
Bioanalytical Method Development and Validation Study of Neuroprotective Extract of Kashmiri Saffron Using Ultra-Fast Liquid Chromatography-Tandem Mass Spectrometry (UFLC-MS/MS): In Vivo Pharmacokinetics of Apocarotenoids and Carotenoids

 , ,
, , 
Abstract
:
1. Introduction
2. Results
2.1. Optimization of Chromatographic Conditions
2.2. Optimization of Mass Spectrometric Conditions
2.2.1. Carotenoids–Apocarotenoids: Mass Fragmentation Analysis by UFLC-MS/MS
2.2.2. Linearity and Lower Limit of Quantification (LLOQ)
2.3. Bioanalytical Method Validation
2.3.1. Optimization of Analytes Extraction from the Plasma
2.3.2. System Suitability Test
2.3.3. Specificity and Sensitivity
2.3.4. Quality Control Samples
2.3.5. Accuracy and Precision
2.3.6. Extraction Recovery, Dilution Integrity, and Carryover
2.3.7. Matrix Effect and Stability Studies
2.3.8. Application of Bioanalytical Method to Oral Pharmacokinetics Study
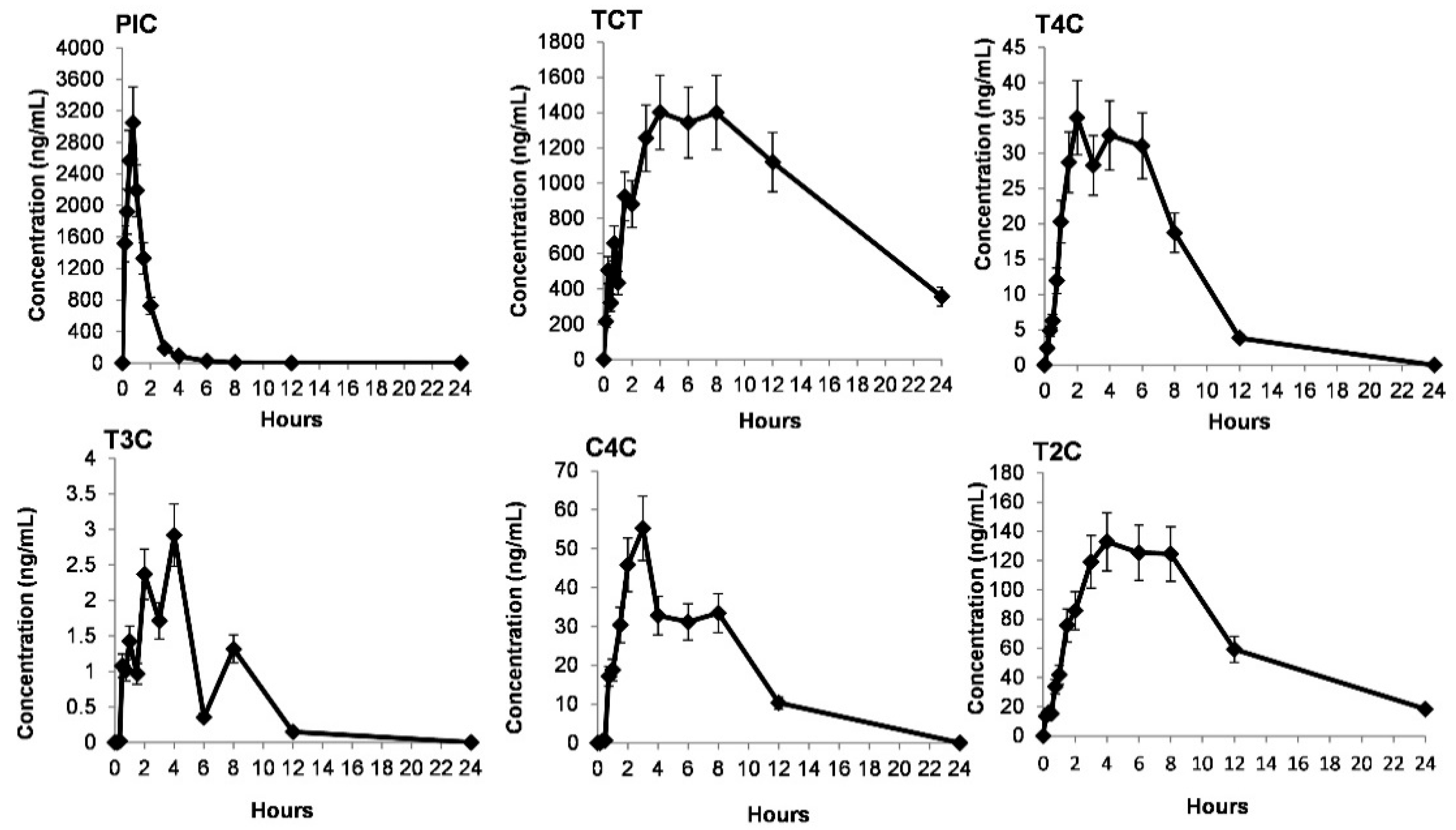
2.4. Pharmacokinetic Results
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Preparation and Standardization of Kashmir Saffron Extract (CSE)
4.3. Animals
4.4. Ultrafast Liquid Chromatography-Mass Spectrometry (UFLC-MS/MS)
4.5. Preparation of Standard Solution, Calibration Standards, and Quality Control Samples
4.5.1. Preparation of Plasma Samples
4.5.2. Linearity and Calibration Curve (CC)
4.6. Bioanalytical Validation: UFLC-MS/MS Method
4.6.1. System Suitability Test
4.6.2. Specificity, Selectivity, and Sensitivity (LLOQ)
4.6.3. Quality Control Samples
4.6.4. Accuracy and Precision
4.6.5. Extraction Recovery, Matrix Effect, and Carryover
4.6.6. Stability Studies of Carotenoids and Apocarotenoids
4.7. Application of Validated Methodology in Investigation of Pharmacokinetic Parameters of CSE
4.8. Pharmacokinetic Study
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hosseinzadeh, H.; Nassiri-Asl, M. Avicenna’s (Ibn Sina) the Canon of Medicine and Saffron (Crocus sativus): A review. Phytother. Res. 2013, 27, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.; Ahmed, H.; Dixit, R.K. Crocus sativus L.: A comprehensive review. Pharmacogn. Rev. 2010, 4, 200. [Google Scholar] [CrossRef] [Green Version]
- Batarseh, Y.S.; Bharate, S.S.; Kumar, V.; Kumar, A.; Vishwakarma, R.A.; Bharate, S.B.; Kaddoumi, A. Crocus sativus extract tightens the blood-brain barrier, reduces amyloid β load and related toxicity in 5XFAD mice. ACS Chem. Neurosci. 2017, 8, 1756–1766. [Google Scholar] [CrossRef]
- Bharate, S.S.; Kumar, V.; Singh, G.; Singh, A.; Gupta, M.; Singh, D.; Kumar, A.; Vishwakarma, R.A.; Bharate, S.B. Preclinical development of Crocus sativus-based botanical lead IIIM-141 for Alzheimer’s disease: Chemical standardization, efficacy, formulation development, pharmacokinetics, and safety pharmacology. ACS Omega 2018, 3, 9572–9585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GI-Geographical Indications Kashmir Saffron. Available online: http://ipindiaservices.gov.in/GIRPublic/Application/Details/635 (accessed on 17 October 2020).
