
Effect of Water Microsolvation on the Excited-State Proton Transfer of 3-Hydroxyflavone Enclosed in γ-Cyclodextrin

 , , , , and
, , , , and
Abstract
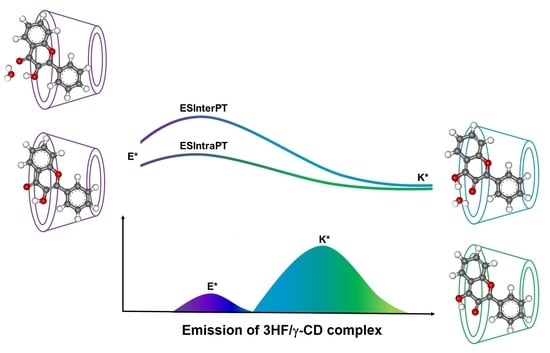
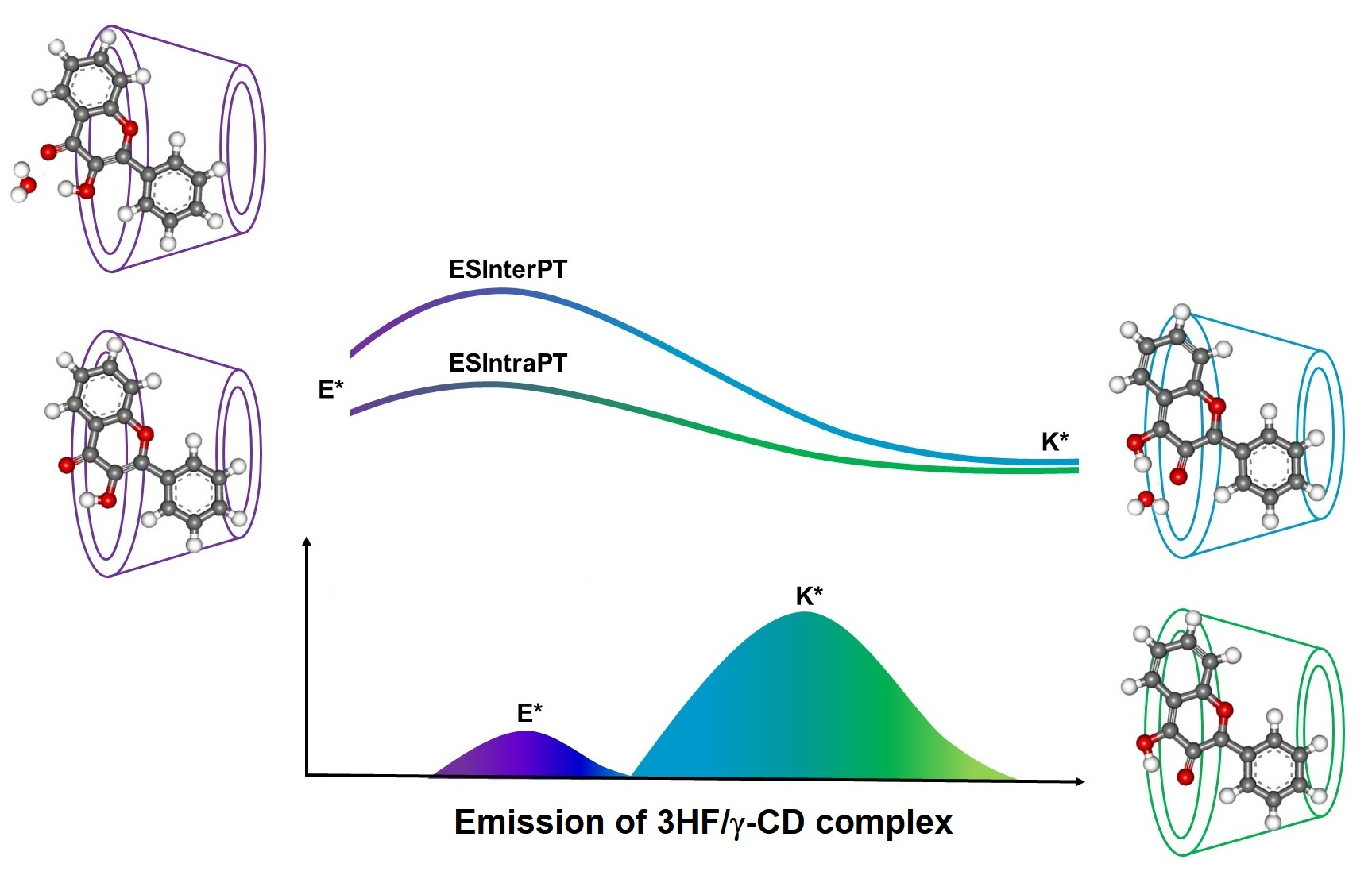
:
1. Introduction
2. Results and Discussion
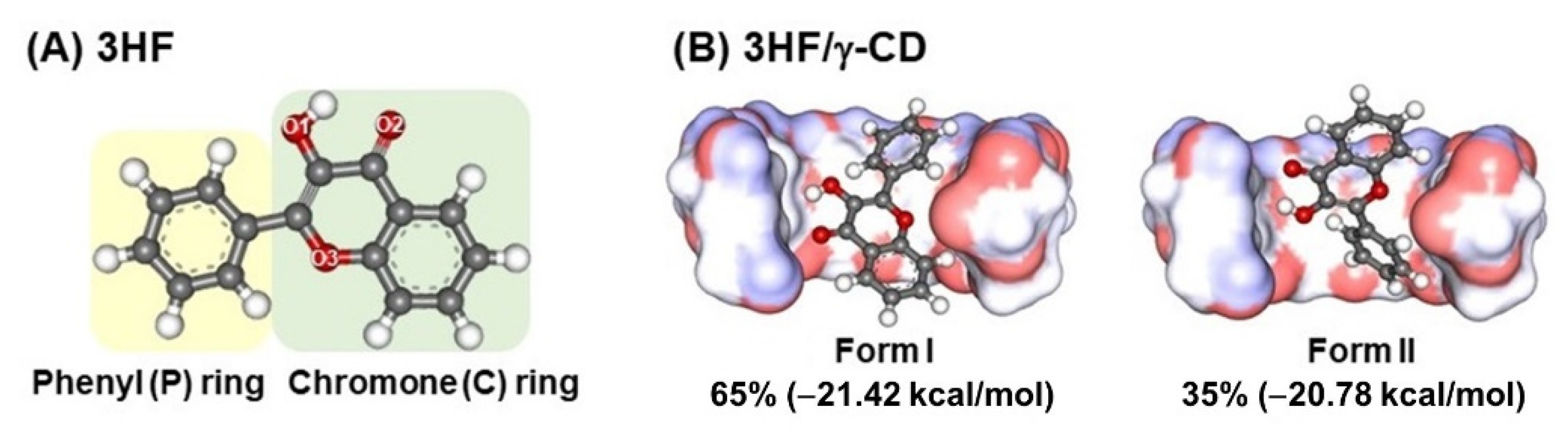
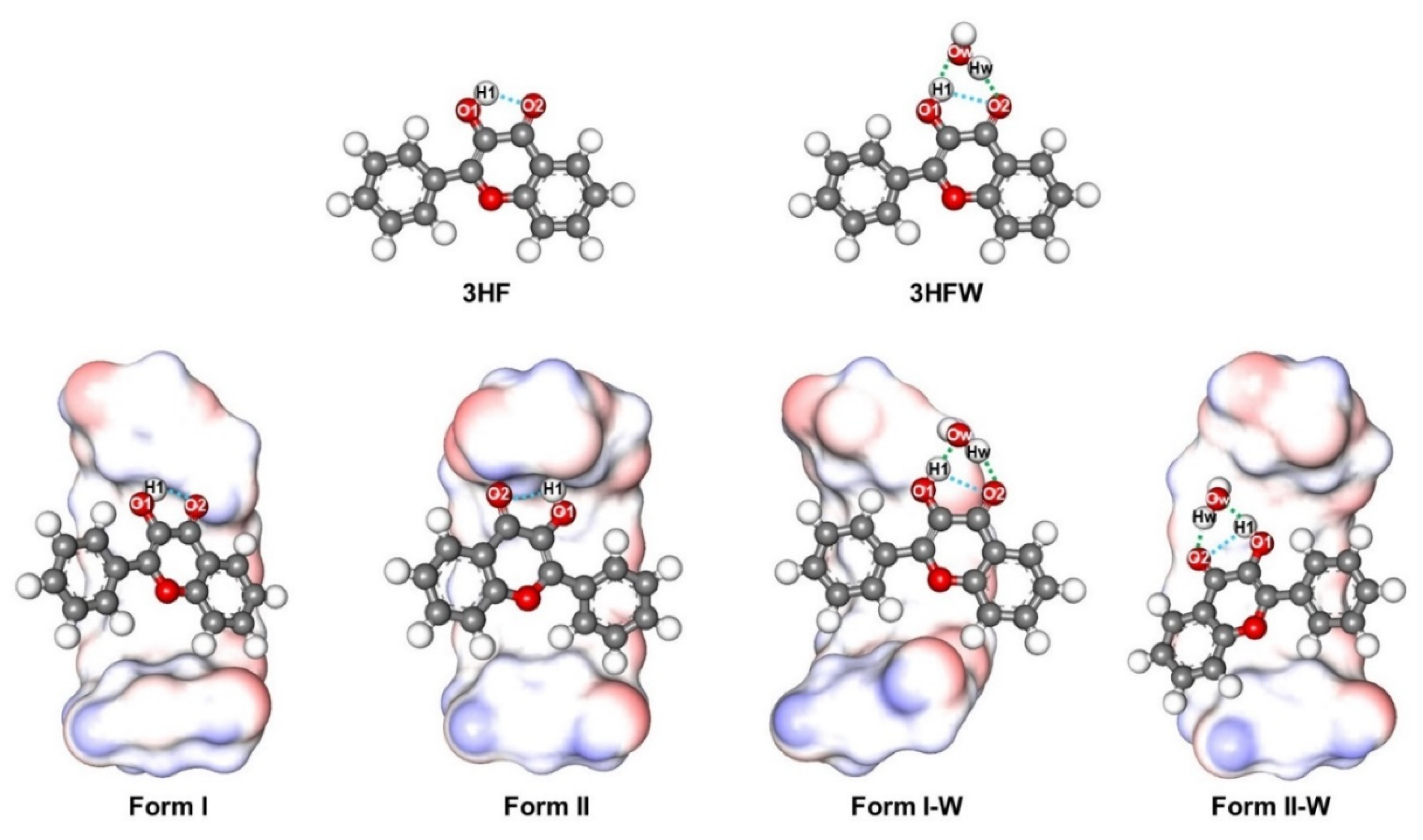
2.1. Possible Inclusion Complexes
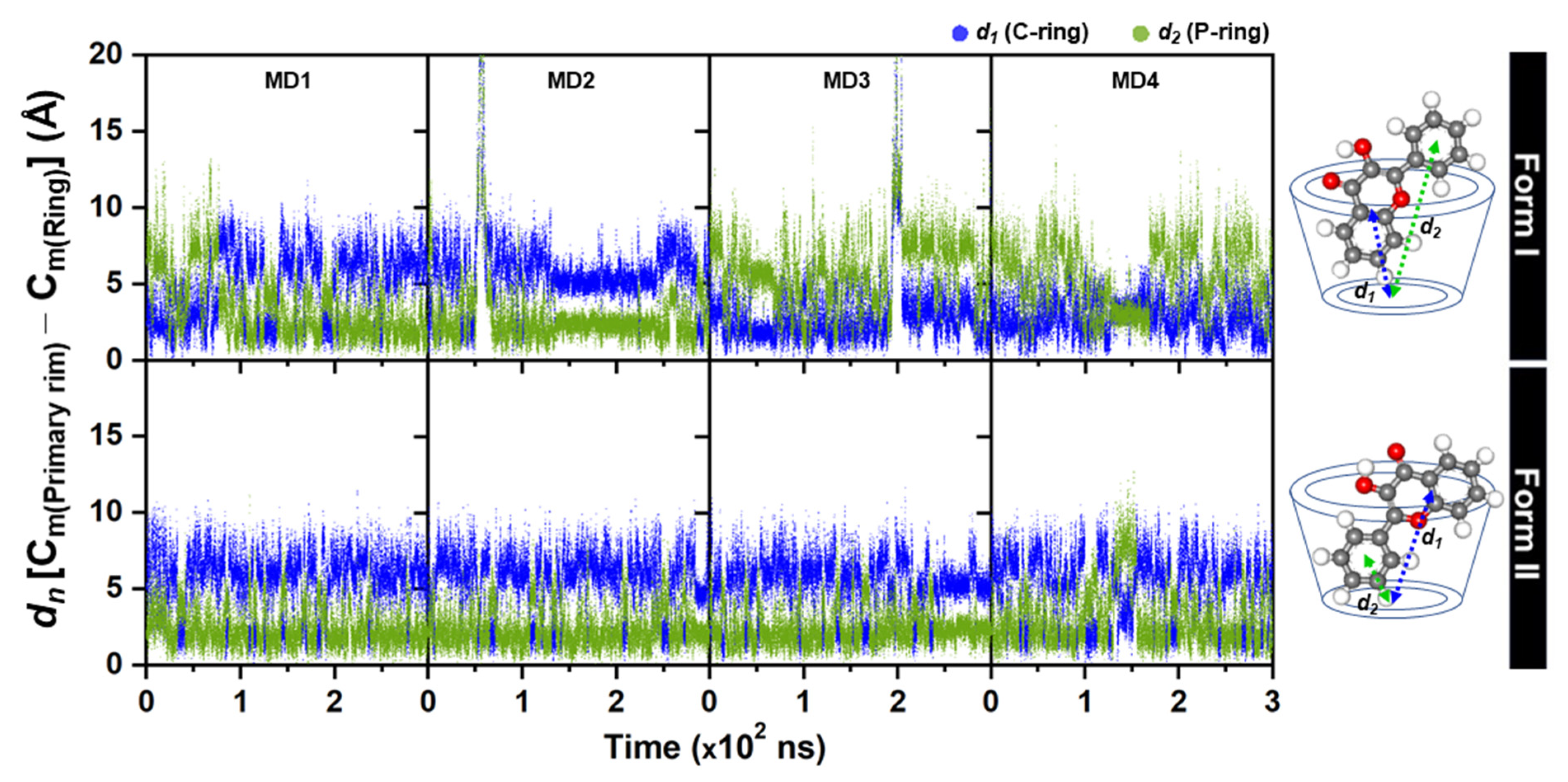
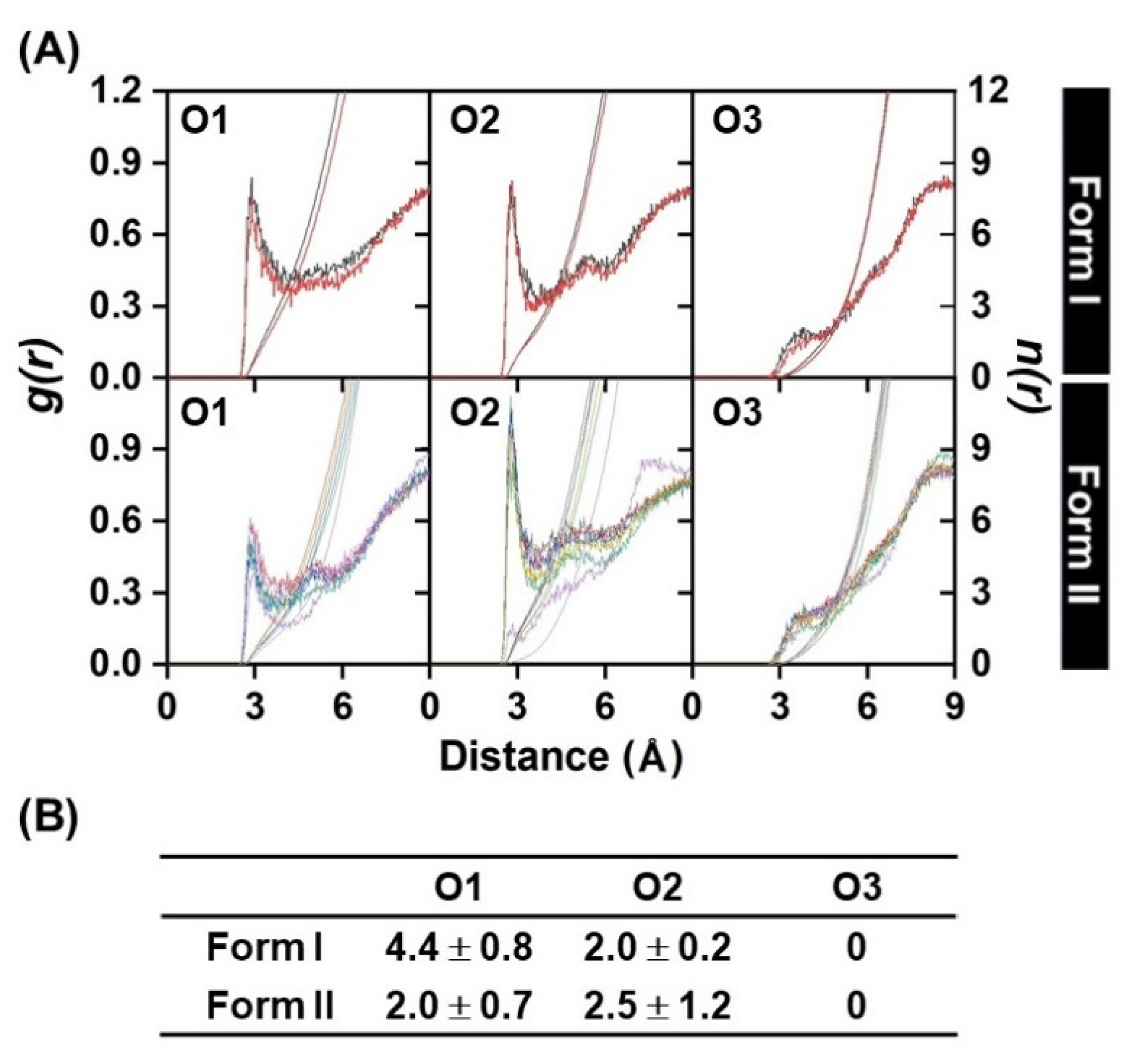
2.2. 3HF Mobility in γ-CD Cavity and Water Accessibility
2.3. Structural Optimizations
2.4. O–H Stretching and Topology Analysis
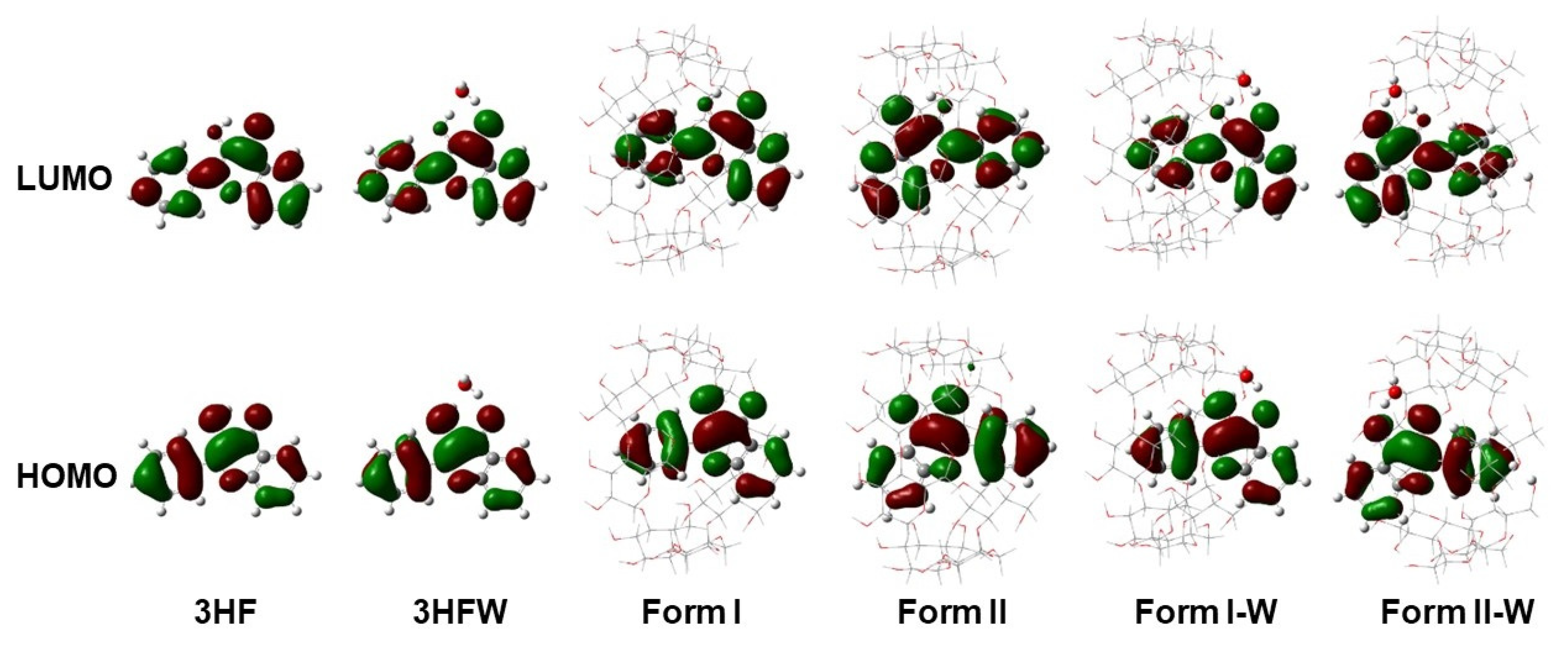
2.5. Frontier MOs Analysis and Simulated Spectra
3. Methodology
3.1. Model Preparation
3.2. Molecular Dynamics Simulations
3.3. Quantum Chemical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Zhou, P.; Han, K. Unraveling the detailed mechanism of excited-state proton transfer. Acc. Chem. Res. 2018, 51, 1681–1690. [Google Scholar] [CrossRef]
- Uzhinov, B.M.; Khimich, M.N. Conformational effects in excited state intramolecular proton transfer of organic compounds. Russ. Chem. Rev. 2011, 80, 553–577. [Google Scholar] [CrossRef]
- Padalkar, V.S.; Seki, S. Excited-state intramolecular proton-transfer (ESIPT)-inspired solid state emitters. Chem. Soc. Rev. 2016, 45, 169–202. [Google Scholar] [CrossRef]
- Weller, A. Innermolekularer protonenübergang im angeregten zustand. Z. Elektrochem. Ber. Bunsenges. Phys. Chem. 1956, 60, 1144–1147. [Google Scholar]
- Goodman, J.; Brus, L.E. Proton transfer and tautomerism in an excited state of methyl salicylate. J. Am. Chem. Soc. 1978, 100, 7472–7474. [Google Scholar] [CrossRef]
- Yoon, M.; Kim, M.; Kim, M.H.; Kang, J.-G.; Sohn, Y.; Kim, I.T. Synthesis and photophysical properties of S, N and Se-modified methyl salicylate derivatives. Inorg. Chim. Acta 2019, 495, 119008. [Google Scholar] [CrossRef]
- Lee, J.; Kwon, J.E.; You, Y.; Park, S.Y. Wholly π-Conjugated low-molecular-weight organogelator that displays triple-channel responses to fluoride ions. Langmuir 2014, 30, 2842–2851. [Google Scholar] [CrossRef]
- Mitra, S.; Tamai, N. Dynamics of photochromism in salicylideneaniline: A femtosecond spectroscopic study. Phys. Chem. Chem. Phys 2003, 5, 4647–4652. [Google Scholar] [CrossRef] [Green Version]
- Borbone, F.; Tuzi, A.; Panunzi, B.; Piotto, S.; Concilio, S.; Shikler, R.; Nabha, S.; Centore, R. On–Off Mechano-responsive Switching of ESIPT Luminescence in Polymorphic N-Salicylidene-4-amino-2-methylbenzotriazole. Cryst. Growth Des. 2017, 17, 5517–5523. [Google Scholar] [CrossRef]
