
2,3-Dihydroferroceno[3,4]pyrrolo[2,1-b]thiazol-5(8b)-ones: Synthesis, Structure and DFT Study on the Mechanism of Chemo- and Diastereoslective Annulations of (Sp)-2-Formylferrocenecarbonyl Fluoride and (Sp)-2-Formylferrocenecarboxylic Acid

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
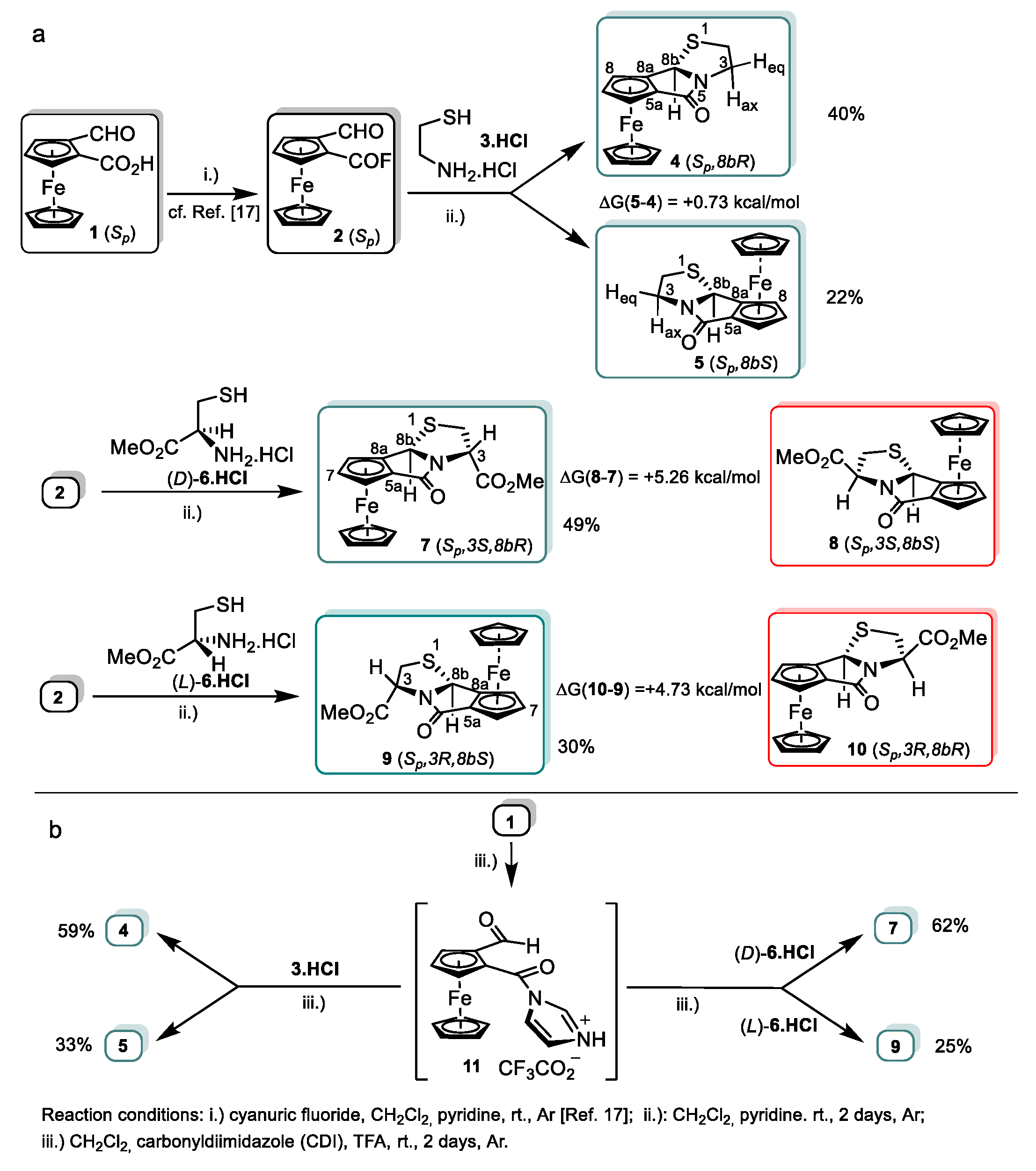
2.1. Synthesis and Structural Elucidation of Diastereomeric Planar Chiral Ferroceno[3,4]pyrrolo[2,1-b]thiazol-5(8bH)-ones
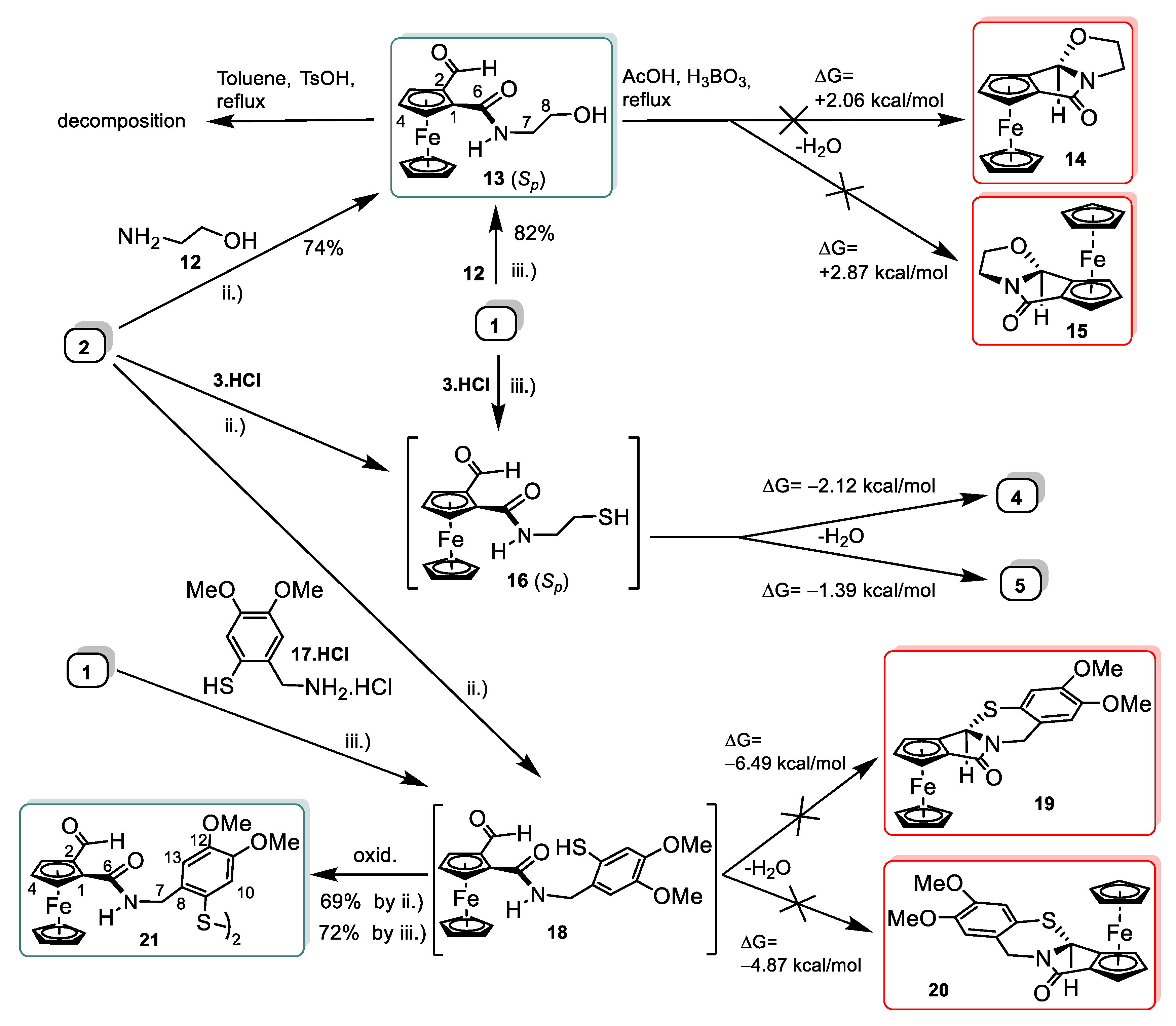
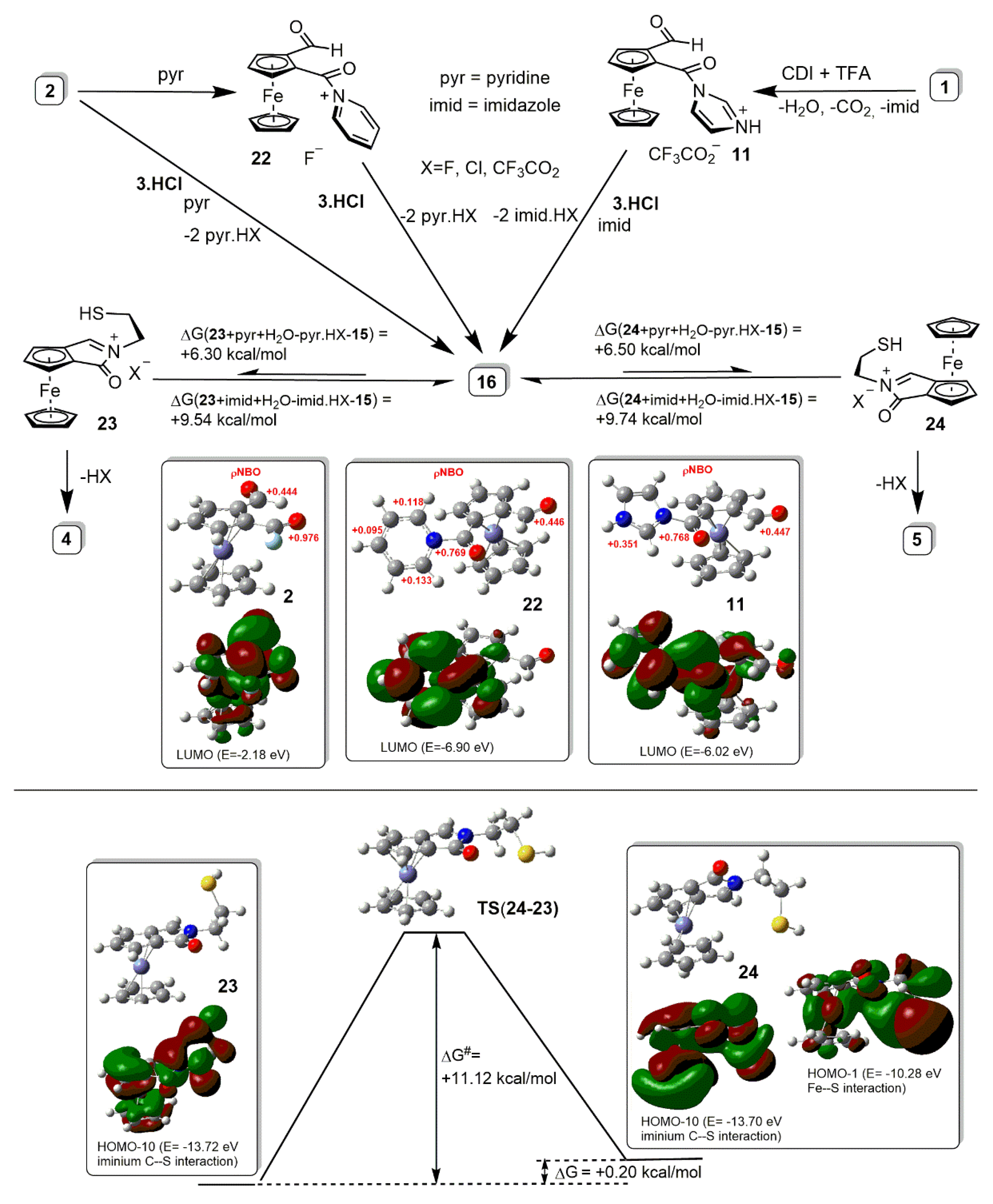
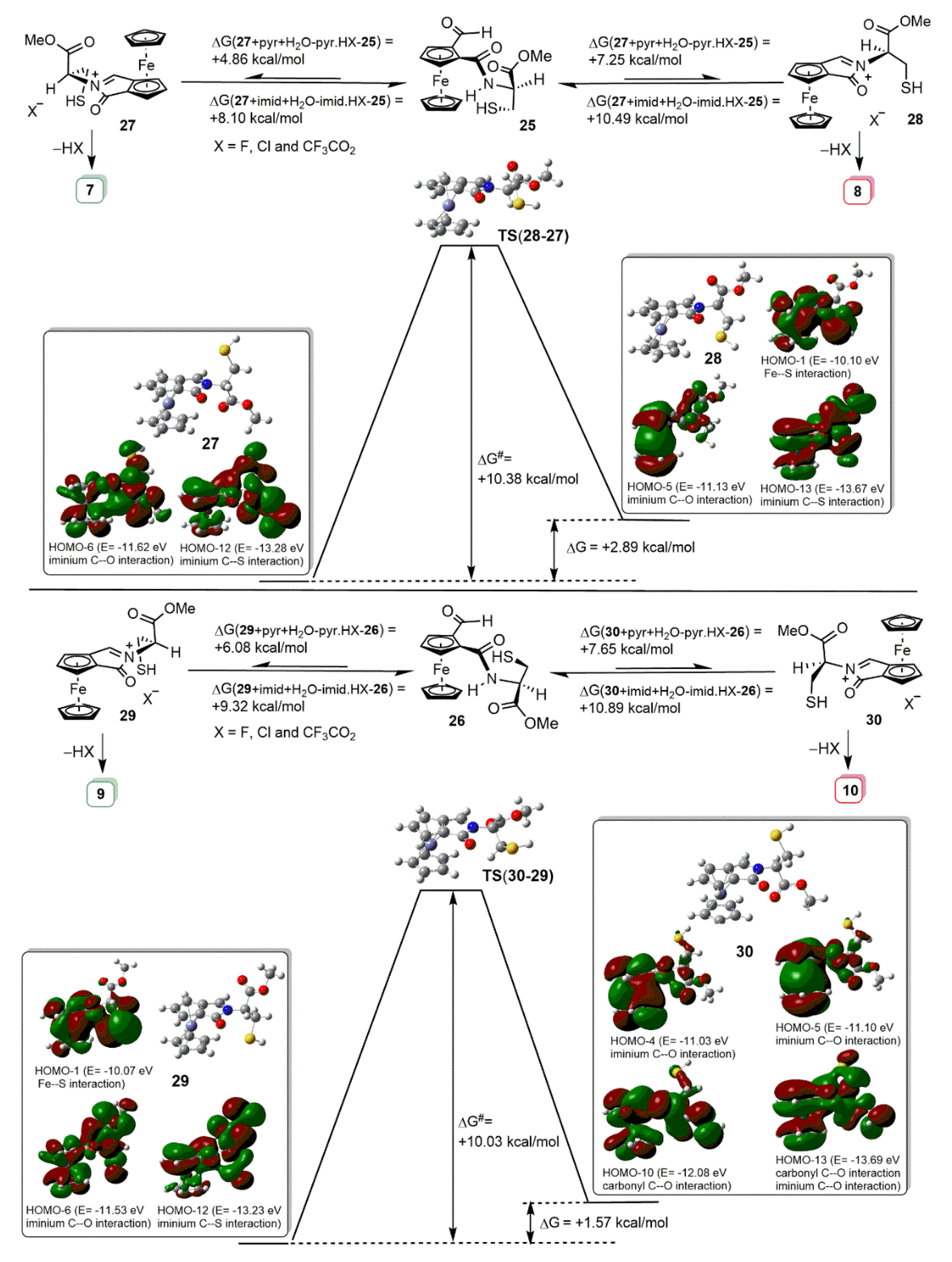
2.2. Synthetic and DFT Study on the Sequence and Diastereoselectivity of the Thiazolidine-Forming Annulation Reactions
3. Materials and Methods
3.1. General Procedure for the Conversions of (Sp)-2-Formylferrocenoylfluoride 2 with N,S- and N,O-Donor Bifunctional Nucleophilic Reagents (3.HCl, (D)-6.HCl, (L)-6.HCl, 12 and 17.HCl) by Method (ii)
3.1.1. (Sp,8bR)-2,3-Dihydroferroceno[3,4]pyrrolo[2,1-b]thiazol-5(8bH)-one (4)
3.1.2. (Sp,8bS)-2,3-Dihydroferroceno[3,4]pyrrolo[2,1-b]thiazol-5(8bH)-one (5)
3.1.3. Methyl (Sp,3S, 8bR)-5-oxo-2,3,5,8b-tetrahydroferroceno[3,4]pyrrolo[2,1-b]thiazol-3-carboxylate (7)
3.1.4. Methyl (Sp,3R, 8bS)-5-oxo-2,3,5,8b-tetrahydroferroceno[3,4]pyrrolo[2,1-b]thiazol-3-carboxylate (9)
3.1.5. (Sp)-2-Formyl-N-(2-hydroxyethyl)ferrocene-carboxamide (13)
3.1.6. (Sp,Sp)-N,N′-((Disulfanediylbis(4,5-dimethoxy-2,1-phenylene))bis(methylene))bis(2-formylferro-cene-carboxamide) (21)