- Carmona, M.; Zalacain, A.; Sánchez, A.M.; Novella, J.L.; Alonso, G.L. Crocetin esters, picrocrocin, and its related compounds present in Crocus sativus stigmas and Gardenia jasminoides fruits. Tentative identification of seven new compounds by LC-ESI-MS. J. Agric. Food Chem. 2006, 54, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Moratalla-López, N.; Bagur, M.J.; Lorenzo, C.; Martínez-Navarro, M.E.; Salinas, M.R.; Alonso, G.L. Bioactivity and bioavailability of the major metabolites of Crocus sativus L. Flower. Molecules 2019, 24, 2827. [Google Scholar] [CrossRef] [Green Version]
- Maggi, L.; Carmona, M.; Sánchez, A.M.; Alonso, G.L. Saffron flavor: Compounds involved, biogenesis, and human perception. Funct. Plant. Biol. 2010, 4, 45–55. [Google Scholar]
- Chrysanthou, A.; Pouliou, E.; Kyriakoudi, A.; Tsimidou, M.Z. Sensory threshold studies of picrocrocin, the major bitter compound of saffron. J. Food Sci. 2016, 81, S189–S198. [Google Scholar] [CrossRef]
- Del Campo, C.P.; Carmona, M.; Maggi, L.; Kanakis, C.D.; Anastasaki, E.G.; Tarantilis, P.A.; Polissiou, M.G.; Alonso, G.L. Picrocrocin content and quality categories in different (345) worldwide samples of Saffron (Crocus sativus L.). J. Agric. Food Chem. 2010, 58, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Alonson, G.L.; Salinas, M.R.; Sánchez-Fernández, M.A.; Garijo, J. Note. Safranal content in spanish Saffron. Food Sci. Technol. Int. 2001, 7, 225–229, . [Google Scholar] [CrossRef]
- Condurso, C.; Cincotta, F.; Tripodi, G.; Verzera, A. Bioactive volatiles in Sicilian (South Italy) saffron: Safranal and its related compounds. J. Essent. Oil Res. 2017, 29, 221–227. [Google Scholar] [CrossRef]
- Alonso, G.L.; Salinas, M.R.; Garijo, J. Method to determine the authenticity of the aroma of Saffron (Crocus sativus L.). J. Food Prot. 1998, 61, 1525–1528. [Google Scholar] [CrossRef] [PubMed]
- Carmona, M.; Zalacain, A.; Salinas, M.R.; Alonso, G.L. A new approach to saffron aroma. Crit. Rev. Food Sci. Nutr. 2007, 47, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, A.M.; Winterhalter, P. Carotenoid cleavage products in saffron (Crocus sativus L.). In Carotenoid Cleavage Products; Winterhalter, P., Ebeler, S.E., Eds.; American Chemical Society: Washington, DC, USA, 2013; Volume 1134, pp. 45–63. [Google Scholar] [CrossRef]
- Kanakis, C.D.; Daferera, D.J.; Tarantilis, P.A.; Polissiou, M.G. Qualitative determination of volatile compounds and quantitative evaluation of safranal and 4-hydroxy-2, 6, 6-trimethyl-1-cyclohexene-1-carboxaldehyde (HTCC) in Greek Saffron. J. Agric. Food Chem. 2004, 52, 4515–4521. [Google Scholar] [CrossRef] [PubMed]
