dilute analytes in micellar electrokinetic chromatography. Science 1998, 282, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Quirino, J.P.; Terabe, S. Sweeping of analyte zones in electrokinetic chromatography. Anal. Chem. 1999, 71, 1638–1644. [Google Scholar] [CrossRef]
- Quirino, J.P.; Terabe, S. On-line concentration of neutral analytes for micellar electrokinetic chromatography. 5. Field-enhanced sample injection with reverse migrating micelles. Anal. Chem. 1998, 70, 1893–1901. [Google Scholar] [CrossRef]
- Liu, Z.; Sam, P.; Sirimanne, S.R.; McClure, P.C.; Grainger, J.; Patterson Jr, D.G. Field-amplified sample stacking in micellar electrokinetic chromatography for on-column sample concentration of neutral molecules. J. Chromatogr. A 1994, 673, 125–132. [Google Scholar] [CrossRef]
- Gursale, A.; Dighe, V.; Parekh, G. Simultaneous Quantitative Determination of Cinnamaldehyde and Methyl Eugenol From Stem Bark of Cinnamomum zeylanicum Blume Using RP-HPLC. J. Chromatogr. Sci. 2010, 48, 59–62. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plants | Alkenylbenzenes | Food | ||||
|---|---|---|---|---|---|---|
 Eugenol Eugenol |  Methyleugenol Methyleugenol |  Myristicin Myristicin |  Safrole Safrole |  Estragole Estragole | ||
| cloves | 28.5 ± 0.4 mg/g [11] | Detected † [12] | Detected † [12] | various curries, jams, cooking rice | ||
| cinnamon | 0.19–1.65 g/kg [13] | Detected † [12] | various curries, churros, donut, various pastries | |||
| nutmeg | 0.32 mg/g [14] | 8 mg/g [15] | 280–420 mg/kg [16] | 109.8 mg/mL of extract [17] | various curries, alfredo pasta, soufflés, beef stew, various baked products | |
| sweet/holy basil, basil | 540 mg/kg [11] | Detected † [18] | Detected † [12] | 710 mg/kg [11] | various pesto sauces, Thai noodle and curry, caprese salad | |
| star anise | 98 mg/g [19] | Detected † [12] | 66 mg/g [19] | Thai noodle, Chinese soups and stews | ||
| dill | 28.1–76.3 mg/g [20] | various curries and soups, salad dressing, sandwich fillings | ||||
| celery | Detected † [21] | chicken noodle soup, various stir fry dishes, various vegetarian dishes | ||||
| ginger | Detected † [22] | 14.0 ± 1.6 µg/g [23] | 500 ± 36 mg/kg [24] | various teas, sushi, congee, various soups, various curries | ||
| tarragon | Detected † [25] | 0.5–28.9% v/v [26] | 17–75% v/v [26] | pasta, mojito, Béarnaise sauce | ||
| rosemary | Detected † [27] | various soups, salads, stews, and pasta sauces | ||||
| thyme | 0.021 mg/g [14] | detected† [28] | seasoned roasted vegetables, various pasta sauces and soups, pizza toppings | |||
| bay leaves | 110–120 mg/g [29] | 90–120 mg/g [29] | beef stew, pate, various curries and soups | |||
| carrot | Detected † [30] | 1.1–16.6 µg/g [31] | various soups, salads, noodle dishes, and curries, cakes, salads, coleslaw | |||
| pepper | 11–120 mg/kg [32] | 0.5–20.1 mg/kg [32] | 0.2–6.1 mg/kg [32] | 955 ± 80 mg/kg [24] | 2.2–45.7 mg/kg [32] | various pasta and curry dishes, and sauces |
