
RNA-Targeting Splicing Modifiers: Drug Development and Screening Assays




Abstract
:1. Introduction
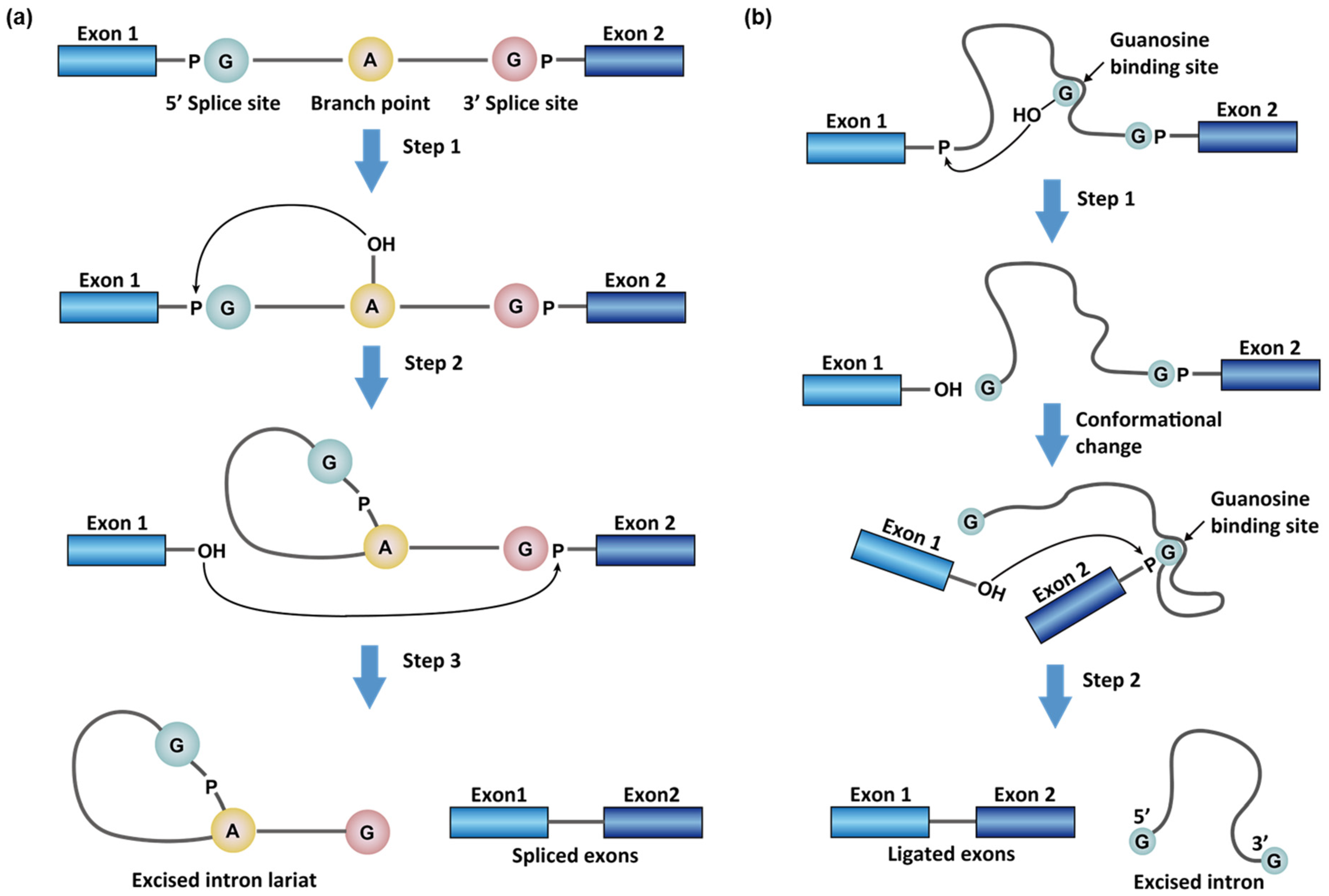
1.1. Chemistry in RNA Splicing Reactions
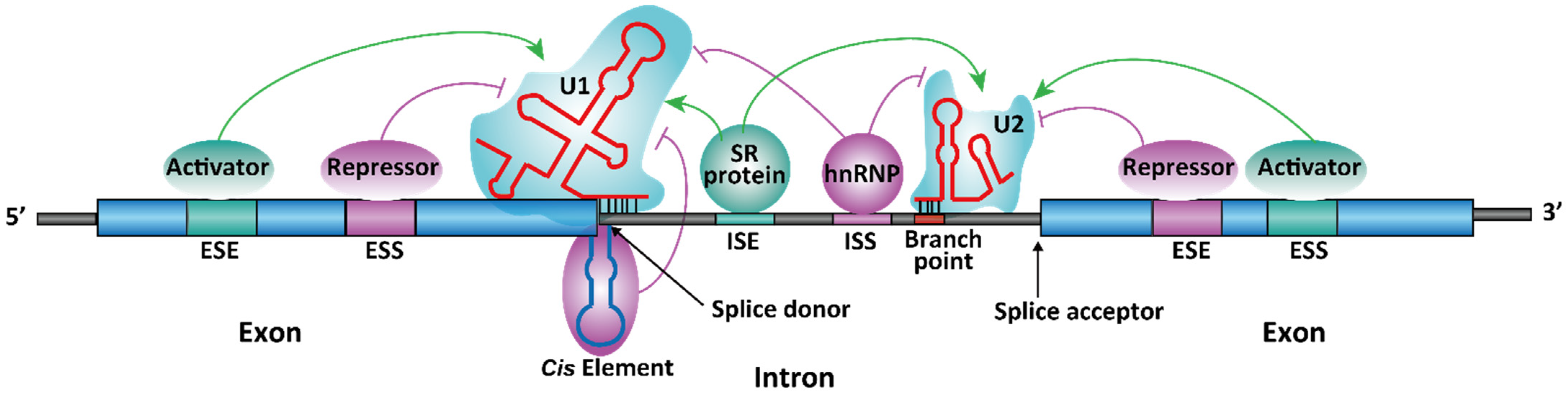
1.2. Spliceosome-Dependent Alternative Splicing
1.3. Spliceosome-Independent Splicing
2. Known RNA-Targeting Splicing Modifiers for the Treatment of Human Diseases
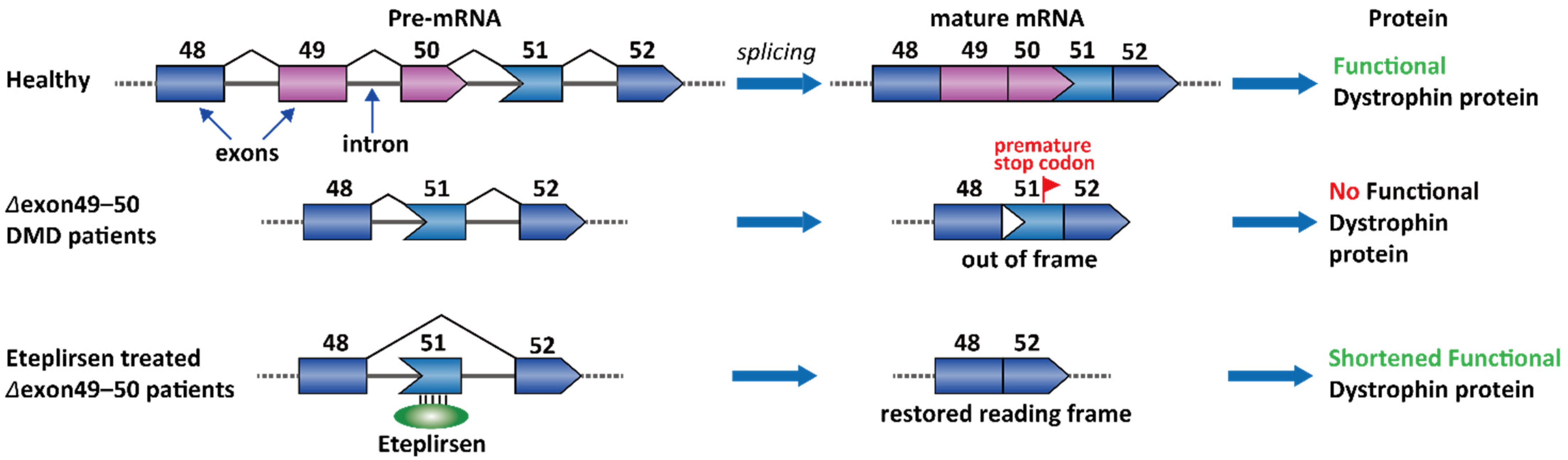
2.1. Duchenne Muscular Dystrophy (DMD)
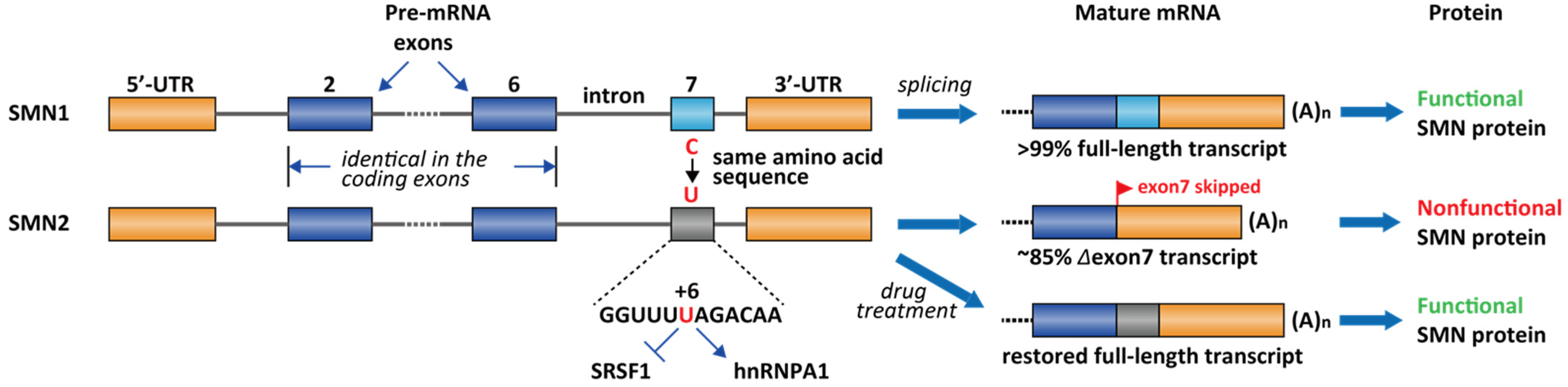
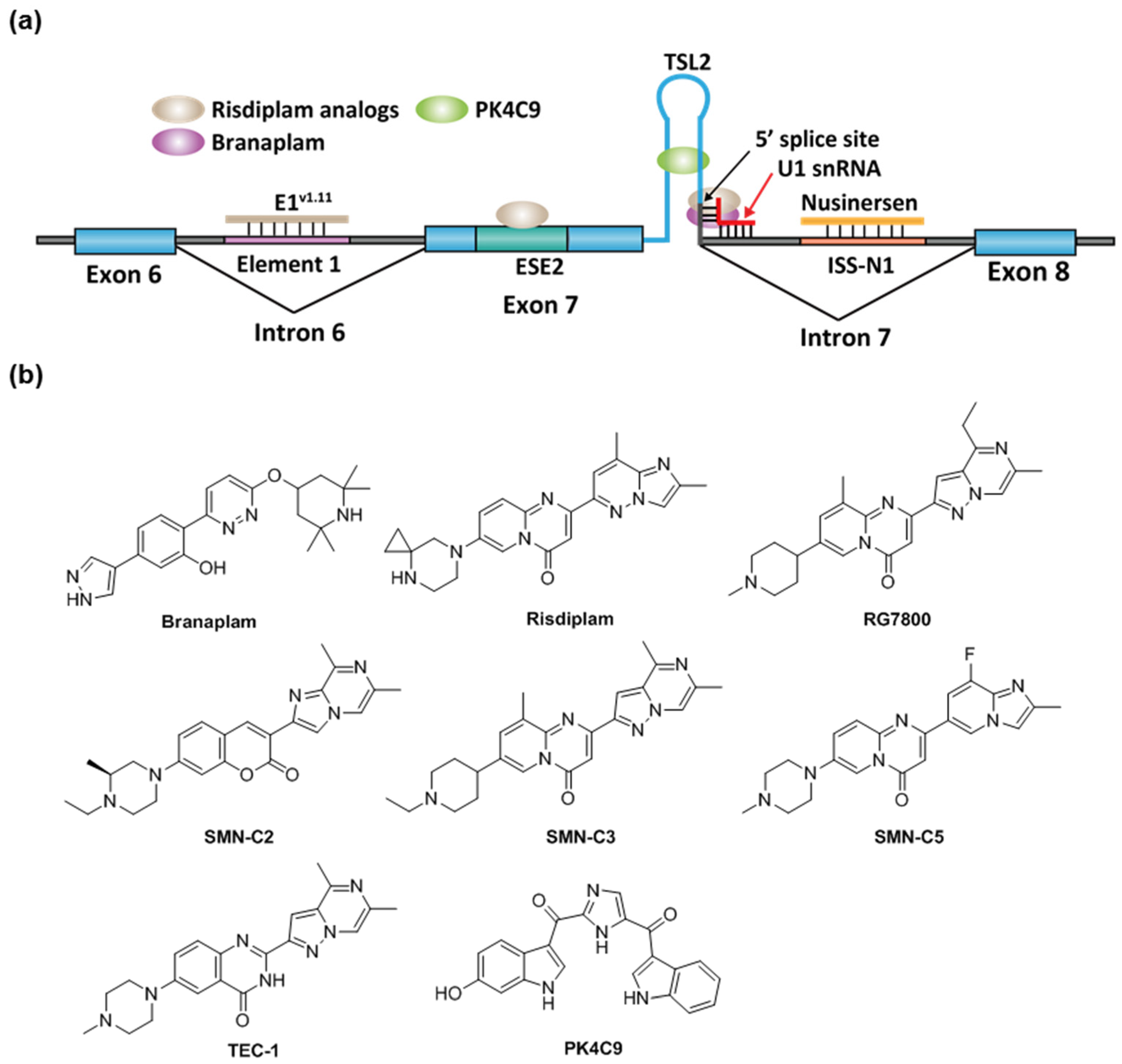
2.2. Spinal Muscular Atrophy
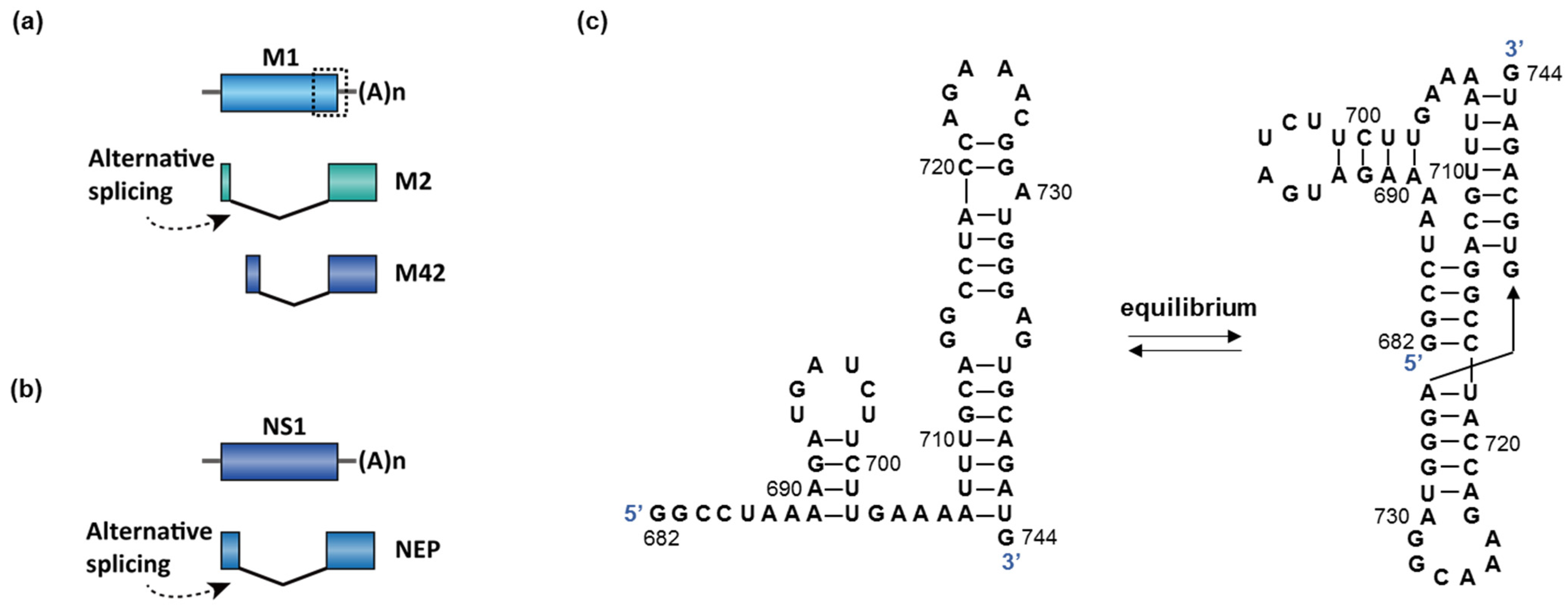
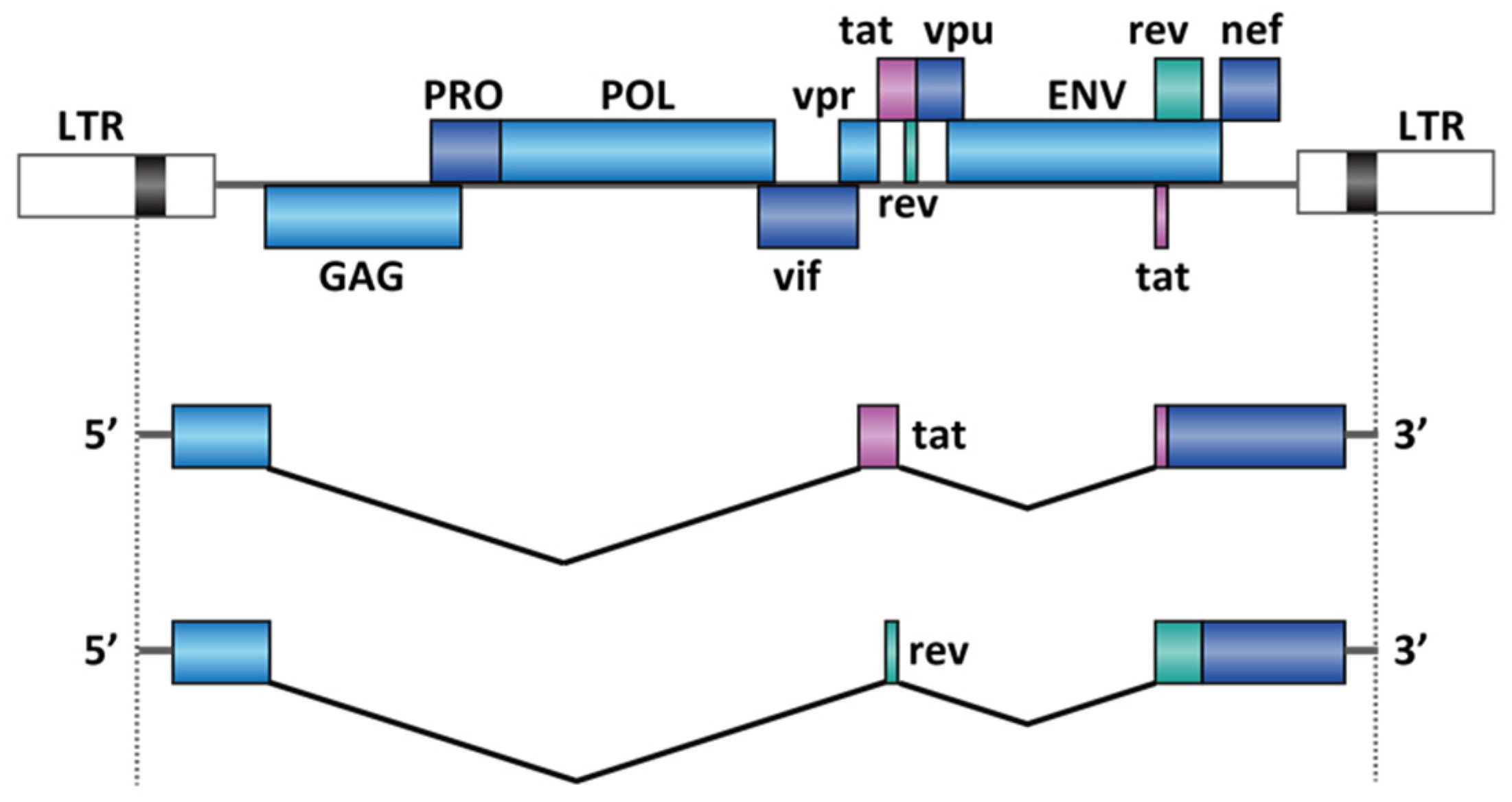
2.3. Virus Infection
2.4. Yeast Infection
2.5. Frontotemporal Dementia and Parkinsonism Linked to Chromosome 17 (FTDP-17)
2.6. Familial Dysautonomia
3. Screening Methods for RNA Splicing Modifiers
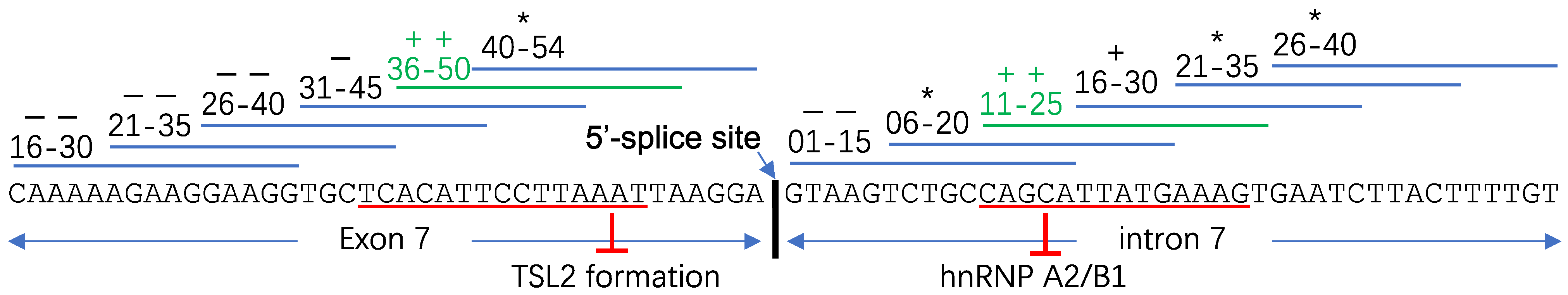
3.1. “ASO Walking” Uncovers Cis-Acting Factors
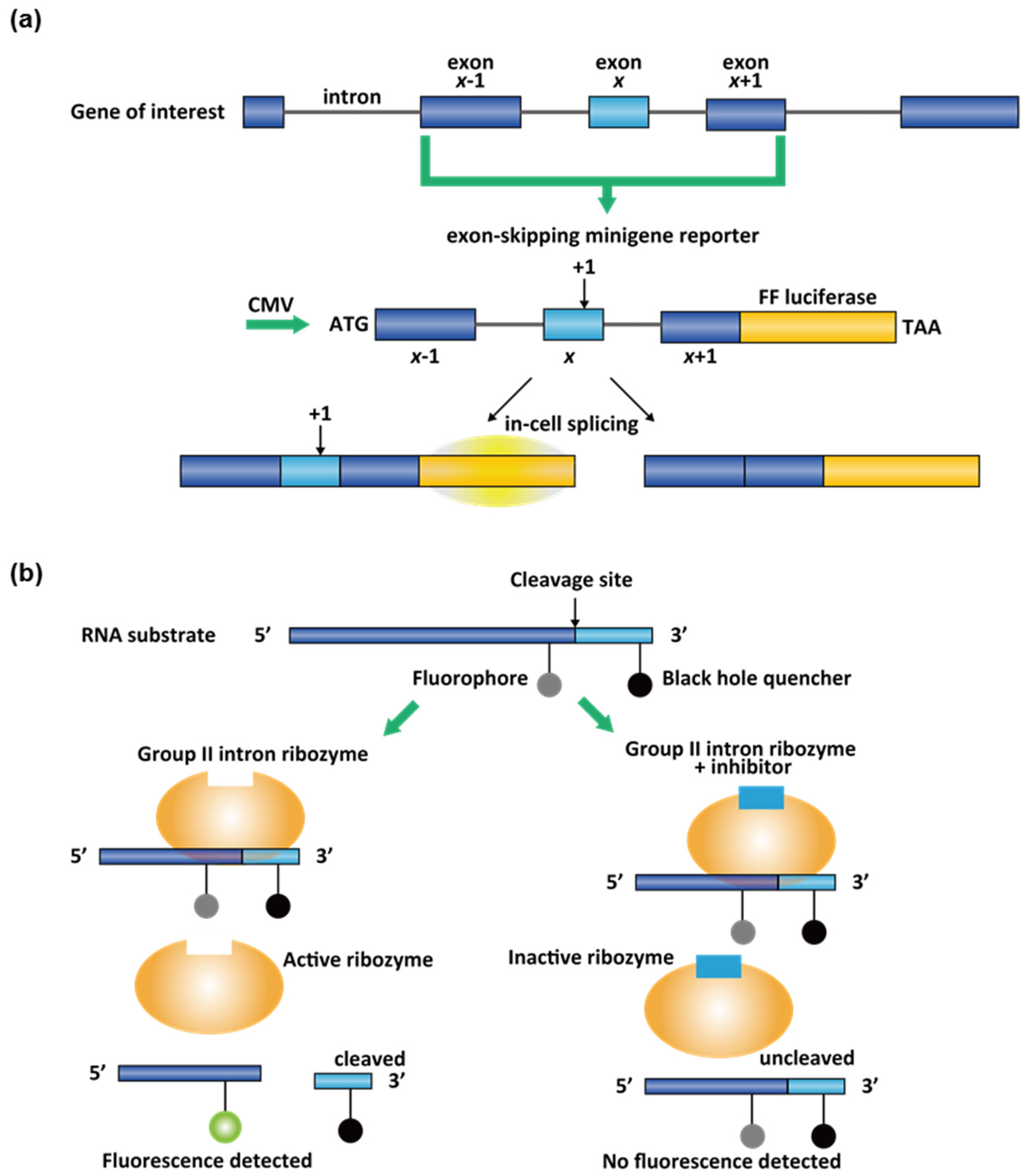
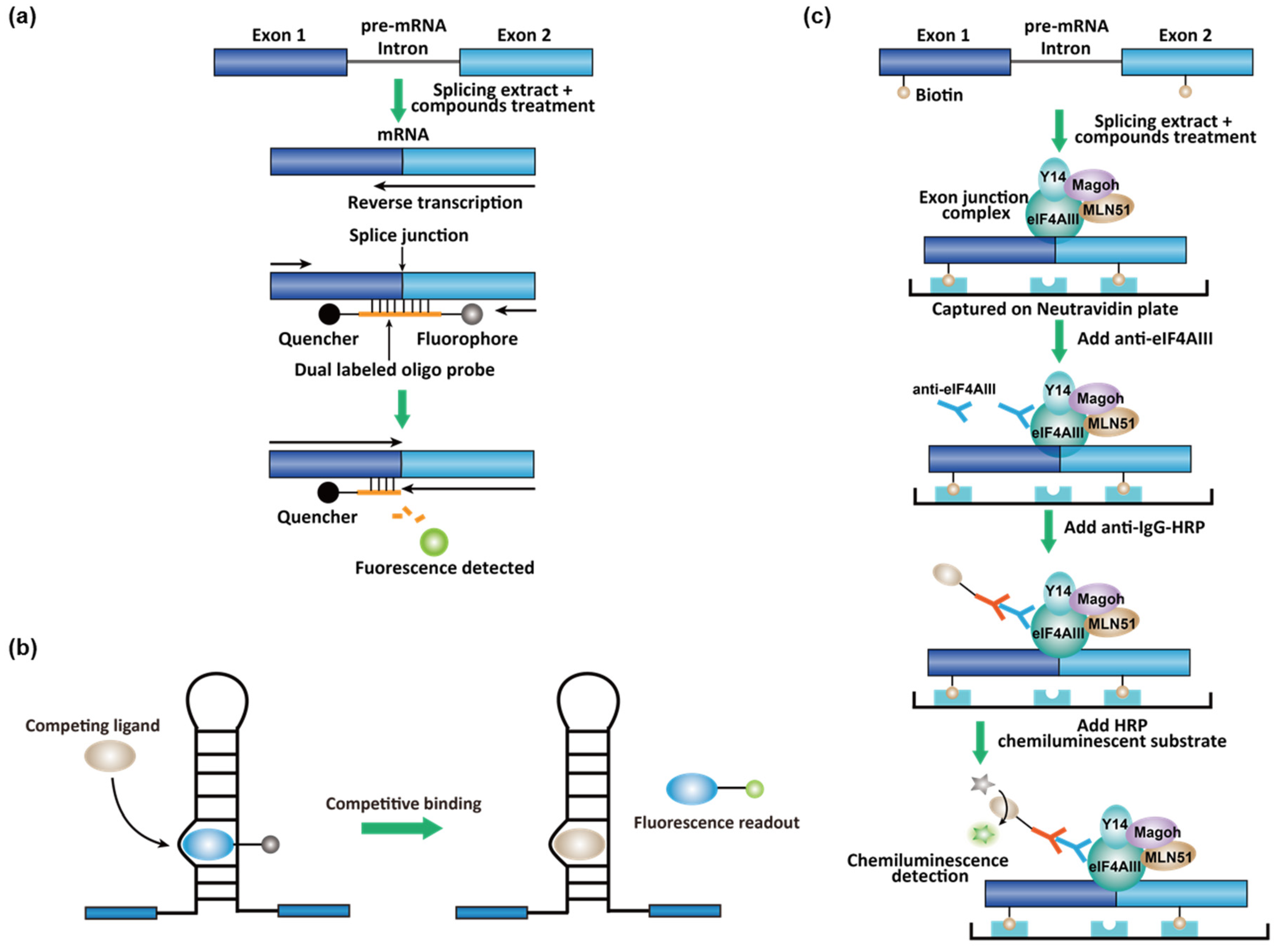
3.2. High-Throughput Screening (HTS) Assays for Small-Molecule Splicing Modifiers
4. The Chemical Space of RNA Splicing Modifiers
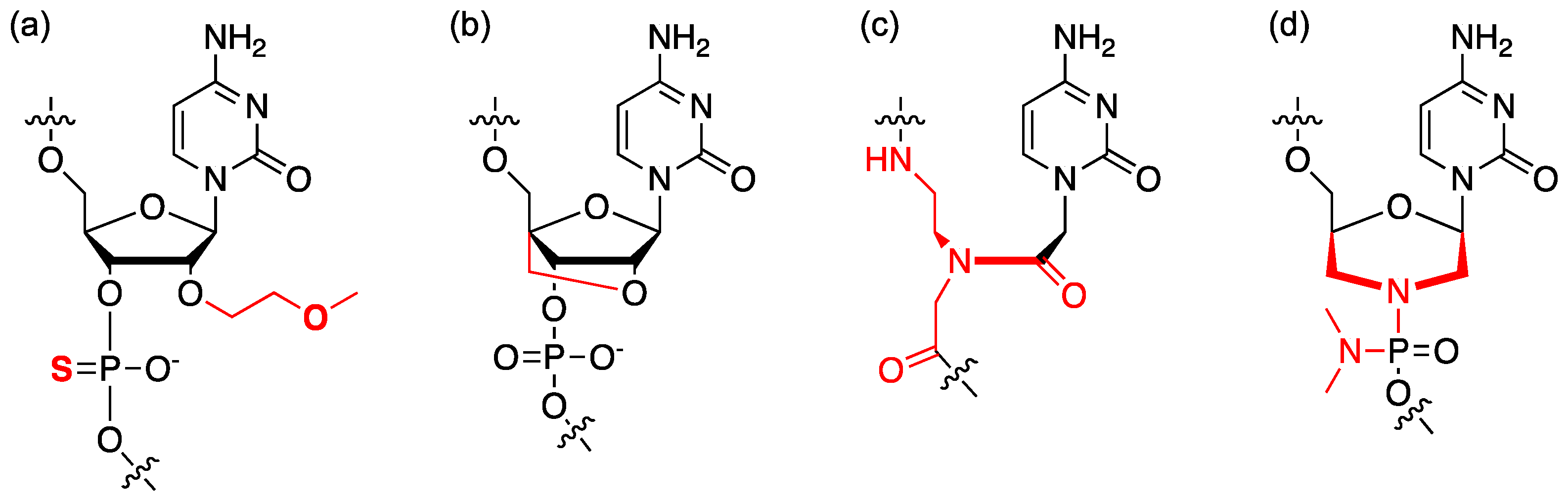
4.1. Chemical Modification of ASO
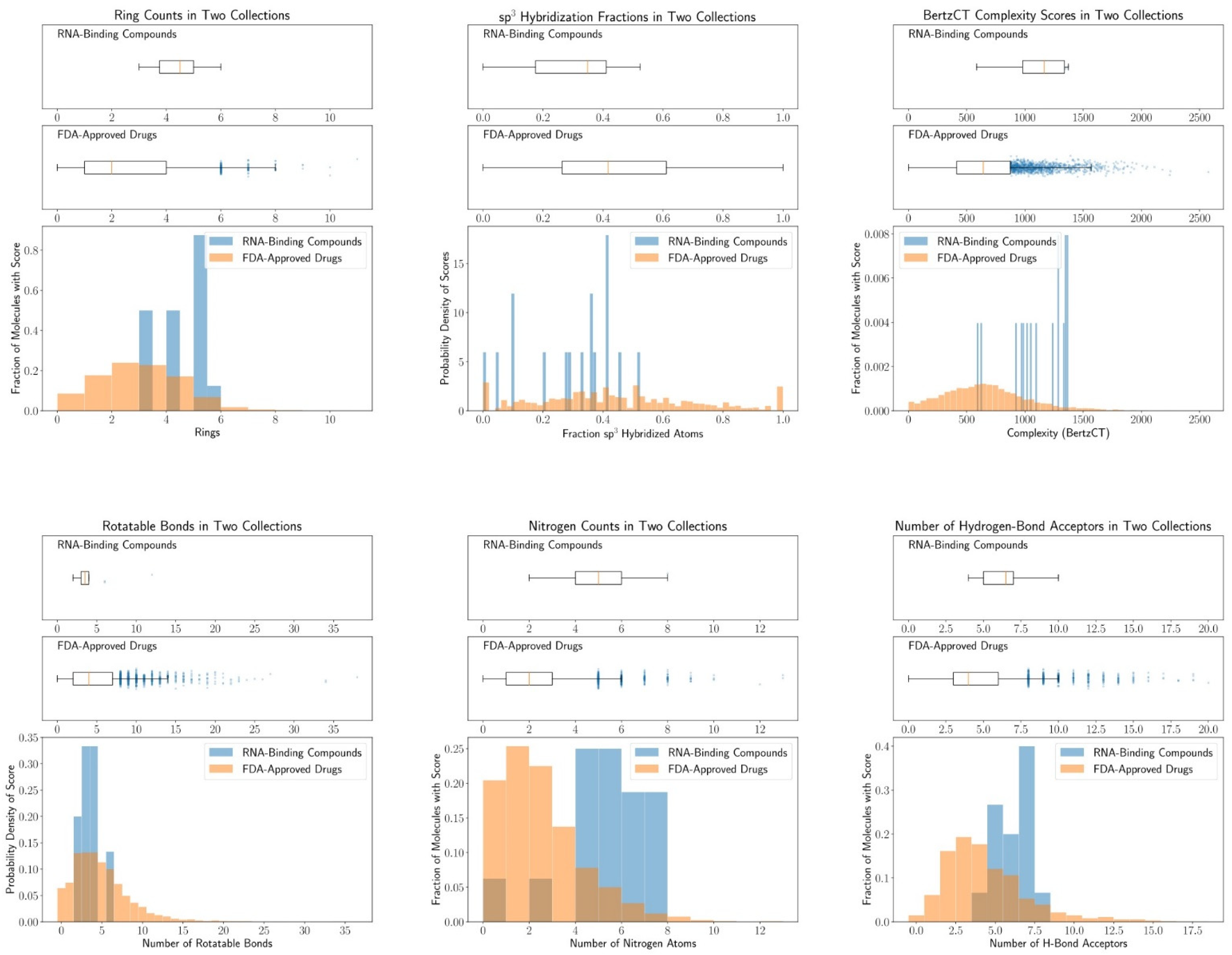
4.2. Chemical Space of RNA-Targeting Small-Molecule Splicing Modifiers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Cheminformatic Analysis
References
- Vosseberg, J.; Snel, B. Domestication of self-splicing introns during eukaryogenesis: The rise of the complex spliceosomal machinery. Biol. Direct 2017, 12, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Lambowitz, A.M.; Zimmerly, S. Mobile group II introns. Annu. Rev. Genet. 2004, 38, 1–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Jaklevic, M.C. Oral Drug Approved for Spinal Muscular Atrophy. J. Am. Med. Assoc. 2020, 324, 1026. [Google Scholar] [CrossRef]
- Clemens, P.R.; Rao, V.K.; Connolly, A.M.; Harper, A.D.; Mah, J.K.; Smith, E.C.; McDonald, C.M.; Zaidman, C.M.; Morgenroth, L.P.; Osaki, H.; et al. Safety, tolerability, and efficacy of viltolarsen in boys with duchenne muscular dystrophy amenable to exon 53 skipping: A phase 2 randomized clinical trial. JAMA Neurol. 2020, 77, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, S.; Kota, J.; et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.A. Golodirsen: First Approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Beigel, J.H.; Voell, J.; Muñoz, P.; Kumar, P.; Brooks, K.M.; Zhang, J.; Iversen, P.; Heald, A.; Wong, M.; Davey, R.T. Safety, tolerability, and pharmacokinetics of radavirsen (AVI-7100), an antisense oligonucleotide targeting influenza a M1/M2 translation. Br. J. Clin. Pharmacol. 2018, 84, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.L.; Zhang, P.; Abe, M.; Aikawa, H.; Zhang, L.; Frank, A.J.; Zembryski, T.; Hubbs, C.; Park, H.; Withka, J.; et al. Design, Optimization, and Study of Small Molecules That Target Tau Pre-mRNA and Affect Splicing. J. Am. Chem. Soc. 2020, 142, 8706–8727. [Google Scholar] [CrossRef]
- Fedorova, O.; Jagdmann, G.E.; Adams, R.L.; Yuan, L.; Van Zandt, M.C.; Pyle, A.M. Small molecules that target group II introns are potent antifungal agents. Nat. Chem. Biol. 2018, 14, 1073–1078. [Google Scholar] [CrossRef]
- Sinha, R.; Kim, Y.J.; Nomakuchi, T.; Sahashi, K.; Hua, Y.; Rigo, F.; Bennett, C.F.; Krainer, A.R. Antisense oligonucleotides correct the familial dysautonomia splicing defect in IKBKAPtransgenic mice. Nucleic Acids Res. 2018, 46, 4833–4844. [Google Scholar] [CrossRef]
- Hausner, G.; Hafez, M.; Edgell, D.R. Bacterial group i introns: Mobile RNA catalysts. Mob. DNA 2014, 5, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, M.E.; Charenton, C.; Nagai, K. RNA Splicing by the Spliceosome. Annu. Rev. Biochem. 2020, 89, 359–388. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Mechanistic insights into precursor messenger RNA splicing by the spliceosome. Nat. Rev. Mol. Cell Biol. 2017, 18, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Talhouarne, G.J.S.; Gall, J.G. Lariat intronic RNAs in the cytoplasm of vertebrate cells. Proc. Natl. Acad. Sci. USA 2018, 115, E7970–E7977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef]
