
Discovery of Post-Translational Modifications in Emiliania huxleyi



Abstract
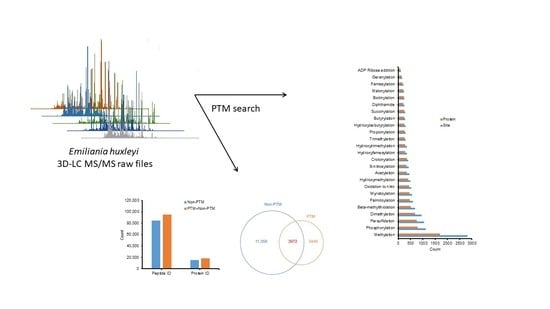
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Global PTM Discovery from MS/MS Spectra
2.2. Effect of PTMs on the Identification of Peptides and Proteins
- Peptide 1: R.n[4 4.01]SN[144.05]LGMEVMHER.N (hydroxylmethylation on Asn).
- Peptide 2: R.n[44.01]E[143.06]TTENESANYGYK.F (methylation on Glu).
- Peptide 3: R.n[44.01]E[143.06]WELSYR.L (methylation on Glu).
- Peptide 4: K.FGQEEETY[208.05]NIVAAHGYFGR.L (oxidation on Tyr).
2.3. Effects of PTMs on Quantitative Analysis
2.4. Biological Relevance of Modified Proteins of Emiliania huxleyi
3. Materials and Methods
3.1. Data Set
3.2. Non-PTM and PTM Search
3.3. Peptide and Protein Quantification
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Li, Q.; Shortreed, M.R.; Wenger, C.D.; Frey, B.L.; Schaffer, L.V.; Scalf, M.; Smith, L.M. Global Post-Translational Modification Discovery. J. Proteome Res. 2017, 16, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.N.; Vitorino, R.; Domingues, M.R.M.; Spickett, C.M.; Domingues, P. Post-translational Modifications and Mass Spectrometry Detection. Free Radic. Biol. Med. 2013, 65, 925–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nørregaard Jensen, O. Modification-specific proteomics: Characterization of post-translational modifications by mass spectrometry. Curr. Opin. Chem. Biol. 2004, 8, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.E.; Mocanu, V.; Mocanu, M.; Dicheva, N.; Warren, M.R. Mass spectrometry for post-translational modifications. In Neuroproteomics; Alzate, O., Ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2010. [Google Scholar] [PubMed]
- Hersman, E.; Nelson, D.M.; Griffith, W.P.; Jelinek, C.; Cotter, R.J. Analysis of histone modifications from tryptic peptides of deuteroacetylated isoforms. Int. J. Mass Spectrom. 2012, 312, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.-k.; Yang, Y.-h.; Chen, Z.; Zhang, J.; Lin, Y.; Wang, Y.; Xiong, Q.; Li, T.; Ge, F.; Bryant, D.A.; et al. Proteogenomic analysis and global discovery of posttranslational modifications in prokaryotes. Proc. Natl. Acad. Sci. USA 2014, 111, E5633–E5642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanner, S.; Shu, H.; Frank, A.; Wang, L.-C.; Zandi, E.; Mumby, M.; Pevzner, P.A.; Bafna, V. InsPecT: Identification of Posttranslationally Modified Peptides from Tandem Mass Spectra. Anal. Chem. 2005, 77, 4626–4639. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, R.; Yang, Z.; Lee, J.; Liu, Y.; Tian, J.; Qin, X.; Ren, Z.; Ding, H.; Chen, Q.; et al. Comparative Effectiveness and Safety of Oral Phosphodiesterase Type 5 Inhibitors for Erectile Dysfunction: A Systematic Review and Network Meta-analysis. Eur. Urol. 2013, 63, 902–912. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Mendoza, L.; Shteynberg, D.; Farrah, T.; Lam, H.; Tasman, N.; Sun, Z.; Nilsson, E.; Pratt, B.; Prazen, B.; et al. A guided tour of the Trans-Proteomic Pipeline. Proteomics 2010, 10, 1150–1159. [Google Scholar] [CrossRef] [Green Version]
- Fíla, J.; Honys, D. Enrichment techniques employed in phosphoproteomics. Amino Acids 2012, 43, 1025–1047. [Google Scholar] [CrossRef] [Green Version]
- Andersson, L.; Porath, J. Isolation of phosphoproteins by immobilized metal (Fe3+) affinity chromatography. Anal. Biochem. 1986, 154, 250–254. [Google Scholar] [CrossRef]