- García-Rodríguez, M.V.; López-Córcoles, H.; Alonso, G.L.; Pappas, C.S.; Polissiou, M.G.; Tarantilis, P.A. Comparative evaluation of an ISO 3632 method and an HPLC-DAD method for safranal quantity determination in Saffron. Food Chem. 2017, 221, 838–843. [Google Scholar] [CrossRef]
- Hatziagapiou, K.; Kakouri, E.; Lambrou, G.I.; Bethanis, K.; Tarantilis, P.A. Antioxidant properties of Crocus sativus L. and its constituents and relevance to neurodegenerative diseases; focus on Alzheimer’s and Parkinson’s disease. Curr. Neuropharmacol. 2019, 17, 377–402. [Google Scholar] [CrossRef]
- Singh, D. Neuropharmacological aspects of Crocus sativus L.: A review of preclinical studies and ongoing clinical research. CNS Neurol. Disord. Drug Targets 2015, 14, 880–902. [Google Scholar] [CrossRef]
- Tsolaki, M.; Karathanasi, E.; Lazarou, I.; Dovas, K.; Verykouki, E.; Karakostas, A.; Georgiadis, K.; Tsolaki, A.; Adam, K.; Kompatsiaris, I.; et al. Efficacy and safety of Crocus sativus L. in patients with mild cognitive impairment: One year single-blind randomized, with parallel groups, clinical trial. J. Alzheimers Dis. 2016, 54, 129–133. [Google Scholar] [CrossRef]
- Akhondzadeh, S.; Sabet, M.S.; Harirchian, M.H.; Togha, M.; Cheraghmakani, H.; Razeghi, S.; Hejazi, S.S.; Yousefi, M.H.; Alimardani, R.; Jamshidi, A.; et al. A 22-week, multicenter, randomized, double-blind controlled trial of Crocus sativus in the treatment of mild-to-moderate Alzheimer’s disease. Psychopharmacology 2010, 207, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, T.N.; Rajasekharan, S.; Badola, D.P.; Shah, D.C. Important medicinal plants of Jammu and Kashmir, I. Kesar (Saffron). Anc. Sci. Life. 1985, 5, 68–73. [Google Scholar]
- Kafi, A.; Koocheki, A.; Rashed, M.H.; Nassiri, M. Saffron (Crocus sativus): Production and Processing; Science Publisher; CRC Press: Boca Raton, FL, USA, 2006; pp. 7–8. [Google Scholar]
- Ballabh, B.; Chaurasia, O.P.; Ahmed, Z.; Singh, S.B. Traditional medicinal plants of cold desert Ladakh-used against kidney and urinary disorders. J. Ethanopharmacol. 2008, 118, 331–339. [Google Scholar] [CrossRef]
- Dash, B. Saffron in Ayurveda and Tibetan medicine. Tibet J. 1976, 1, 59–66. Available online: https://jstor.org/stable/43299808 (accessed on 25 November 2020).
- Sahni, M. Parenting in Saffron. J. Intellect. Prop. Rights 2002, 7, 530–535. Available online: http://nopr.niscair.res.in/bitstream/123456789/4937/1/JIPR%207%286%29%20530-535.pdf (accessed on 25 November 2020).
- National Medicinal Plants Board, Ministry of AYUSH (Ayurveda, Yoga & Naturopathy, Unani, Siddha & Homoeopathy), Government of India, Indian Medicinal plants List. Available online: http://www.medicinalplants.in/ (accessed on 25 November 2020).