- Hopper, A.K.; Nostramo, R.T.; Bao, A.; Peltier, L.M. tRNA: Splicing and Intron Turnover. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef]
- Chen, M.; Manley, J.L. Mechanisms of alternative splicing regulation: Insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol. 2009, 10, 741–754. [Google Scholar] [CrossRef]
- Lee, Y.; Rio, D.C. Mechanisms and regulation of alternative Pre-mRNA splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [Green Version]
- Black, D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef] [Green Version]
- Romero-Barrios, N.; Legascue, M.F.; Benhamed, M.; Ariel, F.; Crespi, M. Survey and summary Splicing regulation by long noncoding RNAs. Nucleic Acids Res. 2018, 46, 2169–2184. [Google Scholar] [CrossRef] [Green Version]
- Geuens, T.; Bouhy, D.; Timmerman, V. The hnRNP family: Insights into their role in health and disease. Hum. Genet. 2016, 135, 851–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Fu, X.D. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma 2013, 122, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.G.; Singh, R.K.; Mohammad, F.; Bebee, T.W.; Chandler, D.S. The splicing factor FUBP1 is required for the efficient splicing of oncogene MDM2 pre-mRNA. J. Biol. Chem. 2014, 289, 17350–17364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Wang, Z.; Zhou, X.; Cheng, Y.; Xie, Z.; Manley, J.L.; Feng, Y. Far upstream element-binding protein 1 and RNA secondary structure both mediate second-step splicing repression. Proc. Natl. Acad. Sci. USA 2013, 110, E2687–E2695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miro, J.; Laaref, A.M.; Rofidal, V.; Lagrafeuille, R.; Hem, S.; Thorel, D.; Mechin, D.; Mamchaoui, K.; Mouly, V.; Claustres, M.; et al. FUBP1: A new protagonist in splicing regulation of the DMD gene. Nucleic Acids Res. 2015, 43, 2378–2389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Schultz, P.G.; Johnson, K.A. Mechanistic studies of a small-molecule modulator of SMN2 splicing. Proc. Natl. Acad. Sci. USA 2018, 115, E4604–E4612. [Google Scholar] [CrossRef] [Green Version]
- Solnick, D.; Lee, S.I. Amount of RNA secondary structure required to induce an alternative splice. Mol. Cell. Biol. 1987, 7, 3194–3198. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.L.; Stahley, M.R.; Kosek, A.B.; Wang, J.; Strobel, S.A. Crystal structure of a self-splicing group I intron with both exons. Nature 2004, 430, 45–50. [Google Scholar] [CrossRef]
- Stahley, M.R.; Strobel, S.A. Structural biology: Structural evidence for a two-metal-ion mechanism of group I intron splicing. Science 2005, 309, 1587–1590. [Google Scholar] [CrossRef] [Green Version]
- Zimmerly, S.; Hausner, G.; Wu, X.C. Phylogenetic relationships among group II intron ORFs. Nucleic Acids Res. 2001, 29, 1238–1250. [Google Scholar] [CrossRef] [Green Version]
- Toor, N.; Keating, K.S.; Taylor, S.D.; Pyle, A.M. Crystal structure of a self-spliced group II intron. Science 2008, 320, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Fujishima, K.; Kanai, A. tRNA gene diversity in the three domains of life. Front. Genet. 2014, 5, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abelson, J.; Trotta, C.R.; Li, H. tRNA splicing. J. Biol. Chem. 1998, 273, 12685–12688. [Google Scholar] [CrossRef] [Green Version]
- Christopher, D.A.; Hallick, R.B. Euglena gracilis chloroplast ribosomal protein operon: A new chloroplast gene for ribosomal protein L5 and description of a novel organelle intron category designated group III. Nucleic Acids Res. 1989, 17, 7591–7608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montandon, P.E.; Stutz, E. Nucleotide sequence of a Euglena gracilis chloroplast genome region coding for the elongation factor Tu; evidence for a spliced mRNA. Nucleic Acids Res. 1983, 11, 5877–5892. [Google Scholar] [CrossRef] [Green Version]
- Paushkin, S.V.; Patel, M.; Furia, B.S.; Peltz, S.W.; Trotta, C.R. Identification of a human endonuclease complex reveals a link between tRNA splicing and pre-mRNA 3′ end formation. Cell 2004, 117, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Haugen, P.; Simon, D.M.; Bhattacharya, D. The natural history of group I introns. Trends Genet. 2005, 21, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Belfort, M. Two for the price of one: A bifunctional intron-encoded DNA endonuclease-RNA maturase. Genes Dev. 2003, 17, 2860–2863. [Google Scholar] [CrossRef] [Green Version]
