
A Mathematical Radiobiological Model (MRM) to Predict Complex DNA Damage and Cell Survival for Ionizing Particle Radiations of Varying Quality

 , ,
, , 
Abstract
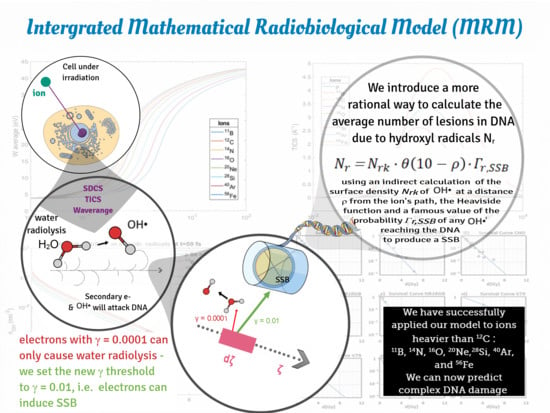
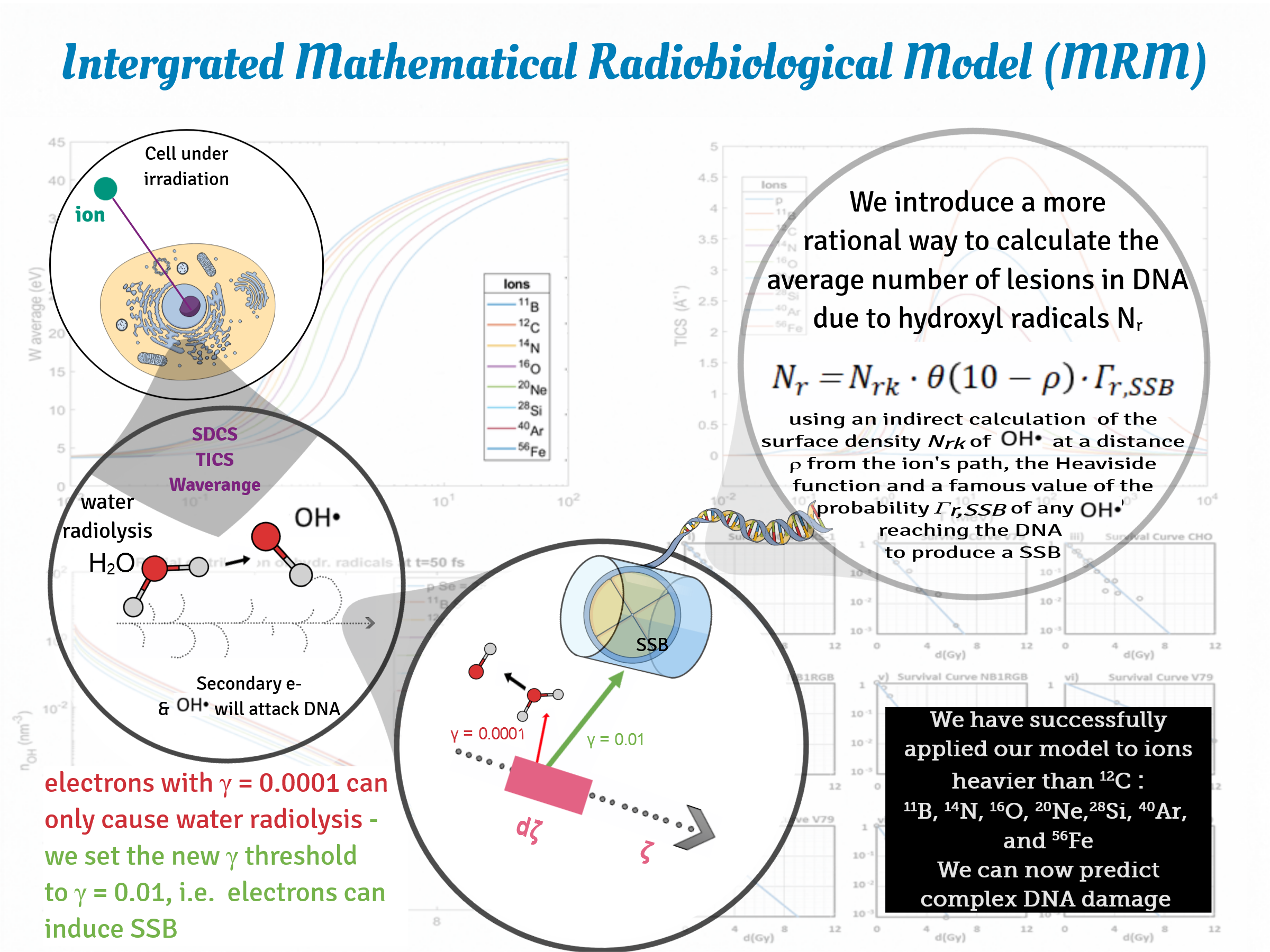
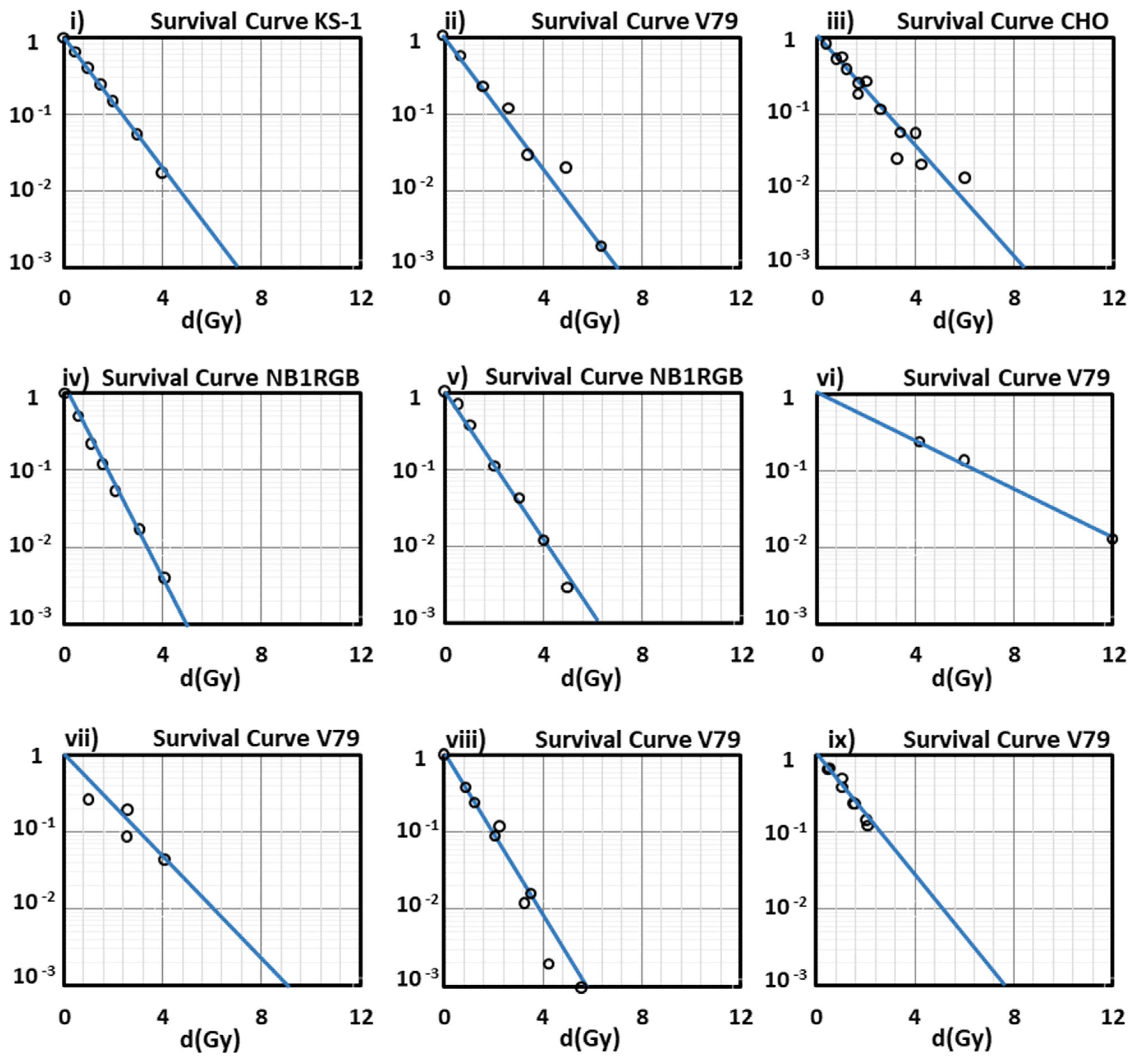
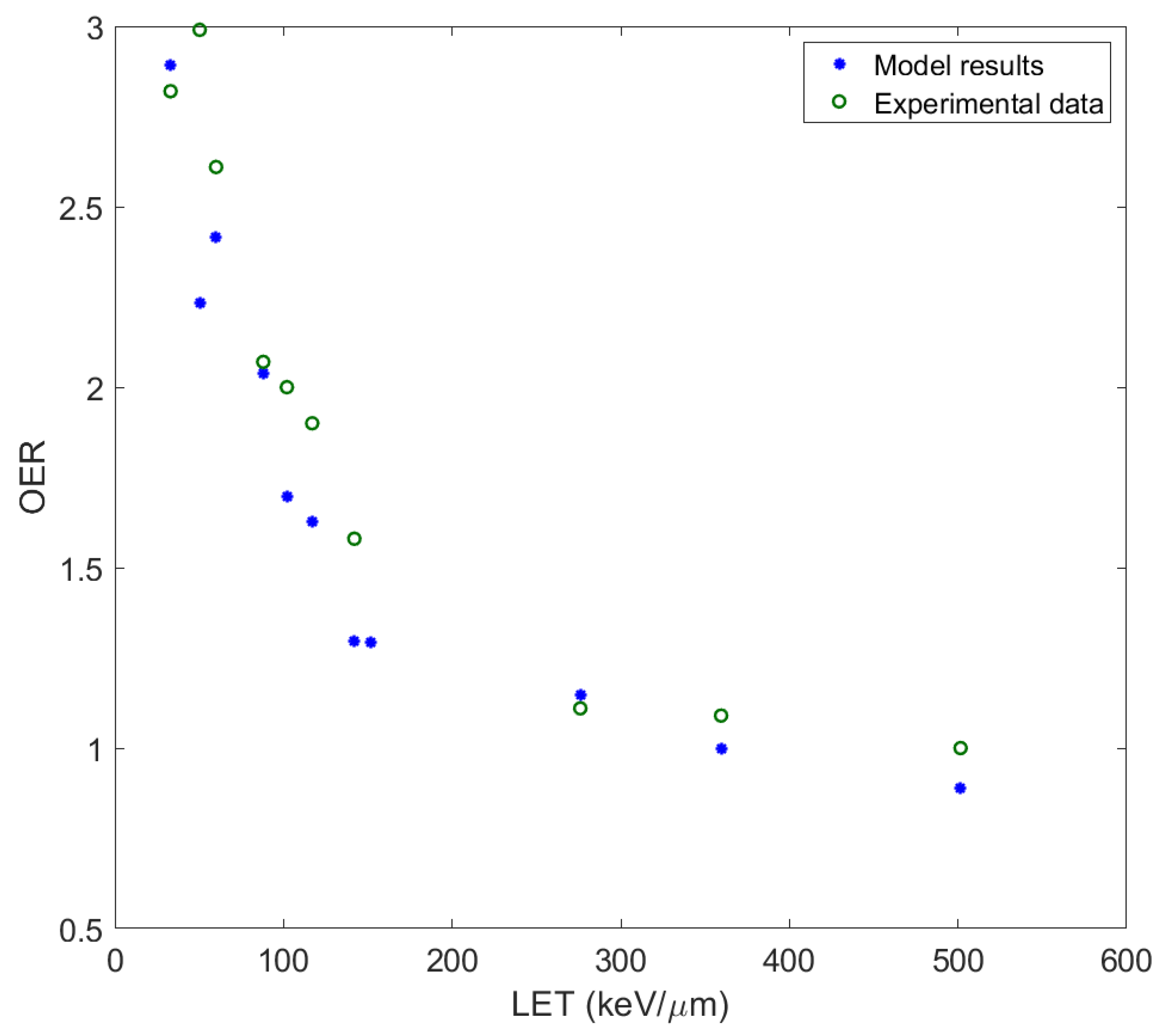
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
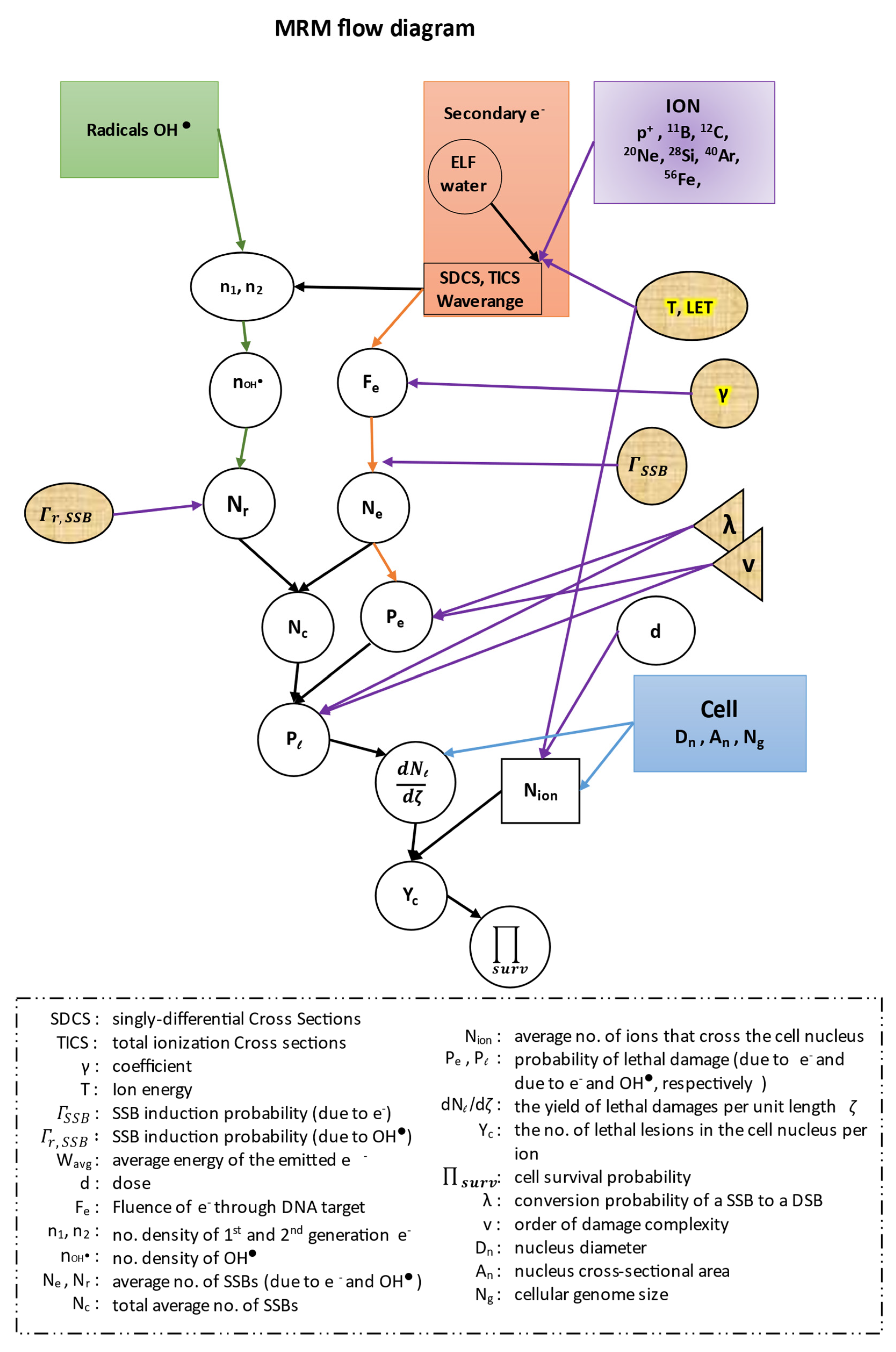
Calculation of Complex DNA Damage and Survival in an Irradiated Cell
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harrabi, S.B.; Bougatf, N.; Mohr, A.; Haberer, T.; Herfarth, K.; Combs, S.E.; Debus, J.; Adeberg, S. Dosimetric advantages of proton therapy over conventional radiotherapy with photons in young patients and adults with low-grade glioma. Strahlenther. Und Onkol. 2016, 192, 759–769. [Google Scholar] [CrossRef] [Green Version]
- Funayama, T. Heavy-Ion Microbeams for Biological Science: Development of System and Utilization for Biological Experiments in QST-Takasaki. Quantum Beam Sci. 2019, 3, 13. [Google Scholar] [CrossRef] [Green Version]
- Hamada, N. What are the intracellular targets and intratissue target cells for radiation effects? Radiat. Res. 2014, 181, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Byrne, H.L.; Domanova, W.; McNamara, A.L.; Incerti, S.; Kuncic, Z. The cytoplasm as a radiation target: An in silico study of microbeam cell irradiation. Phys. Med. Biol. 2015, 60, 2325–2337. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.D.; Yu, V.K.; Georgakilas, A.G.; Koumenis, C.; Park, J.H.; Carlson, D.J. Effects of radiation quality and oxygen on clustered DNA lesions and cell death. Radiat. Res. 2011, 176, 587–602. [Google Scholar] [CrossRef] [PubMed]
- Mihailescu, D. Biophysical models in hadrontherapy. J. Adv. Res. Phys. 2012, 3. [Google Scholar]
- Scholz, M.; Elsässer, T. Biophysical models in ion beam radiotherapy. Adv. Space Res. 2007, 40, 1381–1391. [Google Scholar] [CrossRef]
- Nikjoo, H.; Emfietzoglou, D.; Liamsuwan, T.; Taleei, R.; Liljequist, D.; Uehara, S. Radiation track, DNA damage and response-a review. Rep. Prog. Phys. Phys. Soc. 2016, 79, 116601. [Google Scholar] [CrossRef]
- Sanche, L. Low energy electron-driven damage in biomolecules. Eur. Phys. J. D. 2005, 35, 367–390. [Google Scholar] [CrossRef]
- Kellerer, A.M.; Rossi, H.H. The theory of dual radiation action. Curr. Top. Radiat. Res. Q. 1972, 8, 85–158. [Google Scholar]
- Chadwick, K.H.; Leenhouts, H.P. A molecular theory of cell survival. Phys. Med. Biol. 1973, 18, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.J. The linear-quadratic model is an appropriate methodology for determining isoeffective doses at large doses per fraction. Semin. Radiat. Oncol. 2008, 18, 234–239. [Google Scholar] [CrossRef] [Green Version]
- Belkić, D.; Belkić, K. Padé–Froissart exact signal-noise separation in nuclear magnetic resonance spectroscopy. J. Phys. B At. Mol. Opt. Phys. 2011, 44, 125003. [Google Scholar] [CrossRef] [Green Version]
- Park, C.; Papiez, L.; Zhang, S.; Story, M.; Timmerman, R.D. Universal survival curve and single fraction equivalent dose: Useful tools in understanding potency of ablative radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, M.; Carlone, M. Mechanistic formulation of a lineal-quadratic-linear (LQL) model: Split-dose experiments and exponentially decaying sources. Med. Phys. 2010, 37, 4173–4181. [Google Scholar] [CrossRef] [PubMed]