- Liu, B.; Wang, J.; Zhang, G.; Bai, R.; Pang, Y. Flavone-based ESIPT ratiometric chemodosimeter for detection of cysteine in living cells. ACS Appl. Mater. Interfaces 2014, 6, 4402–4407. [Google Scholar] [CrossRef] [PubMed]
- Chou, P.-T.; Chen, Y.-C.; Yu, W.-S.; Cheng, Y.-M. Spectroscopy and dynamics of excited-state intramolecular proton-transfer reaction in 5-hydroxyflavone. Chem. Phys. Lett. 2001, 340, 89–97. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, J.; Li, Y. Theoretical study of the ESIPT process for a new natural product quercetin. Sci. Rep. 2016, 6, 32152. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Deng, Y.; Ji, N.; Zhang, J.; Fan, C.; Ding, T.; Cao, Z.; Li, Y.; Fang, Y. A rationally designed flavone-based ESIPT fluorescent chemodosimeter for highly selective recognition towards fluoride and its application in live-cell imaging. Dyes Pigm. 2019, 166, 473–479. [Google Scholar] [CrossRef]
- Henary, M.M.; Fahrni, C.J. Excited state intramolecular proton transfer and metal ion complexation of 2-(2‘-Hydroxyphenyl)benzazoles in aqueous solution. J. Phys. Chem. A 2002, 106, 5210–5220. [Google Scholar] [CrossRef]
- Das, S.; Chattopadhyay, N. Heteroatom controlled probe-water cluster formation of a series of ESIPT probes: An exploration with fluorescence anisotropy. Chem. Phys. Lett. 2018, 708, 37–41. [Google Scholar] [CrossRef]
- Liu, C.; Wang, F.; Xiao, T.; Chi, B.; Wu, Y.; Zhu, D.; Chen, X. The ESIPT fluorescent probes for N2H4 based on benzothiazol and their applications for gas sensing and bioimaging. Sens. Actuators B 2018, 256, 55–62. [Google Scholar] [CrossRef]
- Mai, V.T.N.; Shukla, A.; Mamada, M.; Maedera, S.; Shaw, P.E.; Sobus, J.; Allison, I.; Adachi, C.; Namdas, E.B.; Lo, S.-C. Low amplified spontaneous emission threshold and efficient electroluminescence from a carbazole derivatized excited-state intramolecular proton transfer dye. ACS Photonics 2018, 5, 4447–4455. [Google Scholar] [CrossRef]
- Munch, M.; Curtil, M.; Vérité, P.M.; Jacquemin, D.; Massue, J.; Ulrich, G. Ethynyl-Tolyl Extended 2-(2′-Hydroxyphenyl)benzoxazole Dyes: Solution and Solid-state Excited-State Intramolecular Proton Transfer (ESIPT) Emitters. Eur. J. Org. Chem. 2019, 2019, 1134–1144. [Google Scholar] [CrossRef]