3.2. General Procedure for the Conversions of (Sp)-2-Formylferrocenecarboxylic acid 1 with N,S- and N,O-Donor Bifunctional Nucleophilic Reagents (3.HCl, (D)-6.HCl, (L)-6.HCl, 12 and 17.HCl) by Method (iii)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Togni, A.; Hayashi, T. (Eds.) Ferrocenes (Homogeneous Catalysis, Organic Synthesis, Materials Science); VCH Verlagsgessellschaft: Weinheim, Germany, 1995. [Google Scholar]
- Štěpnička, P. (Ed.) Ferrocenes: Ligands, Material and Biomolecules; John Wiley & Sons: West Sussex, UK, 2008. [Google Scholar]
- Astruc, D. Why is Ferrocene so Exceptional? Eur. J. Inorg. Chem. 2017, 6–29. [Google Scholar] [CrossRef]
- Moriuchi, T.; Hirao, T.; Top, S. Ferrocene–Peptide Bioconjugates. Bioorg. Chem. 2006, 17, 143–175. [Google Scholar]
- Biot, C.; Glorian, G.; Maciejewski, L.A.; Brocard, J.S. Synthesis and antimalarial activity in vitro and in vivo of a new ferrocene-chloroquine analogue. J. Med. Chem. 1997, 40, 3715–3718. [Google Scholar] [CrossRef]
- Delhaes, L.; Abessolo, H.; Biot, C.; Berry, L.; Delcourt, P.; Maciejewski, L.; Brocard, J.; Camus, D.; Dive, D. In vitro and in vivo antimalarial activity of ferrochloroquine, a ferrocenyl analogue of chloroquine against chloroquine-resistant malaria parasites. Parasitol. Res. 2001, 87, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Ornelas, C. Application of ferrocene and its derivatives in cancer research. New J. Chem. 2011, 35, 1973–1985. [Google Scholar] [CrossRef]
- Braga, S.S.; Silva, A.M.S. A new age for iron: Antitumoral ferrocenes. Organometallics 2013, 32, 5626–5639. [Google Scholar] [CrossRef]
- Patra, M.; Gasser, G. The medicinal chemistry of ferrocene and its derivatives. Nat. Rev. Chem. 2017, 66. [Google Scholar]
- Jaouen, G.; Top, S.; Vessicres, A.; Alberto, R. New paradigms for synthetic pathways inspired by bioorganometallic chemistry. J. Organomet. Chem. 2000, 600, 23–26. [Google Scholar] [CrossRef]