- Mazumder, A.G.; Sharma, P.; Patial, V.; Singh, D. Crocin attenuates kindling development and associated cognitive impairments in mice via inhibiting reactive oxygen species-mediated NF-κB activation. Basic Clin. Pharmacol.Toxicol. 2017, 120, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Bani, S.; Pandey, A.; Agnihotri, V.K.; Pathania, V.; Singh, B. Selective Th2 upregulation by Crocus sativus: A neutraceutical spice. Evid. Based Complement Alternat. Med. 2011. [Google Scholar] [CrossRef] [Green Version]
- Sachdeva, J.; Tanwar, V.; Golechha, M.; Siddiqui, K.M.; Nag, T.C.; Ray, R.; Kumari, S.; Arya, D.S. Crocus sativus L.(Saffron) attenuates isoproterenol-induced myocardial injury via preserving cardiac functions and strengthening antioxidant defense system. Exp. Toxicol.Pathol. 2012, 64, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Sarwat, M.; Khan, T.H. Mechanism behind the anti-tumour potential of Saffron (Crocus sativus L.): The molecular perspective. Crit. Rev. Oncol. Hematol. 2017, 115, 27–35. [Google Scholar] [CrossRef]
- Bukhari, S.I.; Pattnaik, B.; Rayees, S.; Kaul, S.; Dhar, M.K. Safranal of Crocus sativus L. inhibits inducible nitric oxide synthase and attenuates asthma in a mouse model of asthma. Phytother. Res. 2015, 29, 617–627. [Google Scholar] [CrossRef]
- Premkumar, K.; Thirunavukkarasu, C.; Abraham, S.K.; Santhiya, S.T.; Ramesh, A. Protective effect of Saffron (Crocus sativus L.) aqueous extract against genetic damage induced by anti-tumor agents in mice. Hum. Exp. Toxicol. 2006, 25, 79–84. [Google Scholar] [CrossRef]
- Mir, M.A.; Ganai, S.A.; Mansoor, S.; Jan, S.; Mani, P.; Masoodi, K.Z.; Amin, H.; Rehman, M.U.; Ahmad, P. Isolation, purification and characterization of naturally derived crocetin beta-d-glucosyl ester from Crocus sativus L. against breast cancer and its binding chemistry with ER-alpha/HDAC2. Saudi J. Biol. Sci. 2020, 27, 975–984. [Google Scholar] [CrossRef]
- Xi, L.; Qian, Z.; Du, P.; Fu, J. Pharmacokinetic properties of crocin (crocetin digentiobiose ester) following oral administration in rats. Phytomedicine 2007, 14, 633–636. [Google Scholar] [CrossRef]
- Christodoulou, E.; Grafakou, M.E.; Skaltsa, E.; Kadoglou, N.; Kostomitsopoulos, N.; Valsami, G. Preparation, chemical characterization, and determination of crocetin’s pharmacokinetics after oral and intravenous administration of Saffron (Crocus sativus L.) aqueous extract to C57/BL 6J mice. J. Pharm. Pharmacol. 2019, 71, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Umigai, N.; Murakami, K.; Ulit, M.V.; Antonio, L.S.; Shirotori, M.; Morikawa, H.; Nakano, T. Thepharmacokinetic profile of crocetin in healthy adult human volunteers after a single oral administration. Phytomedicine 2011, 18, 575–578. [Google Scholar] [CrossRef]
- Almodóvar, P.; Briskey, D.; Rao, A.; Prodanov, M.; Inarejos-García, A.M. Bioaccessibility and Pharmacokinetics of a Commercial Saffron (Crocus sativus L.) Extract. Evid. Based Complementary Altern. Med. 2020, 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karkoula, E.; Dagla, I.V.; Baira, E.; Kokras, N.; Dalla, C.; Skaltsounis, A.L.; Gikas, E.; Tsarbopoulos, A. A novel UHPLC-HRMS based metabolomics strategy enables the discovery of potential neuroactive metabolites in mice plasma, following i.p. administration of the main Crocus sativus L. bioactive component. J. Pharmaceut. Biomed. 2020, 177, 112878. [Google Scholar] [CrossRef]
- United States Food and Drug Administration (USFDA)—Department of Health and Human Services.Bioanalytical Method Validation, Guidance for Industry. 2018. Available online: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf/ (accessed on 17 December 2019).
- European Medicines Agency. Guideline on Bioanalytical Method Validation. 2011. Available online: www.ema.europa.eu/docs/en_GB/document_library/Scientifc_guideline/2011/08/WC500109686.pdf/ (accessed on 17 December 2019).