| fennel | Detected † [12] | 2.0–3.0% v/v [26] | various pasta dishes, various salads and slaws, sausages | |||
| Column. | Specifications (Particle Size) | Flow Rate (mL/min) | Injection Volume (µL) |
|---|---|---|---|
| Thermo Fischer Hypersil GOLD C18 | 50 mm × 2.1 mm i.d. (3 µm) | 0.2 | 5 |
| Acclaim C18 | 100 mm × 2.1 mm i.d. (2.2 µm) | 0.5 | 10 |
| Phenomenex GEMINI C18 | 150 mm × 4.6 mm i.d. (5 µm) | 1.0 | 20 |
| Method | Eugenol | Methyleugenol | Myristicin | Safrole | Estragole | |
|---|---|---|---|---|---|---|
| HPLC | concentration range (µg/mL) | 0.18–12 | 0.07–4.3 | 0.79–50 | 0.36–23 | 0.73–46 |
| slope of the line | 251.27 | 1005.70 | 84.26 | 261.06 | 108.30 | |
| intercept of the line | −0.0169 | −0.0171 | −0.0162 | −0.0310 | −0.0362 | |
| correlation coefficient (R2) | 0.9970 | 0.9967 | 0.9966 | 0.9968 | 0.9965 | |
| LOD (µg/mL) | 0.09 | 0.03 | 0.39 | 0.18 | 0.36 | |
| LOQ (µg/mL) | 0.18 | 0.07 | 0.79 | 0.36 | 0.73 | |
| stacking | concentration range (µg/mL) | 1.6–50 | 1.5–24 | 3.7–29 | 7.1–21 | 6.0–37 |
| MEKC | slope of the line | 8590.2 | 6087.6 | 595.87 | 3020.2 | 1537.7 |
| intercept of the line | −13.801 | +3.279 | +0.374 | −22.81 | −10.36 | |
| correlation coefficient (R2) | 0.9972 | 0.9947 | 0.9964 | 0.9724 | 0.9975 | |
| LOD (µg/mL) | 0.41 | 0.38 | 1.8 | 2.7 | 4.3 | |
| LOQ (µg/mL) | 1.6 | 1.5 | 3.7 | 7.1 | 6.0 |
| Method | Analyte | Concentration Levels (µg/mL) | % RSD (Retention Time 1/Migration Time 2) | % RSD (Peak Area/Corrected Peak Area 5) | |||
|---|---|---|---|---|---|---|---|
| Intra-Day (n = 10) 3 | Inter-Day (n = 15) 4 | Intra-Day (n = 10) 3 | 1nter-Day (n = 15) 4 | ||||
| HPLC | LOQ | Eugenol | 0.2 | 0.2 | 0.4 | 4.0 | 3.3 |
| methyleugenol | 0.1 | 0.1 | 0.4 | 3.8 | 3.9 | ||
| myristicin | 0.8 | 0.2 | 0.5 | 0.0 | 0.0 | ||
| safrole | 0.4 | 0.1 | 0.4 | 0.0 | 3.3 | ||
| estragole | 0.7 | 0.1 | 0.4 | 3.2 | 1.7 | ||
| 8 × LOQ | eugenol | 1.4 | 0.4 | 0.3 | 0.7 | 1.1 | |
| methyleugenol | 5.4 | 0.4 | 0.3 | 0.5 | 0.9 | ||
| myristicin | 6.3 | 0.3 | 0.3 | 0.0 | 0.1 | ||
| safrole | 2.9 | 0.3 | 0.3 | 0.0 | 3.5 | ||
| estragole | 5.8 | 0.3 | 0.3 | 0.5 | 7.0 | ||
| stacking-MEKC | LOQ | eugenol | 1.6 | 2.9 | 4.4 | 0.6 | 0.8 |
| methyleugenol | 1.5 | 2.9 | 5.2 | 0.4 | 0.3 | ||
| myristicin | 3.7 | 2.4 | 2.0 | 1.1 | 3.0 | ||
| safrole | 7.1 | 1.5 | 1.4 | 3.4 | 3.6 | ||
| estragole | 6.0 | 1.8 | 1.6 | 5.0 | 6.3 | ||
| 8 × LOQ | eugenol | 13.0 | 1.3 | 1.6 | 4.8 | 1.1 | |
| methyleugenol | 6.1 | 3.2 | 1.7 | 0.7 | 1.9 | ||
| myristicin | 15.0 | 1.9 | 3.8 | 0.4 | 2.1 | ||
| safrole | 11.0 | 1.4 | 1.9 | 4.4 | 6.1 | ||
| estragole | 18.0 | 2.4 | 2.5 | 4.8 | 7.8 | ||
| Analyte | Sample Concentration (µg/mL) | Added Concentration (µg/mL) | Nominal Concentration (µg/mL) | Found Concentration (µg/mL) | % Recovery |
|---|---|---|---|---|---|
| eugenol | 1.08 | 0.54 | 1.62 | 1.32 ± 0.00 | 81.5 ± 0.0 |