- Song, Z.; Kwok, R.T.K.; Zhao, E.; He, Z.; Hong, Y.; Lam, J.W.Y.; Liu, B.; Tang, B.Z. A ratiometric fluorescent probe based on ESIPT and AIE processes for alkaline phosphatase activity assay and visualization in living cells. ACS Appl. Mater. Interfaces 2014, 6, 17245–17254. [Google Scholar] [CrossRef]
- Dommett, M.; Crespo-Otero, R. Excited state proton transfer in 2′-hydroxychalcone derivatives. Phys. Chem. Chem. Phys. 2017, 19, 2409–2416. [Google Scholar] [CrossRef]
- Luo, Z.; Liu, B.; Qin, T.; Zhu, K.; Zhao, C.; Pan, C.; Wang, L. Cyclization of chalcone enables ratiometric fluorescence determination of hydrazine with a high selectivity. Sens. Actuators B 2018, 263, 229–236. [Google Scholar] [CrossRef]
- Kwon, J.E.; Park, S.Y. Advanced organic optoelectronic materials: Harnessing excited-state intramolecular proton transfer (ESIPT) process. Adv. Mater. 2011, 23, 3615–3642. [Google Scholar] [CrossRef]
- Ashton, T.D.; Jolliffe, K.A.; Pfeffer, F.M. Luminescent probes for the bioimaging of small anionic species in vitro and in vivo. Chem. Soc. Rev. 2015, 44, 4547–4595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.H.; Kim, J.S.; Sessler, J.L. Small molecule-based ratiometric fluorescence probes for cations, anions, and biomolecules. Chem. Soc. Rev. 2015, 44, 4185–4191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azarias, C.; Budzák, Š.; Laurent, A.D.; Ulrich, G.; Jacquemin, D. Tuning ESIPT fluorophores into dual emitters. Chem. Sci. 2016, 7, 3763–3774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedgwick, A.C.; Wu, L.; Han, H.-H.; Bull, S.D.; He, X.-P.; James, T.D.; Sessler, J.L.; Tang, B.Z.; Tian, H.; Yoon, J. Excited-state intramolecular proton-transfer (ESIPT) based fluorescence sensors and imaging agents. Chem. Soc. Rev. 2018, 47, 8842–8880. [Google Scholar] [CrossRef] [Green Version]
- Massue, J.; Pariat, T.M.; Vérité, P.; Jacquemin, D.; Durko, M.; Chtouki, T.; Sznitko, L.; Mysliwiec, J.; Ulrich, G. Natural born laser dyes: Excited-State Intramolecular Proton Transfer (ESIPT) Emitters and their use in random lasing studies. Nanomaterials 2019, 9, 1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunduz, S.; Goren, A.C.; Ozturk, T. Facile Syntheses of 3-Hydroxyflavones. Org. Lett. 2012, 14, 1576–1579. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, P.K.; Kasha, M. Excited state proton-transfer spectroscopy of 3-hydroxyflavone and quercetin. Chem. Phys. Lett. 1979, 68, 382–385. [Google Scholar] [CrossRef]