- Csókás, D.; Zupkó, I.; Károlyi, B.I.; Drahos, L.; Holczbauer, T.; Palló, A.; Czugler, M.; Csámpai, A. Synthesis, spectroscopy, x-ray analysis and in vitro antiproliferative effect of ferrocenylmethylene-hydrazinylpyridazin-3(2H)-ones and related ferroceno[d]pyridazin-1(2H)-ones. J. Organomet. Chem. 2013, 743, 130–138. [Google Scholar] [CrossRef]
- Csókás, D.; Károlyi, B.I.; Bősze, S.; Szabó, I.; Báti, G.; Drahos, L.; Csámpai, L. 2,3-Dihydroimidazo[1,2-b]ferroceno[d]pyridazines and a 3,4-Dihydro-2H-pyrimido[1,2-b]ferroceno[d]pyridazine: Synthesis, Structure and in Vitro Antiproliferation Activity on Selected Human Cancer Cell Lines. J. Organomet. Chem. 2014, 750, 41–48. [Google Scholar] [CrossRef]
- Jernei, T.; Bősze, S.; Szabó, R.; Hudecz, F.; Majrik, K.; Csámpai, A. N-ferrocenylpyridazinones and new organic analogues: Synthesis, cyclic voltammetry, DFT analysis and in vitro antiproliferative activity associated with ROS-generation. Tetrahedron 2017, 73, 6181–6192. [Google Scholar] [CrossRef] [Green Version]
- Károlyi, B.I.; Bősze, S.; Orbán, E.; Sohár, P.; Drahos, L.; Gál, E.; Csámpai, A. Acylated mono-, bis- and tris-Cinchona-Based Amines Containing Ferrocene or Organic Residues: Synthesis, Structure and in Vitro Antitumor Activity on Selected Human Cancer Cell Lines. Molecules 2010, 17, 2316–2329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocsis, L.; Szabó, I.; Bősze, S.; Jernei, T.; Hudecz, F.; Csámpai, A. Synthesis, structure and in vitro cytostatic activity of ferrocene—Cinchona hybrids. Bioorg. Med. Chem. Lett. 2015, 26, 946–949. [Google Scholar] [CrossRef] [Green Version]