- Lambowitz, A.M.; Zimmerly, S. Group II introns: Mobile ribozymes that invade DNA. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.; Hess, W.R. Complete 5′ and 3′ end maturation of group ii intron-containing tRNA precursors. RNA 2001, 7, 285–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tocchini-Valentini, G.D.; Fruscoloni, P.; Tocchini-Valentini, G.P. Evolution of introns in the archaeal world. Proc. Natl. Acad. Sci. USA 2011, 108, 4782–4787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iben, J.R.; Maraia, R.J. tRNA gene copy number variation in humans. Gene 2014, 536, 376–384. [Google Scholar] [CrossRef] [Green Version]
- Popow, J.; Schleiffer, A.; Martinez, J. Diversity and roles of (t)RNA ligases. Cell. Mol. Life Sci. 2012, 69, 2657–2670. [Google Scholar] [CrossRef] [Green Version]
- Piovesan, A.; Caracausi, M.; Antonaros, F.; Pelleri, M.C.; Vitale, L. GeneBase 1.1: A tool to summarize data from NCBI gene datasets and its application to an update of human gene statistics. Database 2016, 2016. [Google Scholar] [CrossRef]
- Tourasse, N.J.; Stabell, F.B.; Kolstø, A.B. Structural and functional evolution of group II intron ribozymes: Insights from unusual elements carrying a 3′ extension. N. Biotechnol. 2010, 27, 204–211. [Google Scholar] [CrossRef]
- Yoshihisa, T. Handling tRNA introns, archaeal way and eukaryotic way. Front. Genet. 2014, 5, 213. [Google Scholar] [CrossRef] [Green Version]
- Pratt, A.J.; MacRae, I.J. The RNA-induced silencing complex: A versatile gene-silencing machine. J. Biol. Chem. 2009, 284, 17897–17901. [Google Scholar] [CrossRef] [Green Version]
- Crooke, S.T. Molecular Mechanisms of Antisense Oligonucleotides. Nucleic Acid Ther. 2017, 27, 70–77. [Google Scholar] [CrossRef]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef]
- Summerton, J. Morpholino antisense oligomers: The case for an RNase H-independent structural type. Biochim. Biophys. Acta-Gene Struct. Expr. 1999, 1489, 141–158. [Google Scholar] [CrossRef]
- Faria, M.; Spiller, D.G.; Dubertret, C.; Nelson, J.S.; White, M.R.H.; Scherman, D.; Hélène, C.; Giovannangeli, C. Phosphormidate oligonucleotides as potent antisense molecules in cells and in vivo. Nat. Biotechnol. 2001, 19, 40–44. [Google Scholar] [CrossRef]
- Devi, G.R.; Beer, T.M.; Corless, C.L.; Arora, V.; Weller, D.L.; Iversen, P.L. In vivo bioavailability and pharmacokinetics of a c-MYC antisense phosphorodiamidate morpholino oligomer, AVI-4126, in solid tumors. Clin. Cancer Res. 2005, 11, 3930–3938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Warner, K.D.; Hajdin, C.E.; Weeks, K.M. Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug Discov. 2018, 17, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Morgan, B.S.; Forte, J.E.; Culver, R.N.; Zhang, Y.; Hargrove, A.E. Discovery of Key Physicochemical, Structural, and Spatial Properties of RNA-Targeted Bioactive Ligands. Angew. Chem. Int. Ed. 2017, 56, 13498–13502. [Google Scholar] [CrossRef]
- Hargrove, A.E. Small molecule–RNA targeting: Starting with the fundamentals. Chem. Commun. 2020, 56, 14744–14756. [Google Scholar] [CrossRef]
- Disney, M.D. Targeting RNA with Small Molecules To Capture Opportunities at the Intersection of Chemistry, Biology, and Medicine. J. Am. Chem. Soc. 2019, 141, 6776–6790. [Google Scholar] [CrossRef]
- Terns, M.P. CRISPR-Based Technologies: Impact of RNA-Targeting Systems. Mol. Cell 2018, 72, 404–412. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Wang, L.; Zou, X.; Duan, S.; Li, Z.; Deng, Z.; Luo, J.; Lee, S.Y.; Chen, S. Advances in CRISPR-Cas systems for RNA targeting, tracking and editing. Biotechnol. Adv. 2019, 37, 708–729. [Google Scholar] [CrossRef]