- Wani, A.; Al Rihani, S.B.; Sharma, A.; Weadick, B.; Govindarajan, R.; Khan, S.U.; Sharma, S.U.; Sharma, P.R.; Dogra, A.; Nandi, U.; et al. Crocetin promotes clearance of amyloid-β by inducing autophagy via the STK11/LKB1-mediated AMPK pathway. Autophagy 2021, 1–20. [Google Scholar] [CrossRef]
- Van Calsteren, M.R.; Bissonnette, M.C.; Cormier, F.; Dufresne, C.; Ichi, T.; LeBlanc, J.Y.; Perreault, D.; Roewer, I. Spectroscopic characterization of crocetin derivatives from Crocus sativus and Gardenia jasminoides. J. Agric. Food Chem. 1997, 45, 1055–1061. [Google Scholar] [CrossRef]
- Ekor, M. The growing use of herbal medicines: Issues relating to adverse reactions and challenges in monitoring safety. Front Phamacol. 2014, 4, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.Z.; Qian, Z.Y. Pharmacokinetics of crocetin in rats. Yao XueXue Bao 2002, 37, 367–369. [Google Scholar]
- Jafarisani, M.; Bathaie, S.Z.; Mousavi, M.F. Saffron carotenoids (crocin and crocetin) binding to human serum albumin as investigated by different spectroscopic methods and molecular docking. J. Biomol. Struct. Dyn. 2018, 36, 1681–1690. [Google Scholar] [CrossRef]
- Lautenschlager, M.; Sendker, J.; Huwel, S.; Galla, H.; Brandt, S.; Dufer, M. Intestinal formation of trans-crocetin from saffron extract (Crocus sativus L.) and in vitro permeation through intestinal and blood-brain barrier. Phytomedicine 2015, 22, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Reddy, C.N.; Bharate, S.B.; Vishwakarma, R.A.; Bharate, S.S. Chemical analysis of Saffron by HPLC based crocetin estimation. J. Pharm. Biomed. Anal. 2020, 181, 113094. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, H.; Tian, X.; Zhao, C.; Cai, L.; Liu, Y.; Jia, L.; Yin, H.X.; Chen, C. Antioxidant potential of crocins and ethanol extracts of Gardenia jasminoides ELLIS and Crocus sativus L. A relationship investigation between antioxidant activity and crocin contents. Food Chem. 2008, 109, 484–492. [Google Scholar] [CrossRef]
- Tong, Y.; Yan, Y.; Zhu, X.; Liu, R.; Gong, F.; Zhang, L.; Wang, P. Simultaneous quantification of crocetin esters and picrocrocin changes in Chinese Saffron by high-performance liquid chromatography-diode array detector during 15 years of storage. Pharmacogn. Mag. 2015, 11, 540–545. [Google Scholar] [CrossRef] [Green Version]
- D’Archivio, A.A.; Di Donato, F.; Foschi, M.; Maggi, M.A.; Ruggieri, F. UHPLC analysis of Saffron (Crocus sativus L.): Optimization of separation using chemometrics and detection of minor crocetin esters. Molecules 2018, 23, 1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samarghandian, S.; Azimi-Nezhad, M.; Farkhondeh, T. Immunomodulatory and antioxidant effects of saffron aqueous extract (Crocus sativus L.) on streptozotocin-induced diabetes in rats. Indian Heart J. 2017, 69, 151–159. [Google Scholar] [CrossRef] [Green Version]
- Brown, P.N.; Chan, M.; Betz, J.M.; Shahid, M.; Cannon, S.; Palissery, J.; Puglisi, M.P. Regulation of nutraceuticals in Canada and the United States. In Nutraceuticals and Human Health: The Food-to-Supplement Paradigm; Spagnuolo, P.A., Ed.; Royal Society of Chemistry: Piccadilly, London, UK, 2020; pp. 7–26. [Google Scholar]