- Bárány, P.; Szabó, R.; Kovács, I.; Czuczi, T.; Szabó, C.L.; Takács, A.; Lajkó, E.; Láng, O.; Kőhidai, L.; Schlosser, G.; et al. Ferrocene-Containing Impiridone (ONC201) Hybrids: Synthesis, DFT Modelling, In Vitro Evaluation, and Structure–Activity Relationships. Molecules 2018, 23, 2248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyömöre, Á.; Csámpai, A. Synthesis and structure of planar chiral ferroceno [d] pyridazinones, the first representatives of a novel class of fused metallocenes. J. Organomet. Chem. 2011, 696, 1626–1631. [Google Scholar] [CrossRef]
- Mroczek, A.; Erre, G.; Taras, R.; Gladiali, S. Chiral ferrocenyl ligands with bidentate pyridine donors. Synthesis and application in Pd-catalyzed asymmetric allylic alkylation of 1,3-diphenylpropenyl-1-esters. Tetrahedron Asymm. 2010, 21, 1921–1927. [Google Scholar] [CrossRef]
- Korotaev, V.Y.; Kutyashev, I.B.; Barkov, A.Y.; Rozhkova, Y.S.; Plekhanova, I.V.; Shklyaev, Y.V.; Sosnovskikh, V.Y. Synthesis of ferrocene annulated trifluoromethylated heterocycles with crispine and lamellarin skeletons. Tetrahedron Lett. 2019, 60, 150916. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, A.-A.; Zhao, R.-J.; Li, F.; Meng, T.-J.; Ishida, N.; Murakami, M.; Zhao, W.-X. Asymmetric synthesis of planar chiral ferrocenes by enantioselective intramolecular C–H arylation of N-(2-Haloaryl) ferrocenecarboxamides. Org. Lett. 2014, 16, 5336–5338. [Google Scholar] [CrossRef]
- Liu, H.-Y.; Mou, R.-Q.; Shun, C.-Z.; Zhang, S.-Y.; Guo, D.-S. Synthesis of ferrocene[c]pyridin-2(1H)-one derivatives via Pd(II)-catalyzed C–H activation reaction under air. Tetrahedron Lett. 2016, 57, 4676–4679. [Google Scholar] [CrossRef]