| 1.08 | 2.16 | 1.79 ± 0.00 | 82.9 ± 0.0 | ||
| 2.16 | 3.24 | 2.75 ± 0.01 | 84.9 ± 0.0 | ||
| 4.32 | 5.40 | 4.92 ± 0.00 | 91.1 ± 0.0 | ||
| 8.64 | 9.72 | 8.02 ± 0.01 | 82.5 ± 0.0 | ||
| 17.28 | 18.36 | 16.19 ± 0.04 | 88.2 ± 0.0 | ||
| 34.56 | 35.64 | 24.73 ± 0.06 | 69.4 ± 0.1 | ||
| methyleugenol | 0 | 0.21 | 0.21 | 0.12 ± 0.00 | 57.1 ± 0.0 |
| 0.42 | 0.42 | 0.25 ± 0.00 | 59.5 ± 0.0 | ||
| 0.84 | 0.84 | 0.56 ± 0.01 | 66.7 ± 0.0 | ||
| 1.68 | 1.68 | 1.25 ± 0.00 | 74.4 ± 0.0 | ||
| 3.36 | 3.36 | 2.46 ± 0.01 | 73.2 ± 0.0 | ||
| 6.72 | 6.72 | 4.18 ± 0.04 | 62.2 ± 0.0 | ||
| 13.44 | 13.44 | 7.72 ± 0.07 | 57.4 ± 0.1 | ||
| myristicin | 0 | 2.37 | 2.37 | 1.93 ± 0.00 | 81.4 ± 0.0 |
| 4.74 | 4.74 | 2.98 ± 0.00 | 62.9 ± 0.0 | ||
| 9.48 | 9.48 | 6.38 ± 0.00 | 67.3 ± 0.0 | ||
| 18.96 | 18.96 | 12.72 ± 0.03 | 67.1 ± 0.0 | ||
| 37.92 | 37.92 | 22.22 ± 0.01 | 58.6 ± 0.0 | ||
| 75.84 | 75.84 | 47.66 ± 0.03 | 62.8 ± 0.0 | ||
| 151.68 | 151.68 | 95.85 ± 0.05 | 63.2 ± 0.1 | ||
| safrole | 0 | 1.08 | 1.08 | 0.84 ± 0.00 | 77.8 ± 0.0 |
| 2.16 | 2.16 | 1.38 ± 0.01 | 63.9 ± 0.0 | ||
| 4.32 | 4.32 | 2.76 ± 0.00 | 63.9 ± 0.0 | ||
| 8.64 | 8.64 | 5.63 ± 0.03 | 65.2 ± 0.0 | ||
| 17.28 | 17.28 | 10.32 ± 0.03 | 59.7 ± 0.0 | ||
| 34.56 | 34.56 | 23.37 ± 0.04 | 67.6 ± 0.0 | ||
| 69.12 | 69.12 | 45.55 ± 0.07 | 65.9 ± 0.1 | ||
| estragole | 0.59 | 2.19 | 2.78 | 2.06 ± 0.00 | 74.1 ± 0.0 |
| 4.38 | 4.97 | 3.83 ± 0.01 | 77.1 ± 0.0 | ||
| 8.76 | 9.35 | 7.07 ± 0.01 | 75.6 ± 0.0 | ||
| 17.52 | 18.11 | 14.00 ± 0.02 | 77.3 ± 0.0 | ||
| 35.04 | 35.63 | 27.68 ± 0.02 | 77.7 ± 0.0 | ||
| 70.08 | 70.67 | 56.14 ± 0.05 | 79.4 ± 0.1 | ||
| 140.16 | 140.75 | 106.17 ± 0.06 | 75.4 ± 0.1 |
| Sample | Amount Found (mg/g) | ||||
|---|---|---|---|---|---|
| Eugenol | Methyleugenol | Myristicin | Safrole | Estragole | |
| basil leaves | 0.27 ± 0.01 | 0.07 ± 0.01 | ND | ND | 0.72 ± 0.11 |
| Galiko basil sauce | 0.12 ± 0.01 | 0.004 ± 0.00 | ND | ND | 0.07 ± 0.01 |
| cloves | 8.71 ± 0.09 | ND | ND | ND | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dang, H.N.P.; Quirino, J.P. High Performance Liquid Chromatography versus Stacking-Micellar Electrokinetic Chromatography for the Determination of Potentially Toxic Alkenylbenzenes in Food Flavouring Ingredients. Molecules 2022, 27, 13. https://doi.org/10.3390/molecules27010013
Dang HNP, Quirino JP. High Performance Liquid Chromatography versus Stacking-Micellar Electrokinetic Chromatography for the Determination of Potentially Toxic Alkenylbenzenes in Food Flavouring Ingredients. Molecules. 2022; 27(1):13. https://doi.org/10.3390/molecules27010013
Chicago/Turabian StyleDang, Huynh N. P., and Joselito P. Quirino. 2022. "High Performance Liquid Chromatography versus Stacking-Micellar Electrokinetic Chromatography for the Determination of Potentially Toxic Alkenylbenzenes in Food Flavouring Ingredients" Molecules 27, no. 1: 13. https://doi.org/10.3390/molecules27010013