- Osman, E.Y.; Washington, C.W., III; Kaifer, K.A.; Mazzasette, C.; Patitucci, T.N.; Florea, K.M.; Simon, M.E.; Ko, C.-P.; Ebert, A.D.; Lorson, C.L. Optimization of Morpholino Antisense Oligonucleotides Targeting the Intronic Repressor Element1 in Spinal Muscular Atrophy. Mol. Ther. 2016, 24, 1592–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartegni, L.; Krainer, A.R. Correction of disease-associated exon skipping by synthetic exon-specific activators. Nat. Struct. Biol. 2003, 10, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Ratni, H.; Ebeling, M.; Baird, J.; Bendels, S.; Bylund, J.; Chen, K.S.; Denk, N.; Feng, Z.; Green, L.; Guerard, M.; et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 ( SMN2 ) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 61, 6501–6517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, A.K.; Hurley, B.; Kerrigan, R.; Shu, L.; Chin, D.N.; Shen, Y.; O’Brien, G.; Sung, M.J.; Hou, Y.; Axford, J.; et al. Discovery of Small Molecule Splicing Modulators of Survival Motor Neuron-2 (SMN2) for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 61, 11021–11036. [Google Scholar] [CrossRef]
- Garcia-Lopez, A.; Tessaro, F.; Jonker, H.R.A.; Wacker, A.; Richter, C.; Comte, A.; Berntenis, N.; Schmucki, R.; Hatje, K.; Petermann, O.; et al. Targeting RNA structure in SMN2 reverses spinal muscular atrophy molecular phenotypes. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Osman, E.Y.; Rietz, A.; Kline, R.A.; Cherry, J.J.; Hodgetts, K.J.; Lorson, C.L.; Androphy, E.J. Intraperitoneal delivery of a novel drug-like compound improves disease severity in severe and intermediate mouse models of Spinal Muscular Atrophy. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Porensky, P.N.; Mitrpant, C.; McGovern, V.L.; Bevan, A.K.; Foust, K.D.; Kaspar, B.K.; Wilton, S.D.; Burghes, A.H.M. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum. Mol. Genet. 2012, 21, 1625–1638. [Google Scholar] [CrossRef]
- Mendell, J.R.; Goemans, N.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Shao, J.; Kaye, E.M.; Mercuri, E. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 2016, 79, 257–271. [Google Scholar] [CrossRef]
- Goemans, N.; Mercuri, E.; Belousova, E.; Komaki, H.; Dubrovsky, A.; McDonald, C.M.; Kraus, J.E.; Lourbakos, A.; Lin, Z.; Campion, G.; et al. A randomized placebo-controlled phase 3 trial of an antisense oligonucleotide, drisapersen, in duchenne muscular dystrophy. Neuromuscul. Disord. 2018, 28, 4–15. [Google Scholar] [CrossRef] [Green Version]
- Frank, D.E.; Schnell, F.J.; Akana, C.; El-Husayni, S.H.; Desjardins, C.A.; Morgan, J.; Charleston, J.S.; Sardone, V.; Domingos, J.; Dickson, G.; et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology 2020, 94, e2270–e2282. [Google Scholar] [CrossRef] [Green Version]
- Pascual-Morena, C.; Cavero-Redondo, I.; Álvarez-Bueno, C.; Mesas, A.E.; Pozuelo-Carrascosa, D.; Martínez-Vizcaíno, V. Restorative treatments of dystrophin expression in Duchenne muscular dystrophy: A systematic review. Ann. Clin. Transl. Neurol. 2020, 7, 1738–1752. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Awano, H.; Yagi, M.; Matsumoto, M.; Watanabe, N.; Goda, R.; Koizumi, M.; Takeshima, Y.; Matsuo, M. 2′-O-Methyl RNA/Ethylene-Bridged Nucleic Acid Chimera Antisense Oligonucleotides to Induce Dystrophin Exon 45 Skipping. Genes 2017, 8, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sud, R.; Geller, E.T.; Schellenberg, G.D. Antisense-mediated exon skipping decreases Tau protein expression: A potential therapy for tauopathies. Mol. Ther.-Nucleic Acids 2014, 3, e180. [Google Scholar] [CrossRef] [PubMed]
- Kalbfuss, B.; Mabon, S.A.; Misteli, T. Correction of Alternative Splicing of Tau in Frontotemporal Dementia and Parkinsonism Linked to Chromosome 17. J. Biol. Chem. 2001, 276, 42986–42993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peacey, E.; Rodriguez, L.; Liu, Y.; Wolfe, M.S. Targeting a pre-mRNA structure with bipartite antisense molecules modulates tau alternative splicing. Nucleic Acids Res. 2012, 40, 9836–9849. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Rodriguez, L.; Wolfe, M.S. Template-directed synthesis of a small molecule-antisense conjugate targeting an mRNA structure. Bioorg. Chem. 2014, 54, 7–11. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Disney, M.D. Bottom-up design of small molecules that stimulate exon 10 skipping in mutant MAPT pre-mRNA. ChemBioChem 2015, 16, 2041–2044. [Google Scholar] [CrossRef]