- Kasha, M. Proton-transfer spectroscopy. Perturbation of the tautomerization potential. J. Chem. Soc. Faraday Trans. 2 1986, 82, 2379–2392. [Google Scholar] [CrossRef]
- Ash, S.; De, S.P.; Pyne, S.; Misra, A. Excited state intramolecular proton transfer in 3-hydroxy flavone and 5-hydroxy flavone: A DFT based comparative study. J. Mol. Model. 2010, 16, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Chakrabarty, S.; Chattopadhyay, N. Origin of unusually high fluorescence anisotropy of 3-hydroxyflavone in water: Formation of probe–solvent cage-like cluster. J. Phys. Chem. B 2020, 124, 173–180. [Google Scholar] [CrossRef]
- Lazzaroni, S.; Dondi, D.; Mezzetti, A.; Protti, S. Role of solute-solvent hydrogen bonds on the ground state and the excited state proton transfer in 3-hydroxyflavone. A systematic spectrophotometric study. Photochem. Photobiol. Sci. 2018, 17, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Klymchenko, A.S.; Demchenko, A.P. Multiparametric probing of intermolecular interactions with fluorescent dye exhibiting excited state intramolecular proton transfer. Phys. Chem. Chem. Phys 2003, 5, 461–468. [Google Scholar] [CrossRef]
- Klymchenko, A.S.; Pivovarenko, V.G.; Ozturk, T.; Demchenko, A.P. Modulation of the solvent-dependent dual emission in 3-hydroxychromones by substituents. New J. Chem. 2003, 27, 1336–1343. [Google Scholar] [CrossRef]
- Klymchenko, A.S.; Kenfack, C.; Duportail, G.; Mély, Y. Effects of polar protic solvents on dual emissions of 3-hydroxychromones. J. Chem. Sci. 2007, 119, 83–89. [Google Scholar] [CrossRef]
- Klymchenko, A.S.; Demchenko, A.P. Chapter 3 multiparametric probing of microenvironment with solvatochromic fluorescent dyes. Meth. Enzymol. 2008, 450, 37–58. [Google Scholar]
- Ghosh, D.; Pradhan, A.K.; Mondal, S.; Begum, N.A.; Mandal, D. Proton transfer reactions of 4′-chloro substituted 3-hydroxyflavone in solvents and aqueous micelle solutions. Phys. Chem. Chem. Phys 2014, 16, 8594–8607. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Batuta, S.; Das, S.; Begum, N.A.; Mandal, D. Proton transfer dynamics of 4′-N,N-dimethylamino-3-hydroxyflavone observed in hydrogen-bonding solvents and aqueous micelles. J. Phys. Chem. B 2015, 119, 5650–5661. [Google Scholar] [CrossRef]
- Salaeh, R.; Prommin, C.; Chansen, W.; Kerdpol, K.; Daengngern, R.; Kungwan, N. The effect of protic solvents on the excited state proton transfer of 3-hydroxyflavone: A TD-DFT static and molecular dynamics study. J. Mol. Liq. 2018, 252, 428–438. [Google Scholar] [CrossRef]