- Butts, J.J.; Katz, R. Theory of RBE for heavy ion bombardment of dry enzymes and viruses. Radiat. Res. 1967, 30, 855–871. [Google Scholar] [CrossRef] [Green Version]
- Katz, R.; Ackerson, B.; Homayoonfar, M.; Sharma, S.C. Inactivation of Cells by Heavy Ion Bombardment. Radiat. Res. 1971, 47, 402–425. [Google Scholar] [CrossRef]
- Katz, R.; Zachariah, R.; Cucinotta, F.A.; Zhang, C. Survey of cellular radiosensitivity parameters. Radiat. Res. 1994, 140, 356–365. [Google Scholar] [CrossRef] [Green Version]
- Waligórski, M.P.R.; Grzanka, L.; Korcyl, M. The principles of Katz’s cellular track structure radiobiological model. Radiat. Prot. Dosim. 2015, 166, 49–55. [Google Scholar] [CrossRef]
- Scholz, M.; Kellerer, A.M.; Kraft-Weyrather, W.; Kraft, G. Computation of cell survival in heavy ion beams for therapy. The model and its approximation. Radiat. Environ. Biophys. 1997, 36, 59–66. [Google Scholar] [CrossRef]
- Elsässer, T.; Krämer, M.; Scholz, M. Accuracy of the Local Effect Model for the Prediction of Biologic Effects of Carbon Ion Beams In Vitro and In Vivo. Int. J. Radiat. Oncol. Biol. Phys. 2008, 71, 866–872. [Google Scholar] [CrossRef]
- Elsässer, T.; Weyrather, W.K.; Friedrich, T.; Durante, M.; Iancu, G.; Krämer, M.; Kragl, G.; Brons, S.; Winter, M.; Weber, K.J.; et al. Quantification of the relative biological effectiveness for ion beam radiotherapy: Direct experimental comparison of proton and carbon ion beams and a novel approach for treatment planning. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 1177–1183. [Google Scholar] [CrossRef]
- Friedrich, T.; Scholz, U.; Elsässer, T.; Durante, M.; Scholz, M. Calculation of the biological effects of ion beams based on the microscopic spatial damage distribution pattern. Int. J. Radiat. Biol. 2012, 88, 103–107. [Google Scholar] [CrossRef]
- Stewart, R.D.; Carlson, D.J.; Butkus, M.P.; Hawkins, R.; Friedrich, T.; Scholz, M. A comparison of mechanism-inspired models for particle relative biological effectiveness (RBE). Med. Phys. 2018, 45, e925–e952. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, R.B. A microdosimetric-kinetic model of cell death from exposure to ionizing radiation of any LET, with experimental and clinical applications. Int. J. Radiat. Biol. 1996, 69, 739–755. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, R.B. A Microdosimetric-Kinetic Model for the Effect of Non-Poisson Distribution of Lethal Lesions on the Variation of RBE with LET. Radiat. Res. 2003, 160, 61–69. [Google Scholar] [CrossRef]
- Carlson, D.J.; Stewart, R.D.; Semenenko, V.A.; Sandison, G.A. Combined use of Monte Carlo DNA damage simulations and deterministic repair models to examine putative mechanisms of cell killing. Radiat. Res. 2008, 169, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Semenenko, V.A.; Stewart, R.D. A fast Monte Carlo algorithm to simulate the spectrum of DNA damages formed by ionizing radiation. Radiat. Res. 2004, 161, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Semenenko, V.A.; Stewart, R.D. Fast Monte Carlo simulation of DNA damage formed by electrons and light ions. Phys. Med. Biol. 2006, 51, 1693–1706. [Google Scholar] [CrossRef] [Green Version]