- Bruun, G.H.; Bang, J.M.V.; Christensen, L.L.; Brøner, S.; Petersen, U.S.S.; Guerra, B.; Grønning, A.G.B.; Doktor, T.K.; Andresen, B.S. Blocking of an intronic splicing silencer completely rescues IKBKAP exon 20 splicing in familial dysautonomia patient cells. Nucleic Acids Res. 2018, 46, 7938–7952. [Google Scholar] [CrossRef]
- Donadon, I.; Pinotti, M.; Rajkowska, K.; Pianigiani, G.; Barbon, E.; Morini, E.; Motaln, H.; Rogelj, B.; Mingozzi, F.; Slaugenhaupt, S.A.; et al. Exon-specific U1 snRNAs improve ELP1 exon 20 definition and rescue ELP1 protein expression in a familial dysautonomia mouse model. Hum. Mol. Genet. 2018, 27, 2466–2476. [Google Scholar] [CrossRef] [Green Version]
- Axelrod, F.B.; Liebes, L.; Von Simson, G.G.; Mendoza, S.; Mull, J.; Leyne, M.; Norcliffe-Kaufmann, L.; Kaufmann, H.; Slaugenhaupt, S.A. Kinetin improves IKBKAP mRNA splicing in patients with familial dysautonomia. Pediatr. Res. 2011, 70, 480–483. [Google Scholar] [CrossRef]
- Yoshida, M.; Kataoka, N.; Miyauchi, K.; Ohe, K.; Iida, K.; Yoshida, S.; Nojima, T.; Okuno, Y.; Onogi, H.; Usui, T.; et al. Rectifier of aberrant mRNA splicing recovers tRNA modification in familial dysautonomia. Proc. Natl. Acad. Sci. USA 2015, 112, 2764–2769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, K.; Hasegawa, C.; Kaneko, M.; Tsutsumi, S.; Sone, J.; Ishikawa, T.; Imanishi, T.; Koizumi, M. 2′-O,4′-C-ethylene-bridged nucleic acids (ENA): Highly nuclease-resistant and thermodynamically stable oligonucleotides for antisense drug. Bioorganic Med. Chem. Lett. 2002, 12, 73–76. [Google Scholar] [CrossRef]
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002, 82, 291–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landfeldt, E.; Thompson, R.; Sejersen, T.; McMillan, H.J.; Kirschner, J.; Lochmüller, H. Life expectancy at birth in Duchenne muscular dystrophy: A systematic review and meta-analysis. Eur. J. Epidemiol. 2020, 35, 643–653. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.R.Q.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Devel. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juan-Mateu, J.; Gonzalez-Quereda, L.; Rodriguez, M.J.; Baena, M.; Verdura, E.; Nascimento, A.; Ortez, C.; Baiget, M.; Gallano, P. DMD Mutations in 576 Dystrophinopathy Families: A Step Forward in Genotype-Phenotype Correlations. PLoS ONE 2015, 10, e0135189. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xu, M.; Li, H.; He, R.; Lin, J.; Zhang, C.; Zhu, Y. Genotypes and Phenotypes of DMD Small Mutations in Chinese Patients With Dystrophinopathies. Front. Genet. 2019, 10, 114. [Google Scholar] [CrossRef] [PubMed]
- Alter, J.; Lou, F.; Rabinowitz, A.; Yin, H.F.; Rosenfeld, J.; Wilton, S.D.; Partridge, T.A.; Qi, L.L. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat. Med. 2006, 12, 175–177. [Google Scholar] [CrossRef]
- Arechavala-Gomeza, V.; Graham, I.R.; Popplewell, L.J.; Adams, A.M.; Aartsma-Rus, A.; Kinali, M.; Morgan, J.E.; Van Deutekom, J.C.; Wilton, S.D.; Dickson, G.; et al. Comparative analysis of antisense oligonucleotide sequences for targeted skipping of exon 51 during dystrophin pre-mRNA splicing in human muscle. Hum. Gene Ther. 2007, 18, 798–810. [Google Scholar] [CrossRef] [Green Version]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD global database: Analysis of more than 7,000 duchenne muscular dystrophy mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef]
- Dhillon, S. Viltolarsen: First Approval. Drugs 2020, 80, 1027–1031. [Google Scholar] [CrossRef]
- O’Keefe, L. FDA Approves Targeted Treatment for Rare Duchenne Muscular Dystrophy Mutation. 25 Feburary 2021. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation-0 (accessed on 12 April 2021).
- Lunn, M.R.; Wang, C.H. Spinal muscular atrophy. Lancet 2008, 371, 2120–2133. [Google Scholar] [CrossRef]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.N.; Singh, R.N.; Androphy, E.J. Modulating role of RNA structure in alternative splicing of a critical exon in the spinal muscular atrophy genes. Nucleic Acids Res. 2007, 35, 371–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messina, S.; Sframeli, M. New Treatments in Spinal Muscular Atrophy: Positive Results and New Challenges. J. Clin. Med. 2020, 9, 2222. [Google Scholar] [CrossRef]