- Sano, A.; Nakamura, H. Titania as a Chemo-affinity Support for the Column-switching HPLC Analysis of Phosphopeptides: Application to the Characterization of Phosphorylation Sites in Proteins by Combination with Protease Digestion and Electrospray Ionization Mass Spectrometry. Anal. Sci. 2004, 20, 861–864. [Google Scholar] [CrossRef] [Green Version]
- Abbott, K.L.; Pierce, J.M. Chapter Twenty-One—Lectin-Based Glycoproteomic Techniques for the Enrichment and Identification of Potential Biomarkers. In Methods in Enzymology; Fukuda, M., Ed.; Academic Press: London, UK, 2010; Volume 480, pp. 461–476. [Google Scholar]
- Zhang, H.; Li, X.-j.; Martin, D.B.; Aebersold, R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol. 2003, 21, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duong, V.-A.; Park, J.-M.; Lee, H. Review of Three-Dimensional Liquid Chromatography Platforms for Bottom-Up Proteomics. Int. J. Mol. Sci. 2020, 21, 1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Totti, C.; Romagnoli, T.; Accoroni, S.; Coluccelli, A.; Pellegrini, M.; Campanelli, A.; Grilli, F.; Marini, M. Phytoplankton communities in the northwestern Adriatic Sea: Interdecadal variability over a 30-years period (1988–2016) and relationships with meteoclimatic drivers. J. Mar. Syst. 2019, 193, 137–153. [Google Scholar] [CrossRef]
- Paasche, E. A review of the coccolithophorid Emiliania huxleyi (Prymnesiophyceae), with particular reference to growth, coccolith formation, and calcification-photosynthesis interactions. Phycologia 2001, 40, 503–529. [Google Scholar] [CrossRef]
- Rost, B.; Riebesell, U.; Burkhardt, S.; Sültemeyer, D. Carbon acquisition of bloom-forming marine phytoplankton. Limnol. Oceanogr. 2003, 48, 55–67. [Google Scholar] [CrossRef] [Green Version]
- McKew, B.A.; Metodieva, G.; Raines, C.A.; Metodiev, M.V.; Geider, R.J. Acclimation of Emiliania huxleyi (1516) to nutrient limitation involves precise modification of the proteome to scavenge alternative sources of N and P. Environ. Microbiol. 2015, 17, 4050–4062. [Google Scholar] [CrossRef] [Green Version]
- Araie, H.; Sakamoto, K.; Suzuki, I.; Shiraiwa, Y. Characterization of the Selenite Uptake Mechanism in the Coccolithophore Emiliania huxleyi (Haptophyta). Plant Cell Physiol. 2011, 52, 1204–1210. [Google Scholar] [CrossRef] [Green Version]
- Frada, M.J.; Bidle, K.D.; Probert, I.; de Vargas, C. In situ survey of life cycle phases of the coccolithophore Emiliania huxleyi (Haptophyta). Environ. Microbiol. 2012, 14, 1558–1569. [Google Scholar] [CrossRef]
- Read, B.A.; Kegel, J.; Klute, M.J.; Kuo, A.; Lefebvre, S.C.; Maumus, F.; Mayer, C.; Miller, J.; Monier, A.; Salamov, A.; et al. Pan genome of the phytoplankton Emiliania underpins its global distribution. Nature 2013, 499, 209–213. [Google Scholar] [CrossRef] [Green Version]
- Jones, B.M.; Edwards, R.J.; Skipp, P.J.; O’Connor, C.D.; Iglesias-Rodriguez, M.D. Shotgun Proteomic Analysis of Emiliania huxleyi, a Marine Phytoplankton Species of Major Biogeochemical Importance. Mar. Biotechnol. 2011, 13, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.M.; Iglesias-Rodriguez, M.D.; Skipp, P.J.; Edwards, R.J.; Greaves, M.J.; Young, J.R.; Elderfield, H.; O’Connor, C.D. Responses of the Emiliania huxleyi Proteome to Ocean Acidification. PLoS ONE 2013, 8, e61868. [Google Scholar] [CrossRef]