- Zhao, J.; Ji, S.; Chen, Y.; Guo, H.; Yang, P. Excited state intramolecular proton transfer (ESIPT): From principal photophysics to the development of new chromophores and applications in fluorescent molecular probes and luminescent materials. Phys. Chem. Chem. Phys. 2012, 14, 8803–8817. [Google Scholar] [CrossRef]
- Santos, F.S.; Ramasamy, E.; Ramamurthy, V.; Rodembusch, F.S. Excited state chemistry of flavone derivatives in a confined medium: ESIPT emission in aqueous media. Photochem. Photobiol. Sci. 2014, 13, 992–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartl, K.; Funk, A.; Gerhards, M. IR/UV spectroscopy on jet cooled 3-hydroxyflavone (H2O)n (n = 1,2) clusters along proton transfer coordinates in the electronic ground and excited states. J. Chem. Phys. 2008, 129, 234306. [Google Scholar] [CrossRef] [PubMed]
- Stamm, A.; Maué, D.; Gerhards, M. Structural rearrangement by isomer-specific infrared excitation in the neutral isolated dihydrated cluster of 3-hydroxyflavone. J. Phys. Chem. Lett. 2018, 9, 4360–4366. [Google Scholar] [CrossRef]
- Banerjee, A.; Sengupta, P.K. Encapsulation of 3-hydroxyflavone and fisetin in β-cyclodextrins: Excited state proton transfer fluorescence and molecular mechanics studies. Chem. Phys. Lett. 2006, 424, 379–386. [Google Scholar] [CrossRef]
- Das, S.; Chattopadhyay, N. Supramolecular inclusion-assisted disruption of probe-solvent network. ChemistrySelect 2017, 2, 6078–6081. [Google Scholar] [CrossRef]
- Pahari, B.; Chakraborty, S.; Sengupta, P.K. Encapsulation of 3-hydroxyflavone in γ-cyclodextrin nanocavities: Excited state proton transfer fluorescence and molecular docking studies. J. Mol. Struct. 2011, 1006, 483–488. [Google Scholar] [CrossRef]
- Szejtli, J. Introduction and general overview of cyclodextrin chemistry. Chem. Rev. 1998, 98, 1743–1754. [Google Scholar] [CrossRef]
- Arunkumar, E.; Forbes, C.C.; Smith, B.D. Improving the properties of organic dyes by molecular encapsulation. Eur. J. Org. Chem. 2005, 2005, 4031. [Google Scholar] [CrossRef]
- Hou, X.; Ke, C.; Bruns, C.J.; McGonigal, P.R.; Pettman, R.B.; Stoddart, J.F. Tunable solid-state fluorescent materials for supramolecular encryption. Nat. Commun. 2015, 6, 6884. [Google Scholar] [CrossRef] [Green Version]