- Jeggo, P.A.; Löbrich, M. DNA double-strand breaks: Their cellular and clinical impact? Oncogene 2007, 26, 7717–7719. [Google Scholar] [CrossRef]
- Foray, N.; Monroco, C.; Marples, B.; Hendry, J.H.; Fertil, B.; Goodhead, D.T.; Arlett, C.F.; Malaise, E.P. Repair of radiation-induced DNA double-strand breaks in human fibroblasts is consistent with a continuous spectrum of repair probability. Int. J. Radiat. Biol. 1998, 74, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Joiner, M.C.; Marples, B.; Lambin, P.; Short, S.C.; Turesson, I. Low-dose hypersensitivity: Current status and possible mechanisms. Int. J. Radiat. Oncol. Biol. Phys. 2001, 49, 379–389. [Google Scholar] [CrossRef]
- Ferlazzo, M.L.; Sonzogni, L.; Granzotto, A.; Bodgi, L.; Lartin, O.; Devic, C.; Vogin, G.; Pereira, S.; Foray, N. Mutations of the Huntington’s disease protein impact on the ATM-dependent signaling and repair pathways of the radiation-induced DNA double-strand breaks: Corrective effect of statins and bisphosphonates. Mol. Neurobiol. 2014, 49, 1200–1211. [Google Scholar] [CrossRef] [PubMed]
- Deschavanne, P.J.; Fertil, B. A review of human cell radiosensitivity in vitro. Int. J. Radiat. Oncol. Biol. Phys. 1996, 34, 251–266. [Google Scholar] [CrossRef]
- McMahon, S.J.; McNamara, A.L.; Schuemann, J.; Paganetti, H.; Prise, K.M. A general mechanistic model enables predictions of the biological effectiveness of different qualities of radiation. Sci. Rep. 2017, 7, 10790. [Google Scholar] [CrossRef] [Green Version]
- McMahon, S.J.; Schuemann, J.; Paganetti, H.; Prise, K.M. Mechanistic Modelling of DNA Repair and Cellular Survival Following Radiation-Induced DNA Damage. Sci. Rep. 2016, 6, 33290. [Google Scholar] [CrossRef] [Green Version]
- Carante, M.P.; Aimè, C.; Cajiao, J.J.T.; Ballarini, F. BIANCA, a biophysical model of cell survival and chromosome damage by protons, C-ions and He-ions at energies and doses used in hadrontherapy. Phys. Med. Biol. 2018, 63, 075007. [Google Scholar] [CrossRef]
- Tello Cajiao, J.J.; Carante, M.P.; Bernal Rodriguez, M.A.; Ballarini, F. Proximity effects in chromosome aberration induction: Dependence on radiation quality, cell type and dose. Dna Repair 2018, 64, 45–52. [Google Scholar] [CrossRef]
- Wang, W.; Li, C.; Qiu, R.; Chen, Y.; Wu, Z.; Zhang, H.; Li, J. Modelling of Cellular Survival Following Radiation-Induced DNA Double-Strand Breaks. Sci. Rep. 2018, 8, 16202. [Google Scholar] [CrossRef]
- Plante, I.; Ponomarev, A.; Patel, Z.; Slaba, T.; Hada, M. RITCARD: Radiation-Induced Tracks, Chromosome Aberrations, Repair and Damage. Radiat. Res. 2019, 192, 282–298. [Google Scholar] [CrossRef] [PubMed]
- Surdutovich, E.; Solov’yov, A.V. Multiscale approach to the physics of radiation damage with ions. Eur. Phys. J. D 2014, 68, 353. [Google Scholar] [CrossRef] [Green Version]
- Verkhovtsev, A.; Surdutovich, E.; Solov’yov, A.V. Predictive Assessment of Biological Damage Due to Ion Beams. In Nanoscale Insights into Ion-Beam Cancer Therapy; Solov’yov, A.V., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 359–377. [Google Scholar] [CrossRef]