- Tripathi, C.; Girme, A.; Champaneri, S.; Patel, R.J.; Hingorani, L. Nutraceutical regulations: An opportunity in ASEAN countries. Nutrition 2020, 74, 110729. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytes | T4C | T3C | T2C | C4C | TCT | SAF | PIC |
|---|---|---|---|---|---|---|---|
| CSE | 13.76 ± 0.280 | 5.50 ± 0.418 | 0.875 ± 0.0255 | 0.624 ± 0.011 | 0.0378 ± 0.0015 | 0.033 ± 0.00050 | 18.09 ± 0.586 |
| Analyte | Formula | MW (g/mol) | Ionization Mode | Precursor Ion (m/z) | Product ion (m/z) | Dwell Time (msec) | Q1 Pre-Bias (eV) | CE (eV) | Q3 Pre-Bias (eV) | Retention Time, tR (min) |
|---|---|---|---|---|---|---|---|---|---|---|
| T4C | C44H64O24 | 976.38 | -ve | 975.70 | 651.25 | 53.00 | 22.00 | 23.00 | 34.00 | 3.90 |
| 327.20 | 53.00 | 28.00 | 37.00 | 22.00 | ||||||
| T3C | C38H54O19 | 814.33 | -ve | 813.30 | 327.20 | 53.00 | 18.00 | 28.00 | 16.00 | 4.12 |
| 651.20 | 53.00 | 18.00 | 14.00 | 18.00 | ||||||
| T2C | C32H44O14 | 652.27 | -ve | 651.25 | 327.15 | 53.00 | 14.00 | 20.00 | 16.00 | 4.74 |
| 283.20 | 53.00 | 22.00 | 26.00 | 32.00 | ||||||
| C4C | C44H64O24 | 976.38 | -ve | 975.70 | 651.25 | 53.00 | 22.00 | 23.00 | 34.00 | 4.53 |
| 327.20 | 53.00 | 22.00 | 38.00 | 15.00 | ||||||
| TCT | C20H24O4 | 328.17 | -ve | 327.10 | 283.10 | 53.00 | 12.00 | 9.00 | 13.00 | 5.96 |
| 239.35 | 53.00 | 12.00 | 12.00 | 26.00 | ||||||
| PIC | C16H26O7 | 330.17 | +ve | 151.25 | 81.05 | 62.00 | −16.00 | −22.00 | −14.00 | 1.93 |
| 123.15 | 62.00 | −10.00 | −17.00 | −24.00 | ||||||
| SAF | C10H14O | 150.10 | +ve | 151.25 | 81.05 | 53.00 | −16.00 | −22.00 | −14.00 | 6.19 |
| 123.15 | 53.00 | −10.00 | −17.00 | −24.00 | ||||||
| IS1 | C33H40N2O9 | 608.27 | +ve | 609.70 | 195.10 | 53.00 | −22.00 | −40.00 | −20.00 | 4.82 |
| 448.05 | 53.00 | −22.00 | −31.00 | −22.00 | ||||||
| IS2 | C11H12Cl2N2O5 | 322.01 | -ve | 321.00 | 152.00 | 53.00 | 16.00 | 11.00 | 18.00 | 4.42 |
| 257.00 | 53.00 | 11.00 | 16.00 | 15.00 |
| Analyte | Linear Equation | Range (ng/mL) | R2 | LOD (ng/mL) | LLOQ (ng/mL) |
|---|---|---|---|---|---|
| T4C | 10.00 to 3200.00 | y = 0.001106907x + 0.0007873082 | 0.9968 | 2.00 | 10.00 |
| T3C | 10.00 to 3200.00 | y = 0.002373485x − 0.000003492705 | 0.9954 | 2.00 | 10.00 |
| T2C | 10.00 to 3200.00 | y = 0.002490268x − 0.0001086531 | 0.9990 | 5.00 | 10.00 |
| C4C | 10.00 to 3200.00 | y = 0.0002475676x + 0.0003535316 | 0.9960 | 5.00 | 10.00 |
| TCT | 10.00 to 3200.00 | y = 0.002001102x + 0.002474666 | 0.9952 | 2.00 | 10.00 |
| PIC | 10.00 to 3200.00 | y = 0.002674921x + 0.004357420 | 0.9975 | 5.00 | 10.00 |
| SAF | 10.00 to 3200.00 | y= 0.007429112x + 0.02994744 | 0.9985 | 2.00 | 10.00 |
| Analyte | Nominal Concentration (ng/mL) | Intra-Day 01 (n = 6) | Intra-Day 02 (n = 6) | Intra-Day 03 (n = 6) | Intra-Day 04 (n = 6) | Inter-Day (n = 6 × 3) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Precision (RSD, %) | Accuracy (RE, %) | Precision (RSD, %) | Accuracy (RE, %) | Precision (RSD, %) | Accuracy (RE, %) | Precision (RSD, %) | Accuracy (RE, %) | Precision (RSD, %) | Accuracy (RE, %) | ||
| T4C | 10.00 | 6.76 | −2.78 | 6.76 | −2.78 | 7.96 | −1.09 | 5.72 | −6.03 | 6.93 | 5.53 |
| 30.00 | 6.65 | −2.33 | 6.65 | −2.33 | 6.65 | −3.97 | 2.98 | −1.95 | 5.53 | −7.62 | |