- Pahari, B.; Sengupta, B.; Chakraborty, S.; Thomas, B.; McGowan, D.; Sengupta, P.K. Contrasting binding of fisetin and daidzein in γ-cyclodextrin nanocavity. J. Photochem. Photobiol. B 2013, 118, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Douhal, A. Ultrafast guest dynamics in cyclodextrin nanocavities. Chem. Rev. 2004, 104, 1955–1976. [Google Scholar] [CrossRef]
- Kicuntod, J.; Khuntawee, W.; Wolschann, P.; Pongsawasdi, P.; Chavasiri, W.; Kungwan, N.; Rungrotmongkol, T. Inclusion complexation of pinostrobin with various cyclodextrin derivatives. J. Mol. Graph. Model. 2016, 63, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Nutho, B.; Khuntawee, W.; Rungnim, C.; Pongsawasdi, P.; Wolschann, P.; Karpfen, A.; Kungwan, N.; Rungrotmongkol, T. Binding mode and free energy prediction of fisetin/β-cyclodextrin inclusion complexes. Beilstein J. Org. Chem. 2014, 10, 2789–2799. [Google Scholar] [CrossRef] [Green Version]
- Dennington, R.; Keith, T.A.; Millam, J.M. (Eds.) Gauss View; Version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Gaussian 16, Revision, C.01; Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A. (Eds.) Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Kaiyawet, N.; Rungrotmongkol, T.; Hannongbua, S. Effect of Halogen substitutions on dUMP to stability of thymidylate synthase/dUMP/mTHF ternary complex using molecular dynamics simulation. J. Chem. Inf. Model. 2013, 53, 1315–1323. [Google Scholar] [CrossRef]
- Mahalapbutr, P.; Chusuth, P.; Kungwan, N.; Chavasiri, W.; Wolschann, P.; Rungrotmongkol, T. Molecular recognition of naphthoquinone-containing compounds against human DNA topoisomerase IIα ATPase domain: A molecular modeling study. J. Mol. Liq. 2017, 247, 374–385. [Google Scholar] [CrossRef]
- Mahalapbutr, P.; Wonganan, P.; Charoenwongpaiboon, T.; Prousoontorn, M.; Chavasiri, W.; Rungrotmongkol, T. Enhanced solubility and anticancer potential of mansonone G By β-cyclodextrin-based host-guest complexation: A computational and experimental study. Biomolecules 2019, 9, 545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, K.N.; Yongye, A.B.; Tschampel, S.M.; González-Outeiriño, J.; Daniels, C.R.; Foley, B.L.; Woods, R.J. GLYCAM06: A generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 2008, 29, 622–655. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Luty, B.A.; van Gunsteren, W.F. Calculating electrostatic interactions using the particle−particle particle−mesh method with nonperiodic long-range interactions. J. Phys. Chem. 1996, 100, 2581–2587. [Google Scholar] [CrossRef]