- Solov‘yov, A.V.; Surdutovich, E.; Scifoni, E.; Mishustin, I.; Greiner, W. Physics of ion beam cancer therapy: A multiscale approach. Phys. Rev. E Stat. Nonlin Soft Matter Phys. 2009, 79, 011909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surdutovich, E.; Solov’yov, A.V. Multiscale modeling for cancer radiotherapies. Cancer Nanotechnol. 2019, 10, 6. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Molina, R.; Abril, I.; de Vera, P.; Kyriakou, I.; Emfietzoglou, D. Role of the interaction processes in the depth-dose distribution of proton beams in liquid water. Int. J. Mod. Phys. Conf. Ser. 2012, 373, 012015. [Google Scholar] [CrossRef] [Green Version]
- De Vera, P.; Garcia-Molina, R.; Abril, I. Propagation of swift protons in liquid water and generation of secondary electrons in biomaterials. In Nanoscale Insights into Ion-Beam Cancer Therapy; Springer: Berlin/Heidelberg, Germany, 2017; pp. 61–98. [Google Scholar]
- Haume, K.; de Vera, P.; Verkhovtsev, A.; Surdutovich, E.; Mason, N.J.; Solov’yov, A.V. Transport of secondary electrons through coatings of ion-irradiated metallic nanoparticles. Eur. Phys. J. D 2018, 72, 116. [Google Scholar] [CrossRef] [Green Version]
- De Vera, P.; Mason, N.J.; Surdutovich, E.; Solov’yov, A.V. Thermo-mechanical damage of biomolecules under ion-beam radiation. In Nanoscale Insights into Ion-Beam Cancer Therapy; Springer: Berlin/Heidelberg, Germany, 2017; pp. 339–357. [Google Scholar]
- Surdutovich, E.; Solov’yov, A.V. Multiscale Physics of Ion-Beam Cancer Therapy. In Nanoscale Insights into Ion-Beam Cancer Therapy; Solov’yov, A.V., Ed.; Spinger: Berlin/Heidelberg, Germany, 2017; pp. 1–60. [Google Scholar] [CrossRef]
- Zel’dovich, Y.B.; Raĭzer, Y.P. Shock Waves. In Physics of Shock Waves and High-Temperature Hydrodynamic Phenomena; Academic Press Inc.: New York, NY, USA; San Francisco, CA, USA; London, UK, 1966; Volume 2, pp. 45–67. [Google Scholar]
- Landau, L.; Lifshitz, E. Shock Waves. In Fluid Dynamics, 2nd English ed.; Pergamon Press: Oxford, UK, 1987; Volume 6, pp. 313–360. [Google Scholar]
- Alpen, E.L. Chapter 9—Modification of the Radiation Response. In Radiation Biophysics, 2nd ed.; Alpen, E.L., Ed.; Academic Press: Cambridge, MA, USA, 1998; pp. 194–221. [Google Scholar] [CrossRef]
- Furusawa, Y.; Fukutsu, K.; Aoki, M.; Itsukaichi, H.; Eguchi-Kasai, K.; Ohara, H.; Yatagai, F.; Kanai, T.; Ando, K. Inactivation of aerobic and hypoxic cells from three different cell lines by accelerated (3)He-, (12)C- and (20)Ne-ion beams. Radiat. Res. 2000, 154, 485–496. [Google Scholar] [CrossRef]
- Suzuki, M.; Kase, Y.; Yamaguchi, H.; Kanai, T.; Ando, K. Relative biological effectiveness for cell-killing effect on various human cell lines irradiated with heavy-ion medical accelerator in Chiba (HIMAC) carbon-ion beams. Int. J. Radiat. Oncol. Biol. Phys. 2000, 48, 241–250. [Google Scholar] [CrossRef]
- Antonovic, L.; Brahme, A.; Furusawa, Y.; Toma-Dasu, I. Radiobiological description of the LET dependence of the cell survival of oxic and anoxic cells irradiated by carbon ions. J. Radiat. Res. 2013, 54, 18–26. [Google Scholar] [CrossRef]
- Tsuruoka, C.; Suzuki, M.; Kanai, T.; Fujitaka, K. LET and ion species dependence for cell killing in normal human skin fibroblasts. Radiat. Res. 2005, 163, 494–500. [Google Scholar] [CrossRef]