| 1600.00 | 1.23 | −2.73 | 1.23 | −2.73 | 3.43 | −6.39 | 2.92 | −7.50 | 1.16 | −8.92 | |
| 2400.00 | 5.15 | −0.19 | 5.15 | −0.19 | 5.48 | −1.94 | 2.25 | −7.77 | 1.69 | 2.15 | |
| TCT | 10.00 | 7.40 | 0.92 | 7.40 | 0.92 | 7.18 | −1.16 | 6.97 | −5.52 | 6.07 | 1.12 |
| 30.00 | 4.95 | −0.63 | 4.95 | −0.63 | 5.67 | −3.12 | 6.31 | −3.91 | 5.74 | −4.83 | |
| 1600.00 | 1.98 | 2.29 | 1.98 | 2.29 | 5.63 | −4.44 | 1.29 | −7.14 | 3.49 | −8.48 | |
| 2400.00 | 5.81 | 1.68 | 5.82 | 1.68 | 4.38 | −1.21 | 1.94 | −3.99 | 2.48 | −1.32 | |
| PIC | 10.00 | 7.04 | −11.03 | 7.04 | −11.03 | 9.96 | −3.71 | 6.70 | 0.25 | 5.24 | 4.63 |
| 30.00 | 5.95 | −6.23 | 5.95 | −6.23 | 8.00 | −3.20 | 4.83 | −0.77 | 10.51 | −5.14 | |
| 1600.00 | 2.50 | −1.50 | 2.50 | −1.50 | 3.99 | −5.72 | 2.90 | −3.97 | 2.08 | −8.32 | |
| 2400.00 | 1.57 | 0.24 | 1.57 | 0.24 | 3.35 | −1.83 | 4.09 | −4.98 | 3.34 | −0.61 | |
| SAF | 10.00 | 5.11 | 4.90 | 5.11 | 4.90 | 6.61 | 3.83 | 10.18 | −4.73 | 7.41 | 6.35 |
| 30.00 | 5.15 | −1.26 | 5.15 | −1.26 | 5.45 | −1.18 | 5.21 | 1.77 | 7.14 | −1.49 | |
| 1600.00 | 2.24 | −2.95 | 2.24 | −2.95 | 3.21 | −4.85 | 2.59 | −7.35 | 2.33 | −7.63 | |
| 2400.00 | 2.91 | −0.20 | 2.91 | −0.20 | 3.88 | −1.51 | 1.96 | −5.11 | 1.95 | 0.66 | |
| Analyte | Nominal Concentration (ng/mL) | Extraction Recovery (%, mean ± SD) | Precision (RSD, %) | Accuracy (RE, %) |
|---|---|---|---|---|
| T4C | 30.00 | 91.18 ± 0.73 | 0.80 | −9.29 |
| 1600.00 | 99.09 ± 1.55 | 1.56 | −1.15 | |
| 2400.00 | 101.60 ± 0.46 | 0.45 | 1.56 | |
| TCT | 30.00 | 106.23 ± 3.62 | 3.41 | 5.99 |
| 1600.00 | 105.22 ± 3.48 | 3.30 | 4.45 | |
| 2400.00 | 101.45 ± 1.01 | 1.00 | −2.18 | |
| PIC | 30.00 | 96.74 ± 1.04 | 1.07 | −4.30 |
| 1600.00 | 102.89 ± 1.76 | 1.71 | 2.04 | |
| 2400.00 | 106.86 ± 1.73 | 1.61 | 4.67 | |
| SAF | 30.00 | 100.72 ± 0.58 | 0.57 | 0.91 |
| 1600.00 | 103.05 ± 2.95 | 2.86 | 2.38 | |
| 2400.00 | 105.29 ± 1.83 | 1.74 | 4.98 |
| Analyte | Nominal Concentration (ng/mL) | Two-Fold Dilution (ng/mL) | Precision (RSD, %) | Accuracy (RE, %) | Four-Fold Dilution (ng/mL) | Precision (RSD, %) | Accuracy (RE, %) |
|---|---|---|---|---|---|---|---|
| T4C | 5000.00 | 4663.09 ± 92.81 | 1.99 | −6.74 | 4642.16 ± 118.87 | 2.56 | −7.16 |
| TCT | 4634.89 ± 137.40 | 2.96 | −7.30 | 4689.90 ± 182.80 | 3.90 | −6.20 | |
| PIC | 4740.06 ± 164.85 | 3.48 | −5.20 | 4744.10 ± 118.81 | 2.50 | −5.12 | |
| SAF | 4706.07 ± 57.69 | 1.23 | −5.88 | 4722.58 ± 62.50 | 1.32 | −5.55 |
| Analyte | Nominal Concentration (ng/mL) | Matrix Effect (%, mean ± SD) | Precision (RSD, %) | Accuracy (RE, %) |
|---|---|---|---|---|
| T4C | 30.00 | 100.03 ± 5.21 | 5.21 | −7.63 |
| 2400.00 | 98.98 ± 4.17 | 4.21 | 3.33 | |
| TCT | 30.00 | 97.68 ± 8.35 | 8.55 | −2.25 |
| 2400.00 | 100.78 ± 2.59 | 2.57 | −2.08 | |
| PIC | 30.00 | 95.88 ± 8.44 | 8.81 | 2.14 |
| 2400.00 | 98.91 ± 6.91 | 6.99 | 0.71 | |
| SAF | 30.00 | 96.43 ± 6.51 | 6.75 | 2.18 |
| 2400.00 | 99.19 ± 2.53 | 2.55 | 1.51 |
| Analyte | Nominal Concentration (ng/mL) | Autosampler Stability (%, mean ± SD) | Precision (RSD, %) | Accuracy (RE, %) | Room Temperature Stability (%, mean ± SD) | Precision (RSD, %) | Accuracy (RE, %) | Freeze-Thaw Stability (%, mean ± SD) | Precision (RSD, %) | Accuracy (RE, %) |