- Jacquemin, D.; Perpète, E.A.; Scalmani, G.; Frisch, M.J.; Assfeld, X.; Ciofini, I.; Adamo, C. Time-dependent density functional theory investigation of the absorption, fluorescence, and phosphorescence spectra of solvated coumarins. J. Chem. Phys. 2006, 125, 164324. [Google Scholar] [CrossRef] [PubMed]
- Corni, S.; Cammi, R.; Mennucci, B.; Tomasi, J. Electronic excitation energies of molecules in solution within continuum solvation models: Investigating the discrepancy between state-specific and linear-response methods. J. Chem. Phys. 2005, 123, 134512. [Google Scholar] [CrossRef]
- Jacquemin, D.; Mennucci, B.; Adamo, C. Excited-state calculations with TD-DFT: From benchmarks to simulations in complex environments. Phys. Chem. Chem. Phys 2011, 13, 16987–16998. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wen, K.; Feng, S.; Yuan, H.; An, B.; Zhu, Q.; Guo, X.; Zhang, J. Tunable excited-state intramolecular proton transfer reactions with NH or OH as a proton donor: A theoretical investigation. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2017, 187, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Prommin, C.; Kerdpol, K.; Saelee, T.; Kungwan, N. Effects of π-expansion, an additional hydroxyl group, and substitution on the excited state single and double proton transfer of 2-hydroxybenzaldehyde and its relative compounds: TD-DFT static and dynamic study. New J. Chem. 2019, 43, 19107–19119. [Google Scholar] [CrossRef]
- Sun, C.; Su, X.; Zhou, Q.; Shi, Y. Regular tuning of the ESIPT reaction of 3-hydroxychromone-based derivatives by substitution of functional groups. Org. Chem. Front. 2019, 6, 3093–3100. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Important Bond Distance (Å) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| S0 State | S1 State | |||||||||
| O1–H1 | O2⋯H1 | Ow⋯H1 | Ow–Hw | O2⋯Hw | O1–H1 | O2⋯H1 | Ow⋯H1 | Ow–Hw | O2⋯Hw | |
| 3HF | 0.983 | 1.920 | 1.017 | 1.706 | ||||||
| 3HFW | 1.000 | 2.387 | 1.630 | 0.984 | 1.723 | 1.069 | 2.439 | 1.394 | 1.008 | 1.572 |
| Form I | 0.979 | 1.978 | 1.002 | 1.804 | ||||||
| Form II | 0.981 | 2.177 | 0.996 | 2.080 | ||||||
| Form I-W | 1.009 | 2.390 | 1.585 | 0.977 | 1.808 | 1.030 | 2.415 | 1.503 | 0.978 | 1.903 |
| Form II-W | 1.004 | 2.429 | 1.612 | 0.983 | 1.733 | 1.078 | 2.452 | 1.375 | 1.006 | 1.581 |
| Compound | Wavenumber (cm−1) | |||||
|---|---|---|---|---|---|---|
| O1–H1 | Ow–Hw | |||||
| S0 | S1 | S0 | S1 | |||
| 3HF | 3544 | 3002 | 541 | |||
| 3HFW | 3129 | 2599 | 530 | 3510 | 3074 | 436 |