- Menduti, G.; Rasà, D.M.; Stanga, S.; Boido, M. Drug Screening and Drug Repositioning as Promising Therapeutic Approaches for Spinal Muscular Atrophy Treatment. Front. Pharmacol. 2020, 11, 1696. [Google Scholar] [CrossRef]
- Singh, N.K.; Singh, N.N.; Androphy, E.J.; Singh, R.N. Splicing of a Critical Exon of Human Survival Motor Neuron Is Regulated by a Unique Silencer Element Located in the Last Intron. Mol. Cell. Biol. 2006, 26, 1333–1346. [Google Scholar] [CrossRef] [Green Version]
- Hua, Y.; Vickers, T.A.; Okunola, H.L.; Bennett, C.F.; Krainer, A.R. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am. J. Hum. Genet. 2008, 82, 834–848. [Google Scholar] [CrossRef] [Green Version]
- Frederiksen, S.B.; Holm, L.L.; Larsen, M.R.; Doktor, T.K.; Andersen, H.S.; Hastings, M.L.; Hua, Y.; Krainer, A.R.; Andresen, B.S. Identification of SRSF10 as a regulator of SMN2 ISS-N1. Hum. Mutat. 2021, 42, 246–260. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [Green Version]
- Osman, E.Y.; Miller, M.R.; Robbins, K.L.; Lombardi, A.M.; Atkinson, A.K.; Brehm, A.J.; Lorson, C.L. Morpholino antisense oligonucleotides targeting intronic repressor Element1 improve phenotype in SMA mouse models. Hum. Mol. Genet. 2014, 23, 4832–4845. [Google Scholar] [CrossRef] [Green Version]
- Ratni, H.; Karp, G.M.; Weetall, M.; Naryshkin, N.A.; Paushkin, S.V.; Chen, K.S.; McCarthy, K.D.; Qi, H.; Turpoff, A.; Woll, M.G.; et al. Specific Correction of Alternative Survival Motor Neuron 2 Splicing by Small Molecules: Discovery of a Potential Novel Medicine To Treat Spinal Muscular Atrophy. J. Med. Chem. 2016, 59, 6086–6100. [Google Scholar] [CrossRef]
- Dhillon, S. Risdiplam: First Approval. Drugs 2020, 80, 1853–1858. [Google Scholar] [CrossRef]
- Campagne, S.; Boigner, S.; Rüdisser, S.; Moursy, A.; Gillioz, L.; Knörlein, A.; Hall, J.; Ratni, H.; Cléry, A.; Allain, F.H.-T. Structural basis of a small molecule targeting RNA for a specific splicing correction. Nat. Chem. Biol. 2019, 15, 1191–1198. [Google Scholar] [CrossRef]
- Palacino, J.; Swalley, S.E.; Song, C.; Cheung, A.K.; Shu, L.; Zhang, X.; Van Hoosear, M.; Shin, Y.; Chin, D.N.; Keller, C.G.; et al. SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat. Chem. Biol. 2015, 11, 511–517. [Google Scholar] [CrossRef]
- Sivaramakrishnan, M.; McCarthy, K.D.; Campagne, S.; Huber, S.; Meier, S.; Augustin, A.; Heckel, T.; Meistermann, H.; Hug, M.N.; Birrer, P.; et al. Binding to SMN2 pre-mRNA-protein complex elicits specificity for small molecule splicing modifiers. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.N.; Ottesen, E.W.; Singh, N.N. The First Orally Deliverable Small Molecule for the Treatment of Spinal Muscular Atrophy. Neurosci. Insights 2020, 15. [Google Scholar] [CrossRef] [PubMed]
- Mehedi, M.; Hoenen, T.; Robertson, S.; Ricklefs, S.; Dolan, M.A.; Taylor, T.; Falzarano, D.; Ebihara, H.; Porcella, S.F.; Feldmann, H. Ebola Virus RNA Editing Depends on the Primary Editing Site Sequence and an Upstream Secondary Structure. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, J.A.; Olson, A.N.; Neupane, K.; Munshi, S.; Emeterio, J.S.; Pollack, L.; Woodside, M.T.; Dinman, J.D. Structural and functional conservation of the programmed −1 ribosomal frameshift signal of SARS coronavirus 2 (SARS-CoV-2). J. Biol. Chem. 2020, 295, 10741–10748. [Google Scholar] [CrossRef] [PubMed]
- Bogdanow, B.; Wang, X.; Eichelbaum, K.; Sadewasser, A.; Husic, I.; Paki, K.; Budt, M.; Hergeselle, M.; Vetter, B.; Hou, J.; et al. The dynamic proteome of influenza A virus infection identifies M segment splicing as a host range determinant. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Bui, C.M.; Chughtai, A.A.; Adam, D.C.; MacIntyre, C.R. An overview of the epidemiology and emergence of influenza A infection in humans over time. Arch. Public Heal. 2017, 75, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Moss, W.N.; Dela-Moss, L.I.; Priore, S.F.; Turner, D.H. The influenza A segment 7 mRNA 3′ splice site pseudoknot/hairpin family. RNA 2012, 9, 1305–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Disease Burden of Influenza. Available online: https://www.cdc.gov/flu/about/disease/burden.htm (accessed on 12 April 2021).
- Weekly U.S. Influenza Surveillance Report. Available online: https://www.cdc.gov/flu/weekly/index.htm (accessed on 12 April 2021).