- Hirayama, R.; Ito, A.; Tomita, M.; Tsukada, T.; Yatagai, F.; Noguchi, M.; Matsumoto, Y.; Kase, Y.; Ando, K.; Okayasu, R.; et al. Contributions of direct and indirect actions in cell killing by high-LET radiations. Radiat. Res. 2009, 171, 212–218. [Google Scholar] [CrossRef]
- Hall, E.J.; Giaccia, A.J. Cell Survival Curves. In Radiobiology for the Radiologist, 7th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2012. [Google Scholar]
- Tilly, N.; Brahme, A.; Carlsson, J.; Glimelius, B. Comparison of cell survival models for mixed LET radiation. Int. J. Radiat. Biol. 1999, 75, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Thacker, J.; Stretch, A.; Stephens, M.A. Mutation and inactivation of cultured mammalian cells exposed to beams of accelerated heavy ions. II. Chinese hamster V79 cells. Int. J. Radiat. Biol. Relat Stud. Phys. Chem. Med. 1979, 36, 137–148. [Google Scholar] [CrossRef]
- Barendsen, G.; Walter, H.; Fowler, J.; Bewley, D. Effects of different ionizing radiations on human cells in tissue culture: III. Experiments with cyclotron-accelerated alpha-particles and deuterons. Radiat. Res. 1963, 18, 106–119. [Google Scholar] [CrossRef]
- Gulliford, S.; Prise, K. Relative Biological Effect/Linear Energy Transfer in Proton Beam Therapy: A Primer. Clin. Oncol. 2019, 31, 809–812. [Google Scholar] [CrossRef] [Green Version]
- Alberts, B.; Johnson, A.D.; Lewis, J.D.; Morgan, D.; Raff, M. 4. DNA, Chromosomes, and Genomes. In Molecular Biology of the Cell; NORTON: New York, NY, USA, 2015. [Google Scholar]
- De Vera, P.; Garcia-Molina, R.; Abril, I.; Solov’yov, A.V. Semiempirical model for the ion impact ionization of complex biological media. Phys. Rev. Lett. 2013, 110, 148104. [Google Scholar] [CrossRef] [Green Version]
- De Vera, P.; Abril, I.; Garcia-Molina, R.; Solov‘yov, A.V. Ionization of biomolecular targets by ion impact: Input data for radiobiological applications. J. Phys. Conf. Ser. 2013, 438, 012015. [Google Scholar] [CrossRef]
- Garcia-Molina, R.; Abril, I.; Kyriakou, I.; Emfietzoglou, D. Inelastic scattering and energy loss of swift electron beams in biologically relevant materials. Surf. Interface Anal. 2017, 49, 11–17. [Google Scholar] [CrossRef]
- Abril, I.; Garcia-Molina, R.; Denton, C.D.; Kyriakou, I.; Emfietzoglou, D. Energy Loss of Hydrogen- and Helium-Ion Beams in DNA: Calculations Based on a Realistic Energy-Loss Function of the Target. Radiat. Res. 2010, 175, 247–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abril, I.; Garcia-Molina, R.; de Vera, P.; Kyriakou, I.; Emfietzoglou, D. Chapter six—Inelastic Collisions of Energetic Protons in Biological Media. In Advances in Quantum Chemistry; Belkić, D., Ed.; Academic Press: Cambridge, MA, USA, 2013; Volume 65, pp. 129–164. [Google Scholar]
- Możejko, P.; Sanche, L. Cross section calculations for electron scattering from DNA and RNA bases. Radiat. Environ. Biophys. 2003, 42, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, P.; Paretzke, H. Calculation of electron impact ionization cross sections of DNA using the Deutsch–Märk and Binary–Encounter–Bethe formalisms. Int. J. Mass Spectrom. 2003, 223, 599–611. [Google Scholar] [CrossRef]