| Form I | 3617 | 3250 | 367 | |||
| Form II | 3574 | 3330 | 244 | |||
| Form I-W | 2967 | 2575 | 392 | 3662 | 3499 | 163 |
| Form II-W | 3063 | 2577 | 486 | 3526 | 3111 | 415 |
| Compounds | UV/Vis Absorption | Emission | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| λabs of E (nm) | Eex (eV) | f | MOs (%contribution) | λemis of E* (nm) | Eex (eV) | f | λemis of K* (nm) | Eex (eV) | f | |
| 3HF | 341 | 3.63 | 0.4907 | HOMO→LUMO (98%) | 390 | 3.18 | 0.5454 | 514 | 2.41 | 0.4632 |
| 3HFW | 351 | 3.53 | 0.4857 | HOMO→LUMO (98%) | 408 | 3.04 | 0.5335 | 504 | 2.46 | 0.4619 |
| Form I | 345 | 3.59 | 0.4618 | HOMO→LUMO (98%) | 392 | 3.16 | 0.5123 | 524 | 2.36 | 0.3692 |
| Form II | 345 | 3.59 | 0.4081 | HOMO→LUMO (98%) | 393 | 3.15 | 0.4689 | 528 | 2.35 | 0.3792 |
| Form I-W | 353 | 3.51 | 0.4288 | HOMO→LUMO (98%) | 399 | 3.11 | 0.5213 | 529 | 2.34 | 0.3708 |
| Form II-W | 350 | 3.54 | 0.4174 | HOMO→LUMO (98%) | 412 | 3.01 | 0.4575 | 508 | 2.44 | 0.3899 |
| Complex | Relative Energy (kcal/mol) | |||||
|---|---|---|---|---|---|---|
| S0 | S1 | ΔE at S0 | ΔE at S1 | |||
| E | K | E* | K* | |||
| 3HF | 0 | 10.74 | 75.19 | 65.81 | 10.74 | −9.38 |
| 3HFW | 0 | 10.27 | 74.92 | 63.41 | 10.27 | −11.51 |
| Form I | 0 | 16.85 | 74.79 | 71.16 | 16.85 | −3.63 |
| Form II | 0 | 8.47 | 72.39 | 64.68 | 8.47 | −7.71 |
| Form I-W | 0 | 6.63 | 71.24 | 63.43 | 6.63 | −7.81 |
| Form II-W | 0 | 8.35 | 73.12 | 64.72 | 8.35 | −8.40 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kerdpol, K.; Daengngern, R.; Sattayanon, C.; Namuangruk, S.; Rungrotmongkol, T.; Wolschann, P.; Kungwan, N.; Hannongbua, S. Effect of Water Microsolvation on the Excited-State Proton Transfer of 3-Hydroxyflavone Enclosed in γ-Cyclodextrin. Molecules 2021, 26, 843. https://doi.org/10.3390/molecules26040843
Kerdpol K, Daengngern R, Sattayanon C, Namuangruk S, Rungrotmongkol T, Wolschann P, Kungwan N, Hannongbua S. Effect of Water Microsolvation on the Excited-State Proton Transfer of 3-Hydroxyflavone Enclosed in γ-Cyclodextrin. Molecules. 2021; 26(4):843. https://doi.org/10.3390/molecules26040843
Chicago/Turabian StyleKerdpol, Khanittha, Rathawat Daengngern, Chanchai Sattayanon, Supawadee Namuangruk, Thanyada Rungrotmongkol, Peter Wolschann, Nawee Kungwan, and Supot Hannongbua. 2021. "Effect of Water Microsolvation on the Excited-State Proton Transfer of 3-Hydroxyflavone Enclosed in γ-Cyclodextrin" Molecules 26, no. 4: 843. https://doi.org/10.3390/molecules26040843