- Dubois, J.; Terrier, O.; Rosa-Calatrava, M. Influenza viruses and mRNA splicing: Doing more with less. MBio 2014, 5, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, W.N.; Priore, S.F.; Turner, D.H. Identification of potential conserved RNA secondary structure throughout influenza A coding regions. RNA 2011, 17, 991–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.L.; Kennedy, S.D.; Turner, D.H. Structural features of a 3′ splice site in influenza A. Biochemistry 2015, 54, 3269–3285. [Google Scholar] [CrossRef] [Green Version]
- Dadonaite, B.; Gilbertson, B.; Knight, M.L.; Trifkovic, S.; Rockman, S.; Laederach, A.; Brown, L.E.; Fodor, E.; Bauer, D.L.V. The structure of the influenza A virus genome. Nat. Microbiol. 2019, 4, 1781–1789. [Google Scholar] [CrossRef]
- Simon, L.M.; Morandi, E.; Luganini, A.; Gribaudo, G.; Martinez-Sobrido, L.; Turner, D.H.; Oliviero, S.; Incarnato, D. In vivo analysis of influenza A mRNA secondary structures identifies critical regulatory motifs. Nucleic Acids Res. 2019, 47, 7003–7017. [Google Scholar] [CrossRef] [Green Version]
- Hale, B.G.; Randall, R.E.; Ortin, J.; Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef]
- Fauci, A.S.; Lane, H.C. Four Decades of HIV/AIDS—Much Accomplished, Much to Do. N. Engl. J. Med. 2020, 383, 1–4. [Google Scholar] [CrossRef]
- Emery, A.; Zhou, S.; Pollom, E.; Swanstrom, R. Characterizing HIV-1 Splicing by Using Next-Generation Sequencing. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Bohne, J.; Wodrich, H.; Kräusslich, H.G. Splicing of human immunodeficiency virus RNA is position-dependent suggesting sequential removal of introns from the 5′ end. Nucleic Acids Res. 2005, 33, 825–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dlamini, Z.; Hull, R. Can the HIV-1 splicing machinery be targeted for drug discovery? HIV/AIDS-Res. Palliat. Care 2017, 9, 63–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blissenbach, M.; Grewe, B.; Hoffmann, B.; Brandt, S.; Überla, K. Nuclear RNA Export and Packaging Functions of HIV-1 Rev Revisited. J. Virol. 2010, 84, 6598–6604. [Google Scholar] [CrossRef] [Green Version]
- Daum, T.; Engels, J.W.; Mag, M.; Muth, J.; Lücking, S.; Schröder, H.C.; Matthes, E.; Müller, W.E.G. Antisense oligodeoxynucleotide: Inhibitor of splicing of mRNA of human immunodeficiency virus. Intervirology 1992, 33, 65–75. [Google Scholar] [CrossRef]
- Zhang, L.L.; Wei, J.Y.; Wang, L.; Chen, J.L. Human T-cell lymphotropic virus type 1 and its oncogenesis. Acta Pharmacol. Sin. 2017, 38, 1093–1103. [Google Scholar] [CrossRef] [Green Version]
- Younis, I.; Green, P.L. The human T-cell leukemia virus Rex protein. Front. Biosci. 2005, 10, 431–445. [Google Scholar] [CrossRef] [Green Version]
- Mole, S.; Milligan, S.G.; Graham, S.V. Human Papillomavirus Type 16 E2 Protein Transcriptionally Activates the Promoter of a Key Cellular Splicing Factor, SF2/ASF. J. Virol. 2009, 83, 357–367. [Google Scholar] [CrossRef] [Green Version]
- Roemer, T.; Krysan, D.J. Antifungal drug development: Challenges, unmet clinical needs, and new approaches. Cold Spring Harb. Perspect. Med. 2014, 4, a019703. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Pyle, A.M. Structural Insights into the Mechanism of Group II Intron Splicing. Trends Biochem. Sci. 2017, 42, 470–482. [Google Scholar] [CrossRef]
- Pyle, A.M. Group II Intron Self-Splicing. Annu. Rev. Biophys. 2016, 45, 183–205. [Google Scholar] [CrossRef] [PubMed]
- Wszolek, Z.K.; Tsuboi, Y.; Ghetti, B.; Pickering-Brown, S.; Baba, Y.; Cheshire, W.P. Frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). Orphanet J. Rare Dis. 2006, 1, 30. [Google Scholar] [CrossRef] [Green Version]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Pascale, E.; Di Battista, M.E.; Rubino, A.; Purcaro, C.; Valente, M.; Fattapposta, F.; Ferraguti, G.; Meco, G. Genetic Architecture of MAPT Gene Region in Parkinson Disease Subtypes. Front. Cell. Neurosci. 2016, 10, 96. [Google Scholar] [CrossRef] [Green Version]
- Umeda, T.; Yamashita, T.; Kimura, T.; Ohnishi, K.; Takuma, H.; Ozeki, T.; Takashima, A.; Tomiyama, T.; Mori, H. Neurodegenerative disorder FTDP-17-related tau intron 10 +16C→T mutation increases tau exon 10 splicing and causes tauopathy in transgenic mice. Am. J. Pathol. 2013, 183, 211–225. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.-X.; Grundke-Iqbal, I. Tau in Alzheimer Disease and Related Tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Lozupone, M.; Seripa, D.; Daniele, A.; Watling, M.; Giannelli, G.; Imbimbo, B.P. Development of disease-modifying drugs for frontotemporal dementia spectrum disorders. Nat. Rev. Neurol. 2020, 16, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–704. [Google Scholar] [CrossRef]
- Donahue, C.P.; Ni, J.; Rozners, E.; Glicksman, M.A.; Wolfe, M.S. Identification of tau stem loop RNA stabilizers. J. Biomol. Screen. 2007, 12, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Chen, Y.; Donahue, C.P.; Wolfe, M.S.; Varani, G. Structural Basis for Stabilization of the Tau Pre-mRNA Splicing Regulatory Element by Novantrone (Mitoxantrone). Chem. Biol. 2009, 16, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Maayan, C.; Kaplan, E.; Shachar, S.; Peleg, O.; Godfrey, S. Incidence of familial dysautonomia in Israel 1977-1981. Clin. Genet. 2008, 32, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Norcliffe-Kaufmann, L.; Slaugenhaupt, S.A.; Kaufmann, H. Familial dysautonomia: History, genotype, phenotype and translational research. Prog. Neurobiol. 2017, 152, 131–148. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.L.; Coli, R.; Daly, I.W.; Kichula, E.A.; Rork, M.J.; Volpi, S.A.; Ekstein, J.; Rubin, B.Y. Familial dysautonomia is caused by mutations of the IKAP gene. Am. J. Hum. Genet. 2001, 68, 753–758. [Google Scholar] [CrossRef] [Green Version]
- Slaugenhaupt, S.A.; Blumenfeld, A.; Gill, S.P.; Leyne, M.; Mull, J.; Cuajungco, M.P.; Liebert, C.B.; Chadwick, B.; Idelson, M.; Reznik, L.; et al. Tissue-specific expression of a splicing mutation in the IKBKAP gene causes familial dysautonomia. Am. J. Hum. Genet. 2001, 68, 598–605. [Google Scholar] [CrossRef] [Green Version]
- Slaugenhaupt, S.A.; Mull, J.; Leyne, M.; Cuajungco, M.P.; Gill, S.P.; Hims, M.M.; Quintero, F.; Axelrod, F.B.; Gusella, J.F. Rescue of a human mRNA splicing defect by the plant cytokinin kinetin. Hum. Mol. Genet. 2004, 13, 429–436. [Google Scholar] [CrossRef]
- Hims, M.M.; Chérif Ibrahim, E.; Leyne, M.; Mull, J.; Liu, L.; Lazaro, C.; Shetty, R.S.; Gill, S.; Gusella, J.F.; Reed, R.; et al. Therapeutic potential and mechanism of kinetin as a treatment for the human splicing disease familial dysautonomia. J. Mol. Med. 2007, 85, 149–161. [Google Scholar] [CrossRef]
- Wee, C.D.; Havens, M.A.; Jodelka, F.M.; Hastings, M.L. Targeting SR proteins improves SMN expression in spinal muscular atrophy cells. PLoS ONE 2014, 9, e115205. [Google Scholar] [CrossRef]
- Hua, Y.; Vickers, T.A.; Baker, B.F.; Bennett, C.F.; Krainer, A.R. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 2007, 5, e73. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, Y.; Wirth, B. hnRNP-G promotes exon 7 inclusion of survival motor neuron (SMN) via direct interaction with Htra2-beta1. Hum. Mol. Genet. 2002, 11, 2037–2049. [Google Scholar] [CrossRef] [Green Version]
- Naryshkin, N.A.; Weetall, M.; Dakka, A.; Narasimhan, J.; Zhao, X.; Feng, Z.; Ling, K.K.Y.; Karp, G.M.; Qi, H.; Woll, M.G.; et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 2014, 345, 688–693. [Google Scholar] [CrossRef]
- Makhortova, N.R.; Hayhurst, M.; Cerqueira, A.; Sinor-Anderson, A.D.; Zhao, W.N.; Heiser, P.W.; Arvanites, A.C.; Davidow, L.S.; Waldon, Z.O.; Steen, J.A.; et al. A screen for regulators of survival of motor neuron protein levels. Nat. Chem. Biol. 2011, 7, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Stoilov, P.; Lin, C.H.; Damoiseaux, R.; Nikolic, J.; Black, D.L. A high-throughput screening strategy identifies cardiotonic steroids as alternative splicing modulators. Proc. Natl. Acad. Sci. USA 2008, 105, 11218–11223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherry, J.J.; Evans, M.C.; Ni, J.; Cuny, G.D.; Glicksman, M.A.; Androphy, E.J. Identification of novel compounds that increase SMN protein levels using an improved SMN2 reporter cell assay. J. Biomol. Screen. 2012, 17, 481–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.L.; Lorson, C.L.; Androphy, E.J.; Zhou, J. An in vivo reporter system for measuring increased inclusion of exon 7 in SMN2 mRNA: Potential therapy of SMA. Gene Ther. 2001, 8, 1532–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donahue, C.P.; Muratore, C.; Wu, J.Y.; Kosik, K.S.; Wolfe, M.S. Stabilization of the tau exon 10 stem loop alters pre-mRNA splicing. J. Biol. Chem. 2006, 281, 23302–23306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa-Ankerhold, H.C.; Ankerhold, R.; Drummen, G.P.C. Advanced Fluorescence Microscopy Techniques—FRAP, FLIP, FLAP, FRET and FLIM. Molecules 2012, 17, 4047–4132. [Google Scholar] [CrossRef] [Green Version]
- Baell, J.B.; Nissink, J.W.M. Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017—Utility and Limitations. ACS Chem. Biol. 2018, 13, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Effenberger, K.A.; Perriman, R.J.; Bray, W.M.; Lokey, R.S.; Ares, M.; Jurica, M.S. A high-throughput splicing assay identifies new classes of inhibitors of human and yeast spliceosomes. J. Biomol. Screen. 2013, 18, 1110–1120. [Google Scholar] [CrossRef] [Green Version]