- Huo, W.M.; Dateo, C.E.; Fletcher, G.D. Molecular data for a biochemical model of DNA damage: Electron impact ionization and dissociative ionization cross sections of DNA bases and sugar-phosphate backbone. Radiat. Meas. 2006, 41, 1202–1208. [Google Scholar] [CrossRef]
- Bug, M.U.; Baek, W.Y.; Rabus, H.; Villagrasa, C.; Meylan, S.; Rosenfeld, A.B. An electron-impact cross section data set (10 eV–1 keV) of DNA constituents based on consistent experimental data: A requisite for Monte Carlo simulations. Radiat. Phys. Chem. 2017, 130, 459–479. [Google Scholar] [CrossRef]
- Emfietzoglou, D.; Nikjoo, H. The effect of model approximations on single-collision distributions of low-energy electrons in liquid water. Radiat. Res. 2005, 163, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Emfietzoglou, D. Inelastic cross-sections for electron transport in liquid water: A comparison of dielectric models. Radiat. Phys. Chem. 2003, 66, 373–385. [Google Scholar] [CrossRef]
- Kyriakou, I.; Šefl, M.; Nourry, V.; Incerti, S. The impact of new Geant4-DNA cross section models on electron track structure simulations in liquid water. J. Appl. Phys. 2016, 119, 194902. [Google Scholar] [CrossRef]
- Emfietzoglou, D.; Kyriakou, I.; Abril, I.; Garcia-Molina, R.; Nikjoo, H. Inelastic scattering of low-energy electrons in liquid water computed from optical-data models of the Bethe surface. Int. J. Radiat. Biol. 2012, 88, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Nikjoo, H.; Taleei, R.; Liamsuwan, T.; Liljequist, D.; Emfietzoglou, D. Perspectives in radiation biophysics: From radiation track structure simulation to mechanistic models of DNA damage and repair. Radiat. Phys. Chem. 2016, 128, 3–10. [Google Scholar] [CrossRef]
- Emfietzoglou, D.; Cucinotta, F.A.; Nikjoo, H. A complete dielectric response model for liquid water: A solution of the Bethe ridge problem. Radiat. Res. 2005, 164, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Emfietzoglou, D.; Kyriakou, I.; Garcia-Molina, R.; Abril, I. Inelastic mean free path of low-energy electrons in condensed media: Beyond the standard models. Surf. Interface Anal. 2017, 49, 4–10. [Google Scholar] [CrossRef]
- Emfietzoglou, D.; Papamichael, G.; Nikjoo, H. Monte Carlo Electron Track Structure Calculations in Liquid Water Using a New Model Dielectric Response Function. Radiat. Res. 2017, 188, 355–368. [Google Scholar] [CrossRef]
- Sanche, L. Low-Energy Electron Interaction with DNA: Bond Dissociation and Formation of Transient Anions, Radicals, and Radical Anions. In Radical and Radical Ion Reactivity in Nucleic Acid Chemistry; Greenberg, M., Ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2009; p. 239. [Google Scholar]
- Fabrikant, I.; Caprasecca, S.; Gallup, G.A.; Gorfinkiel, J.D. Electron attachment to molecules in a cluster environment. J. Chem Phys. 2012, 136, 184301. [Google Scholar] [CrossRef] [Green Version]
- Becker, D.; Sevilla, M. The chemical consequences of radiation damage to DNA. Adv. Radiat. Biol. 1993, 17, 121–180. [Google Scholar]
- Gianturco, F.A.; Sebastianelli, F.; Lucchese, R.R.; Baccarelli, I.; Sanna, N. Ring-breaking electron attachment to uracil: Following bond dissociations via evolving resonances. J. Chem. Phys. 2008, 128, 174302. [Google Scholar] [CrossRef]