- Mitsui, T.; Tokoro, Y.; Haraguchi, R.; Sugita, K.; Harada, M.; Fukuzawa, S.; Minami, Y.; Hiyama, T. Synthesis of Ferrocene-Fused Pyrans through Alkynoxy-Directed CH Activation/Cyclization. Bull. Chem. Soc. Jpn. 2018, 91, 839–845. [Google Scholar] [CrossRef]
- Chen, H.; Wang, Y.-X.; Luan, Y.-X.; Ye, M. Enantioselective Twofold C−H Annulation of Formamides and Alkynes without Built-in Chelating Groups. Angew. Chem. Int. Ed. 2020, 59, 9428–9432. [Google Scholar] [CrossRef] [PubMed]
- Fodor, K.J.; Hegedüs, K.; Csomós, P.; Fodor, L.; Gubán, D.; Sohár, P.; Csámpai, A. Synthesis, Structural Elucidation, Cyclic Voltammetry, and Theoretical Modelling of 2-Ferrocenyl-4H-benzo[e][1,3]thiazinesand 2-Aryl-4H-ferroceno[e][1,3]thiazines. Eur. J. Inorg. Chem. 2017, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Xu, J.; Zhang, J.; Xu, X.; Jin, Z. Intramolecular Direct C–H Arylation via a Metallocenic Radical Pathway: Stereospecific Approach to Planar-Chiral Ferrocenes. Org. Lett. 2017, 19, 5709–5712. [Google Scholar] [CrossRef]
- Wurz, R.P.; Lee, E.C.; Ruble, J.C.; Fu, G.C. Synthesis and Resolution of Planar-Chiral Derivatives of 4-(Dimethylamino)pyridine. Adv. Synth. Catal. 2007, 349, 2345–2352. [Google Scholar] [CrossRef]
- Subasinghe, N.L.; Bontems, R.J.; McIntee, E.; Mishra, R.K.; Johnson, R.L. Bicyclic thiazolidine lactam peptidomimetics of the dopamine receptor modulating peptide Pro-Leu-Gly-NH2. J. Med. Chem. 1993, 36, 2356–2361. [Google Scholar] [CrossRef] [PubMed]