- Nam, O.; Park, J.-M.; Lee, H.; Jin, E. De novo transcriptome profile of coccolithophorid alga Emiliania huxleyi CCMP371 at different calcium concentrations with proteome analysis. PLoS ONE 2019, 14, e0221938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, G.; Park, J.-M.; Duong, V.-A.; Mok, J.-H.; Jeon, J.; Nam, O.; Lee, J.; Jin, E.; Lee, H. Proteomic Profiling of Emiliania huxleyi Using a Three-Dimensional Separation Method Combined with Tandem Mass Spectrometry. Molecules 2020, 25, 3028. [Google Scholar] [CrossRef] [PubMed]
- Armengaud, J. Proteogenomics and systems biology: Quest for the ultimate missing parts. Expert Rev. Proteom. 2010, 7, 65–77. [Google Scholar] [CrossRef]
- Rogers, L.D.; Overall, C.M. Proteolytic post-translational modification of proteins: Proteomic tools and methodology. Mol. Cell. Proteom. 2013, 12, 3532–3542. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; Ong, S.-E.; Mann, M. Trypsin Cleaves Exclusively C-terminal to Arginine and Lysine Residues. Mol. Cell. Proteom. 2004, 3, 608–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zee, B.M.; Garcia, B.A. Discovery of lysine post-translational modifications through mass spectrometric detection. Essays Biochem. 2012, 52, 147–163. [Google Scholar] [CrossRef] [Green Version]
- Pang, C.N.I.; Gasteiger, E.; Wilkins, M.R. Identification of arginine- and lysine-methylation in the proteome of Saccharomyces cerevisiae and its functional implications. BMC Genom. 2010, 11, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaffer, L.V.; Millikin, R.J.; Miller, R.M.; Anderson, L.C.; Fellers, R.T.; Ge, Y.; Kelleher, N.L.; LeDuc, R.D.; Liu, X.; Payne, S.H.; et al. Identification and Quantification of Proteoforms by Mass Spectrometry. Proteomics 2019, 19, 1800361. [Google Scholar] [CrossRef] [PubMed]
- Young, N.L.; Plazas-Mayorca, M.D.; Garcia, B.A. Systems-wide proteomic characterization of combinatorial post-translational modification patterns. Expert Rev. Proteom. 2010, 7, 79–92. [Google Scholar] [CrossRef]
- Nagel, T.; Meyer, B. Simultaneous characterization of sequence polymorphisms, glycosylation and phosphorylation of fibrinogen in a direct analysis by LC–MS. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2014, 1844, 2284–2289. [Google Scholar] [CrossRef] [PubMed]
- Sidoli, S.; Schwämmle, V.; Ruminowicz, C.; Hansen, T.A.; Wu, X.; Helin, K.; Jensen, O.N. Middle-down hybrid chromatography/tandem mass spectrometry workflow for characterization of combinatorial post-translational modifications in histones. Proteomics 2014, 14, 2200–2211. [Google Scholar] [CrossRef]
- Santos-Rosa, H.; Kirmizis, A.; Nelson, C.; Bartke, T.; Saksouk, N.; Cote, J.; Kouzarides, T. Histone H3 tail clipping regulates gene expression. Nat. Struct. Mol. Biol. 2009, 16, 17–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Brown, K.A.; Lin, Z.; Ge, Y. Top-Down Proteomics: Ready for Prime Time? Anal. Chem. 2018, 90, 110–127. [Google Scholar] [CrossRef] [PubMed]
- Ryšlavá, H.; Doubnerová, V.; Kavan, D.; Vaněk, O. Effect of posttranslational modifications on enzyme function and assembly. J. Proteom. 2013, 92, 80–109. [Google Scholar] [CrossRef]
- Shental-Bechor, D.; Levy, Y. Folding of glycoproteins: Toward understanding the biophysics of the glycosylation code. Curr. Opin. Struct. Biol. 2009, 19, 524–533. [Google Scholar] [CrossRef]
- Scott, J.D.; Pawson, T. Cell Signaling in Space and Time: Where Proteins Come Together and When They’re Apart. Science 2009, 326, 1220–1224. [Google Scholar] [CrossRef] [Green Version]