- Sanche, L. Nanoscale Dynamics of Radiosensitivity: Role of Low Energy Electrons. In Radiation Damage in Biomolecular Systems; García Gómez-Tejedor, G., Fuss, M.C., Eds.; Springer: Dordrecht, The Netherlands, 2012; pp. 3–43. [Google Scholar] [CrossRef]
- Panajotovic, R.; Martin, F.; Cloutier, P.; Hunting, D.; Sanche, L. Effective cross sections for production of single-strand breaks in plasmid DNA by 0.1 to 4.7 eV electrons. Radiat. Res. 2006, 165, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Nikjoo, H.; O‘Neill, P.; Goodhead, D.T.; Terrissol, M. Computational modelling of low-energy electron-induced DNA damage by early physical and chemical events. Int. J. Radiat. Biol. 1997, 71, 467–483. [Google Scholar] [CrossRef]
- Schipler, A.; Iliakis, G. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res. 2013, 41, 7589–7605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huels, M.A.; Boudaiffa, B.; Cloutier, P.; Hunting, D.; Sanche, L. Single, double, and multiple double strand breaks induced in DNA by 3-100 eV electrons. J. Am. Chem. Soc. 2003, 125, 4467–4477. [Google Scholar] [CrossRef]
- Surdutovich, E.; Solov‘yov, A. Double strand breaks in DNA resulting from double ionization events. Eur. Phys. J. D 2012, 66, 206. [Google Scholar] [CrossRef]
- Symington, L.S.; Gautier, J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beucher, A.; Birraux, J.; Tchouandong, L.; Barton, O.; Shibata, A.; Conrad, S.; Goodarzi, A.A.; Krempler, A.; Jeggo, P.A.; Löbrich, M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. Embo J. 2009, 28, 3413–3427. [Google Scholar] [CrossRef] [Green Version]
- Brandsma, I.; Gent, D.C. Pathway choice in DNA double strand break repair: Observations of a balancing act. Genome Integr. 2012, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, P.; Goedecke, W.; Obe, G. Mechanisms of DNA double-strand break repair and their potential to induce chromosomal aberrations. Mutagenesis 2000, 15, 289–302. [Google Scholar] [CrossRef]
- Von Sonntag, C. Formation of Reactive Free Radicals in an Aqueous Environment. In Free-Radical-Induced DNA Damage and Its Repair: A Chemical Perspective; Springer: Berlin/Heidelberg, Germany, 2006; pp. 7–46. [Google Scholar] [CrossRef]
- Surdutovich, E.; Solov‘yov, A.V. Random walk approximation for the radial dose dependence. Eur. Phys. J. D 2012, 66, 245. [Google Scholar] [CrossRef]
- Bug, M.U.; Surdutovich, E.; Rabus, H.; Rosenfeld, A.B.; Solov‘yov, A.V. Nanoscale characterization of ion tracks: MC simulations versus analytical approach. Eur. Phys. J. D 2012, 66, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Brahme, A. 9.04-Modeling of Radiation Effects in Cells and Tissues. In Comprehensive Biomedical Physics; Brahme, A. Elsevier: Oxford, UK, 2014; pp. 105–142. [Google Scholar] [CrossRef]
- Baak, J.P.; Gudlaugsson, E.; Skaland, I.; Guo, L.H.; Klos, J.; Lende, T.H.; Søiland, H.; Janssen, E.A.; Zur Hausen, A. Proliferation is the strongest prognosticator in node-negative breast cancer: Significance, error sources, alternatives and comparison with molecular prognostic markers. Breast Cancer Res. Treat. 2009, 115, 241–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, E.G. Nuclear Morphology and the Biology of Cancer Cells. Acta Cytol. 2020, 64, 511–519. [Google Scholar] [CrossRef]