- Khalil, E.M.; Pradhan, A.; Ojala, W.H.; Cleason, W.B.; Mishra, R.K.; Johnson, R.L. Synthesis and Dopamine Receptor Modulating Activity of Substituted Bicyclic Thiazolidine Lactam Peptidomimetics of L-Prolyl-L-leucyl-glycinamide. J. Med. Chem. 1999, 42, 2977–2987. [Google Scholar] [CrossRef] [PubMed]
- Aicher, T.D.; Balkan, B.; Bell, P.A.; Brand, L.J.; Cheon, S.H.; Deems, R.O.; Fell, J.B.; Fillers, W.S.; Fraser, J.D.; Gao, J.; et al. Substituted Tetrahydropyrrolo[2,1-b]oxazol-5(6H)-ones and Tetrahydropyrrolo[2,1-b]thiazol-5(6H)-ones as Hypoglycemic Agents. J. Med. Chem. 1998, 41, 4556–4566. [Google Scholar] [CrossRef]
- Schaefer, W.; Friebe, W.-G.; Leinert, H.; Mertens, A.; Poll, T.; von der Saal, W.; Zilch, H.; Nuber, B.; Ziegler, M.L. Non-nucleoside inhibitors of HIV-1 reverse transcriptase: Molecular modeling and x-ray structure investigations. J. Med. Chem. 1993, 36, 726–732. [Google Scholar] [CrossRef]
- Mertens, A.; Zilch, H.; König, B.; Schaefer, W.; Poll, T.; Kampe, W.; Seidel, H.; Leser, U.; Leinert, H. Selective non-nucleoside HIV-1 reverse transcriptase inhibitors. New 2,3-dihydrothiazolo[2,3-a]isoindol-5(9bH)-ones and related compounds with anti-HIV-1 activity. J. Med. Chem. 1993, 36, 2526–2535. [Google Scholar] [CrossRef]
- Allin, S.M.; Vaidya, D.G.; Page, M.I.; Slawin, A.M.Z. Highly diastereoselective synthesis of 2,3-dihydro-9bH-thiazolo[2,3-a]isoindolin-5-ones. Arkivoc 2000, 1, 151–157. [Google Scholar] [CrossRef] [Green Version]
- Pinhoe Melo, T.M.V.D.; Santos, C.I.A.; Rocha Gonsalves, A.M.A.; Paixao, J.A.; Beja, A.M. Synthesis of tricyclic isoindoles and thiazolo[3,2-c][1,3]benzoxazines. Tetrahedron 2004, 60, 3949–3955. [Google Scholar] [CrossRef] [Green Version]
- Mulay, L.N.; Fox, M.E. Magnetic Anisotropy of Ferrocene and Chemical Bonding. J. Chem. Phys. 1963, 38, 760–764. [Google Scholar] [CrossRef]