- Raposo, A.E.; Piller, S.C. Protein arginine methylation: An emerging regulator of the cell cycle. Cell Div. 2018, 13, 3. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, G.; Kudapa, H.; Ramalingam, A.; Choudhary, D.; Sinha, P.; Garg, V.; Singh, V.K.; Patil, G.B.; Pandey, M.K.; Nguyen, H.T.; et al. Epigenetics and epigenomics: Underlying mechanisms, relevance, and implications in crop improvement. Funct. Integr. Genom. 2020, 20, 739–761. [Google Scholar] [CrossRef] [PubMed]
- Aroca, Á.; Serna, A.; Gotor, C.; Romero, L.C. S-Sulfhydration: A Cysteine Posttranslational Modification in Plant Systems. Plant. Physiol. 2015, 168, 334–342. [Google Scholar] [CrossRef] [Green Version]
- Aroca, A.; Benito, J.M.; Gotor, C.; Romero, L.C. Persulfidation proteome reveals the regulation of protein function by hydrogen sulfide in diverse biological processes in Arabidopsis. J. Exp. Bot. 2017, 68, 4915–4927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Wu, F.-H.; Wang, W.-H.; Zheng, C.-J.; Lin, G.-H.; Dong, X.-J.; He, J.-X.; Pei, Z.-M.; Zheng, H.-L. Hydrogen sulphide enhances photosynthesis through promoting chloroplast biogenesis, photosynthetic enzyme expression, and thiol redox modification in Spinacia oleracea seedlings. J. Exp. Bot. 2011, 62, 4481–4493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ubersax, J.A.; Ferrell Jr, J.E. Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 530–541. [Google Scholar] [CrossRef]
- Cohen, P. The origins of protein phosphorylation. Nat. Cell Biol. 2002, 4, E127–E130. [Google Scholar] [CrossRef]
- Pawson, T.; Scott, J.D. Protein phosphorylation in signaling—50 years and counting. Trends Biochem. Sci. 2005, 30, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Paulo, J.A.; Nusinow, D.P.; Huttlin, E.L.; Gygi, S.P. Investigation of Proteomic and Phosphoproteomic Responses to Signaling Network Perturbations Reveals Functional Pathway Organizations in Yeast. Cell Rep. 2019, 29, 2092–2104. [Google Scholar] [CrossRef] [Green Version]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2018, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Pedrioli, P.G.A.; Eng, J.K.; Hubley, R.; Vogelzang, M.; Deutsch, E.W.; Raught, B.; Pratt, B.; Nilsson, E.; Angeletti, R.H.; Apweiler, R.; et al. A common open representation of mass spectrometry data and its application to proteomics research. Nat. Biotechnol. 2004, 22, 1459–1466. [Google Scholar] [CrossRef]
- Van-An, D.; Jeeyun, A.; Na-Young, H.; Jong-Moon, P.; Jeong-Hun, M.; Tae Wan, K.; Hookeun, L. Proteomic Analysis of the Vitreous Body in Proliferative and Non-Proliferative Diabetic Retinopathy. Curr. Proteom. 2021, 18, 143–152. [Google Scholar] [CrossRef]
- Keller, A.; Nesvizhskii, A.I.; Kolker, E.; Aebersold, R. Empirical Statistical Model To Estimate the Accuracy of Peptide Identifications Made by MS/MS and Database Search. Anal. Chem. 2002, 74, 5383–5392. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Nakaya, A. The KEGG databases at GenomeNet. Nucleic Acids Res. 2002, 30, 42–46. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duong, V.-A.; Nam, O.; Jin, E.; Park, J.-M.; Lee, H. Discovery of Post-Translational Modifications in Emiliania huxleyi. Molecules 2021, 26, 2027. https://doi.org/10.3390/molecules26072027
Duong V-A, Nam O, Jin E, Park J-M, Lee H. Discovery of Post-Translational Modifications in Emiliania huxleyi. Molecules. 2021; 26(7):2027. https://doi.org/10.3390/molecules26072027
Chicago/Turabian StyleDuong, Van-An, Onyou Nam, EonSeon Jin, Jong-Moon Park, and Hookeun Lee. 2021. "Discovery of Post-Translational Modifications in Emiliania huxleyi" Molecules 26, no. 7: 2027. https://doi.org/10.3390/molecules26072027