- Boehm, V.; Gehring, N.H. Exon Junction Complexes: Supervising the Gene Expression Assembly Line. Trends Genet. 2016, 32, 724–735. [Google Scholar] [CrossRef]
- Le Hir, H.; Saulière, J.; Wang, Z. The exon junction complex as a node of post-transcriptional networks. Nat. Rev. Mol. Cell Biol. 2016, 17, 41–54. [Google Scholar] [CrossRef]
- Berg, M.G.; Wan, L.; Younis, I.; Diem, M.D.; Soo, M.; Wang, C.; Dreyfuss, G. A Quantitative High-Throughput In Vitro Splicing Assay Identifies Inhibitors of Spliceosome Catalysis. Mol. Cell. Biol. 2012, 32, 1271–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Nielsen, P.; Koshkin, A.A.; Wengel, J. LNA (locked nucleic acids): Synthesis and high-affinity nucleic acid recognition. Chem. Commun. 1998, 90, 455–456. [Google Scholar] [CrossRef]
- Owczarzy, R.; You, Y.; Groth, C.L.; Tataurov, A.V. Stability and Mismatch Discrimination of Locked Nucleic Acid–DNA Duplexes. Biochemistry 2011, 50, 9352–9367. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Mishra, A.; Puri, N. Peptide nucleic acids: Advanced tools for biomedical applications. J. Biotechnol. 2017, 259, 148–159. [Google Scholar] [CrossRef]
- Corey, D.R.; Abrams, J.M. Morpholino antisense oligonucleotides: Tools for investigating vertebrate development. Genome Biol. 2001, 2, reviews1015.1. [Google Scholar] [CrossRef] [PubMed]
- Trabulo, S.; Cardoso, A.L.; Mano, M.; De Lima, M.C.P. Cell-Penetrating Peptides—Mechanisms of Cellular Uptake and Generation of Delivery Systems. Pharmaceuticals 2010, 3, 961–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClorey, G.; Banerjee, S. Cell-penetrating peptides to enhance delivery of oligonucleotide-based therapeutics. Biomedicines 2018, 6, 51. [Google Scholar] [CrossRef] [Green Version]
- Kole, R.; Krainer, A.R.; Altman, S. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012, 11, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Crooke, S.T.; Baker, B.F.; Crooke, R.M.; Liang, X. Antisense technology: An overview and prospectus. Nat. Rev. Drug Discov. 2021. [Google Scholar] [CrossRef]
- Crooke, S.T.; Liang, X.-H.; Baker, B.F.; Crooke, R.M. Antisense technology: A review. J. Biol. Chem. 2021, 296, 100416. [Google Scholar] [CrossRef]
- Bennett, C.F. Therapeutic antisense oligonucleotides are coming of age. Annu. Rev. Med. 2019, 70, 307–321. [Google Scholar] [CrossRef]
- Chenoweth, D.M.; Meier, J.L.; Dervan, P.B. Pyrrole-imidazole polyamides distinguish between double-helical DNA and RNA. Angew. Chem. Int. Ed. 2013, 52, 415–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dock-Bregeon, A.C.; Chevrier, B.; Podjarny, A.; Johnson, J.; de Bear, J.S.; Gough, G.R.; Gilham, P.T.; Moras, D. Crystallographic structure of an RNA helix: [U(UA)6A]2. J. Mol. Biol. 1989, 209, 459–474. [Google Scholar] [CrossRef]
- Batey, R.T.; Rambo, R.P.; Doudna, J.A. Doudna Tertiary Motifs in RNA Structure and Folding. Angew. Chem. Int. Ed. Engl. 1999, 38, 2326–2343. [Google Scholar] [CrossRef]
- Ursu, A.; Childs-Disney, J.L.; Andrews, R.J.; O’Leary, C.A.; Meyer, S.M.; Angelbello, A.J.; Moss, W.N.; Disney, M.D. Design of small molecules targeting RNA structure from sequence. Chem. Soc. Rev. 2020, 49, 7252–7270. [Google Scholar] [CrossRef]
- Velagapudi, S.P.; Gallo, S.M.; Disney, M.D. Sequence-based design of bioactive small molecules that target precursor microRNAs. Nat. Chem. Biol. 2014, 10, 291–297. [Google Scholar] [CrossRef]
- Disney, M.D.; Winkelsas, A.M.; Velagapudi, S.P.; Southern, M.; Fallahi, M.; Childs-Disney, J.L. Inforna 2.0: A Platform for the Sequence-Based Design of Small Molecules Targeting Structured RNAs. ACS Chem. Biol. 2016, 11, 1720–1728. [Google Scholar] [CrossRef] [Green Version]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemons, P.A.; Bodycombe, N.E.; Carrinski, H.A.; Wilson, J.A.; Shamji, A.F.; Wagner, B.K.; Koehler, A.N.; Schreiber, S.L. Small molecules of different origins have distinct distributions of structural complexity that correlate with protein-binding profiles. Proc. Natl. Acad. Sci. USA 2010, 107, 18787–18792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luker, T.; Alcaraz, L.; Chohan, K.K.; Blomberg, N.; Brown, D.S.; Butlin, R.J.; Elebring, T.; Griffin, A.M.; Guile, S.; St-Gallay, S.; et al. Strategies to improve in vivo toxicology outcomes for basic candidate drug molecules. Bioorganic Med. Chem. Lett. 2011, 21, 5673–5679. [Google Scholar] [CrossRef] [PubMed]
- Bertz, S.H. The First General Index of Molecular Complexity. J. Am. Chem. Soc. 1981, 103, 3599–3601. [Google Scholar] [CrossRef]
- Landrum, G. RDKit: Open-Source Cheminformatics. 2006. Available online: https://www.rdkit.org/ (accessed on 12 April 2021).
- Zhu, C.; Chen, Z.; Guo, W. Pre-mRNA mis-splicing of sarcomeric genes in heart failure. Biochim. Biophys. Acta-Mol. Basis Dis. 2017, 1863, 2056–2063. [Google Scholar] [CrossRef]
- Guo, W.; Sun, M. RBM20, a potential target for treatment of cardiomyopathy via titin isoform switching. Biophys. Rev. 2018, 10, 15–25. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spliceosome | Group I Intron | Group II Intron | tRNA 1 | |
|---|---|---|---|---|
| Organisms | Eukaryotes | Bacteria, archaea, bacteriophages, plants, fungi | Fungi, algal plastids, bacteria, archaea | Eukaryotes, archaea |
| Location | Nucleus | Bacterial/archaeal, mitochondria, chloroplast | Bacterial/archaeal, mitochondria, chloroplast | vertebrate/plant nucleus, yeast cytosol, archaea |
| Branch point | A | Exogenous G cofactor | A | (not applicable) |
| Intron size | Dynamic, ~6.5 kb on average [46] | 250–500 nt [39] | 2–3 kb [47] | 6–133 nt [48] |
| Intron format | Lariat | Linear | Lariat | Linear |
| Protein involvement | Spliceosome | Not necessary | Reverse transcriptase | Endonuclease, ligase |
| Disease | Target Gene | Name | Category 1 | Development Stage |
|---|---|---|---|---|
| SMA | SMN2 | Nusinersen [3] | ASO (MOE) | Approved |
| E1v1.11-ASO [63] | ASO (PMO) | Phase I | ||
| SMN-PNA [64] | ASO (PNA) | Cellular | ||
| Risdiplam [65] | small molecule | Approved | ||
| Branaplam [66] | small molecule | Phase II/III (discontinued) | ||
| PK4C9 [67] | small molecule | Preclinical | ||
| LDN-2014 [68] | small molecule | Preclinical | ||
| HSMNEx7D [69] | ASO (PMO) | Preclinical | ||
| DMD | Dystrophin Ex51 | Eteplirsen [70] | ASO (PMO) | Approved |
| Dystrophin Ex51 | Drisapersen [71] | ASO (2′-OMe) | Discontinued | |
| Dystrophin Ex53 | Golodirsen [72] | ASO (PMO) | Approved | |
| Dystrophin Ex53 | Viltolarsen [5] | ASO (PMO) | Approved | |
| Dystrophin Ex45 | Casimersen [73] | ASO (PMO) | Phase III | |
| Dystrophin Ex45 | DS-5141 [74] | ASO (ENA 2) | Phase I/II | |
| Yeast infection | group II intron | Intronistat B [10] | small molecule | Cellular |
| FTDP-17 | MAPT | E1.4, E5.3 [75] | ASO (PMO) | Preclinical |
| E10α, E10β [76] | ASO (2′-OMe) | Cellular | ||
| bipartite ASO [77] | ASO (PNA) | Cellular | ||
| Mitoxantrone– bipartite ASO [78] | small molecule-ASO conjugate | Cellular | ||
| compound 9 [9] | small molecule | Cellular | ||
| compound 2 [79] | small molecule | Cellular | ||
| Familial Dysautonomia | IKBKAP | 7-26S [11] | ASO (MOE) | Preclinical |
| SSO1 [80] | ASO (OMe) | Cellular | ||
| ExSpeU1 [81] | ASO | Preclinical | ||
| Kinetin [82] | small molecule | Phase I (discontinued) | ||
| RECTAS [83] | small molecule | Cellular | ||
| Influenza A virus infection | RNA segment 7 | Radavirsen [8] | ASO (PMO) | Phase I |
| Virus Name | Essential Genes | Viral Regulatory Factors |
|---|---|---|
| Influenza A virus | M1, M2/M42 | NS1 [118] |
| human immunodeficiency virus 1 | tat, rev; env | rev [128] |
| Human T-lymphotropic virus | env, tax, rex | rex [130,131] |
| Parvovirus B19 | VP1, VP2 | |
| Human Papillomavirus 1 | E1, E2, L1, L2 | E2 [132] |
| Molecular Descriptor | RNA-Targeting Splicing Modifier | FDA-Approved Drugs |
|---|---|---|
| Ratio of sp3-hybridization | 0.29 (±0.16) | 0.44 (±0.26) |
| Rotatable bonds | 4.1 (±2.4) | 4.9 (±3.7) |
| Hydrogen bonding donor | 2.3 (±2.3) | 1.7 (±1.7) |
| Hydrogen bonding acceptor | 6.4 (±1.5) | 4.4 (±2.7) |
| Number of nitrogen atoms | 4.9 (±2.0) | 2.0 (±1.8) |
| Topological polar surface area (TPSA) | 89 (±29) | 73 (±49) |
| Number of rings | 4.3 (±0.9) | 2.6 (±1.5) |
| BertzCT | 1113 (±253) | 669 (±363) |
| Molecular weight | 380 (±61) | 332 (±126) |
| Calculated LogP | 2.8 (±1.1) | 2.5 (±2.3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, Z.; Zhao, J.; Pearson, Z.J.; Boskovic, Z.V.; Wang, J. RNA-Targeting Splicing Modifiers: Drug Development and Screening Assays. Molecules 2021, 26, 2263. https://doi.org/10.3390/molecules26082263
Tang Z, Zhao J, Pearson ZJ, Boskovic ZV, Wang J. RNA-Targeting Splicing Modifiers: Drug Development and Screening Assays. Molecules. 2021; 26(8):2263. https://doi.org/10.3390/molecules26082263
Chicago/Turabian StyleTang, Zhichao, Junxing Zhao, Zach J. Pearson, Zarko V. Boskovic, and Jingxin Wang. 2021. "RNA-Targeting Splicing Modifiers: Drug Development and Screening Assays" Molecules 26, no. 8: 2263. https://doi.org/10.3390/molecules26082263