- McGlinchey, M.J.; Nikitin, K. Direct measurement of the diamagnetic anisotropy of the ferrocenyl moiety: The origin of unusual 1H-NMR shifts in ferrocenyl-triptycenes and barrelenes J. Organomet. Chem. 2014, 751, 809–814. [Google Scholar] [CrossRef]
- Csámpai, A.; Győrfi, A.Z.; Turós, G.I.; Sohár, P. Application of Biginelli reaction to the synthesis of ferrocenylpyrimidones and [3]-ferrocenophane-containing pyrimido[4,5-d]pyrimidinediones. J. Organomet. Chem. 2009, 694, 3667–3673. [Google Scholar] [CrossRef]
- Fodor, K.J.; Kocsis, V.L.; Kiss, K.; Károlyi, B.I.; Szabolcs, Á.; Silaghi-Dumitrescu, L.; Csámpai, A. Comparative evaluation of a Pictet–Spengler protocol in microwave-assisted conversions of tryptamine with aryl- and carboxyaryl aldehydes: Role of ring strain in cyclocondensation of the primarily formed carboxyaryl-substituted β-carbolines. Monatsh. Chem. 2013, 144, 1381–1387. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B. 1992, 45, 13244. [Google Scholar] [CrossRef] [PubMed]
- Godbout, N.; Salahub, D.R.; Andzelm, J.; Wimmer, E. Optimization of Gaussian-type basis sets for localspin density functional calculations. Part, I. Boron through neon, optimization technique and validation. Can. J. Chem. 1992, 70, 560–571. [Google Scholar] [CrossRef] [Green Version]
- Paier, J.; Marsman, M.; Kresse, G. Why does the B3LYP hybrid functional fail for metals? J. Chem. Phys. 2007, 127, 024103. [Google Scholar] [CrossRef]