|---|---|---|---|---|---|---|---|---|---|---|
| T4C | 30.00 | 29.01 ± 1.38 | 4.76 | −3.29 | 27.80 ± 1.29 | 4.64 | −7.34 | 28.45 ± 1.06 | 3.74 | −5.18 |
| 2400.00 | 2481.58 ± 86.48 | 3.49 | 3.40 | 2413.86 ± 73.67 | 3.05 | 0.58 | 2369.47 ± 195.39 | 8.25 | −1.27 | |
| TCT | 30.00 | 29.94 ± 1.29 | 4.30 | −0.19 | 28.59 ± 1.18 | 4.12 | −4.69 | 29.79 ± 1.58 | 5.30 | −0.69 |
| 2400.00 | 2467.92 ± 101.06 | 4.09 | 2.83 | 2425.78 ± 112.27 | 4.63 | 1.07 | 2383.90 ± 133.23 | 5.59 | −0.67 | |
| PIC | 30.00 | 29.52 ± 1.26 | 4.26 | −1.61 | 30.40 ± 0.92 | 3.01 | 1.33 | 29.98 ± 1.66 | 5.55 | −0.06 |
| 2400.00 | 2463.69 ± 113.42 | 4.60 | 2.65 | 2406.19 ± 94.01 | 3.91 | −0.26 | 2498.53 ± 111.53 | 4.46 | 4.11 | |
| SAF | 30.00 | 29.97 ± 0.90 | 3.08 | −2.44 | 28.07 ± 0.67 | 2.38 | −6.43 | 29.66 ± 1.42 | 4.78 | −1.13 |
| 2400.00 | 2472.88 ± 0.90 | 4.08 | 3.04 | 2370.67 ± 126.71 | 5.35 | −1.22 | 2499.17 ± 60.75 | 2.43 | 4.13 |
| PK Parameters | T4C | T3C | T2C | C4C | TCT | PIC |
|---|---|---|---|---|---|---|
| Cmax (ng/mL) | 49.27 ± 11.15 | 7.59 ± 4.71 | 160.44 ± 15.17 | 86.14 ± 15.65 | 2076.21 ± 373.61 | 2722.95 ± 231.41 |
| Tmax (h) | 3.5 ± 0.57 | 3.5 ± 1.09 | 4.34 ± 0.51 | 3.25 ± 1.05 | 6.84 ± 1.69 | 0.792 ± 0.10 |
| AUC0-t (h.ng/mL) | 277.04 ± 67.69 | 20.36 ± 9.64 | 1529.1 ± 197.40 | 395.64 ± 113.41 | 23,590 ± 3119.25 | 3691.19 ± 274.38 |
| AUC0-∞ (h.ng/mL) | 370.45 ± 75.64 | 55.46 ± 31.21 | 1676.12 ± 238.38 | 407.21 ± 187.28 | 30,679.13 ± 3706.46 | 3818.76 ± 256.67 |
| t1/2 (h) | 3.36 ± 1.00 | 4.6 ± 2.74 | 5.75 ± 0.73 | 1.57 ± 1.57 | 8.98 ± 2.00 | 0.793 ± 0.078 |
| CL (mL/h/kg) | 19,568.35 ± 4835.41 | 114,958.33 ± 71,000.46 | 234.77 ± 31.12 | 326.37 ± 445.89 | 0.547 ± 0.094 | 1935.88 ± 123.45 |
| Vd (L/kg) | 69.250 ± 16.150 | 325.573 ± 13.099 | 2.006 ± 0.457 | 4.29 ± 1.94 | 0.00617 ± 0.00104 | 2.258 ± 0.3168 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Girme, A.; Pawar, S.; Ghule, C.; Shengule, S.; Saste, G.; Balasubramaniam, A.K.; Deshmukh, A.; Hingorani, L. Bioanalytical Method Development and Validation Study of Neuroprotective Extract of Kashmiri Saffron Using Ultra-Fast Liquid Chromatography-Tandem Mass Spectrometry (UFLC-MS/MS): In Vivo Pharmacokinetics of Apocarotenoids and Carotenoids. Molecules 2021, 26, 1815. https://doi.org/10.3390/molecules26061815
Girme A, Pawar S, Ghule C, Shengule S, Saste G, Balasubramaniam AK, Deshmukh A, Hingorani L. Bioanalytical Method Development and Validation Study of Neuroprotective Extract of Kashmiri Saffron Using Ultra-Fast Liquid Chromatography-Tandem Mass Spectrometry (UFLC-MS/MS): In Vivo Pharmacokinetics of Apocarotenoids and Carotenoids. Molecules. 2021; 26(6):1815. https://doi.org/10.3390/molecules26061815
Chicago/Turabian StyleGirme, Aboli, Sandeep Pawar, Chetana Ghule, Sushant Shengule, Ganesh Saste, Arun Kumar Balasubramaniam, Amol Deshmukh, and Lal Hingorani. 2021. "Bioanalytical Method Development and Validation Study of Neuroprotective Extract of Kashmiri Saffron Using Ultra-Fast Liquid Chromatography-Tandem Mass Spectrometry (UFLC-MS/MS): In Vivo Pharmacokinetics of Apocarotenoids and Carotenoids" Molecules 26, no. 6: 1815. https://doi.org/10.3390/molecules26061815