- Claret, J.; Fernandez, I.; Galvez, C.; Lapouyade, R. Role of dichloromethane in the photocyclization-oxidation of vinylheterocyclic systems. J. Photochem. Photobiol. A Chem. 1991, 55, 347–359. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Eyring, H. The Activated Complex in Chemical Reactions. J. Chem. Phys. 1935, 3, 107–115. [Google Scholar] [CrossRef]
- Peng, C.; Schlegel, H.B. Combining Synchronous Transit and Quasi-Newton Methods for Finding Transition States. Isr. J. Chem. 1993, 33, 449–454. [Google Scholar] [CrossRef]
- Gaussian, M.J.; Frisch, G.W.; Trucks, H.B.; Schlegel, G.E.; Scuseria, M.A.; Robb, J.R.; Cheeseman, G.; Scalmani, V.; Barone, G.A.; Petersson, H.; et al. Fox Gaussian 09, Revision C. 01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovács, Z.; Csámpai, A. 2,3-Dihydroferroceno[3,4]pyrrolo[2,1-b]thiazol-5(8b)-ones: Synthesis, Structure and DFT Study on the Mechanism of Chemo- and Diastereoslective Annulations of (Sp)-2-Formylferrocenecarbonyl Fluoride and (Sp)-2-Formylferrocenecarboxylic Acid. Molecules 2021, 26, 1420. https://doi.org/10.3390/molecules26051420
Kovács Z, Csámpai A. 2,3-Dihydroferroceno[3,4]pyrrolo[2,1-b]thiazol-5(8b)-ones: Synthesis, Structure and DFT Study on the Mechanism of Chemo- and Diastereoslective Annulations of (Sp)-2-Formylferrocenecarbonyl Fluoride and (Sp)-2-Formylferrocenecarboxylic Acid. Molecules. 2021; 26(5):1420. https://doi.org/10.3390/molecules26051420
Chicago/Turabian StyleKovács, Zoltán, and Antal Csámpai. 2021. "2,3-Dihydroferroceno[3,4]pyrrolo[2,1-b]thiazol-5(8b)-ones: Synthesis, Structure and DFT Study on the Mechanism of Chemo- and Diastereoslective Annulations of (Sp)-2-Formylferrocenecarbonyl Fluoride and (Sp)-2-Formylferrocenecarboxylic Acid" Molecules 26, no. 5: 1420. https://doi.org/10.3390/molecules26051420