- Friedrich, T.; Scholz, U.; Elsässer, T.; Durante, M.; Scholz, M. Systematic analysis of RBE and related quantities using a database of cell survival experiments with ion beam irradiation. J. Radiat. Res. 2013, 54, 494–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikjoo, H.; Bolton, C.E.; Watanabe, R.; Terrissol, M.; O’Neill, P.; Goodhead, D.T. Modelling of DNA damage induced by energetic electrons (100 eV to 100 keV). Radiat Prot. Dosim. 2002, 99, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Milligan, J.R.; Aguilera, J.A.; Ward, J.F. Variation of single-strand break yield with scavenger concentration for plasmid DNA irradiated in aqueous solution. Radiat. Res. 1993, 133, 151–157. [Google Scholar] [CrossRef]
- Van Rijn, K.; Mayer, T.; Blok, J.; Verberne, J.B.; Loman, H. Reaction Rate of OH Radicals with ϕX174 DNA: Influence of Salt and Scavenger. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1985, 47, 309–317. [Google Scholar] [CrossRef]
- Smyth, M.; Kohanoff, J. Excess electron interactions with solvated DNA nucleotides: Strand breaks possible at room temperature. J. Am. Chem. Soc. 2012, 134, 9122–9125. [Google Scholar] [CrossRef]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Cui, J. Present and future of cancer immunotherapy: A tumor microenvironmental perspective. Oncol. Lett. 2018, 16, 4105–4113. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Good, J.S.; Harrington, K.J. The hallmarks of cancer and the radiation oncologist: Updating the 5Rs of radiobiology. Clin. Oncol. (R. Coll. Radiol.) 2013, 25, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Shevtsov, M.; Sato, H.; Multhoff, G.; Shibata, A. Novel Approaches to Improve the Efficacy of Immuno-Radiotherapy. Front. Oncol. 2019, 9, 156. [Google Scholar] [CrossRef] [Green Version]
- Rückert, M.; Deloch, L.; Fietkau, R.; Frey, B.; Hecht, M.; Gaipl, U.S. Immune modulatory effects of radiotherapy as basis for well-reasoned radioimmunotherapies. Strahlenther. Und Onkol. 2018, 194, 509–519. [Google Scholar] [CrossRef]
- Peters, L.J.; Withers, H.R.; Thames, H.D., Jr. Radiobiological Bases for Multiple Daily Fractionation; Raven Press: White River Junction, VT, USA, 1982. [Google Scholar]
- Paganetti, H.; Blakely, E.; Carabe-Fernandez, A.; Carlson, D.J.; Das, I.J.; Dong, L.; Grosshans, D.; Held, K.D.; Mohan, R.; Moiseenko, V.; et al. Report of the AAPM TG-256 on the relative biological effectiveness of proton beams in radiation therapy. Med. Phys. 2019, 46, e53–e78. [Google Scholar] [CrossRef] [Green Version]
- Iliakis, G.; Mladenov, E.; Mladenova, V. Necessities in the Processing of DNA Double Strand Breaks and Their Effects on Genomic Instability and Cancer. Cancers (Basel) 2019, 11, 1671. [Google Scholar] [CrossRef] [Green Version]
- Azzam, E.I.; Jay-Gerin, J.-P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [Green Version]
- Rodemann, H.P.; Blaese, M.A. Responses of normal cells to ionizing radiation. Semin. Radiat. Oncol. 2007, 17, 81–88. [Google Scholar] [CrossRef]
- Somosy, Z. Radiation response of cell organelles. Micron 2000, 31, 165–181. [Google Scholar] [CrossRef]
- Singh, G.; Hauswirth, W.; Ross, W.; Neims, A. A method for assessing damage to mitochondrial DNA caused by radiation and epichlorohydrin. Mol. Pharm. 1985, 27, 167–170. [Google Scholar]
- Leach, J.K.; Van Tuyle, G.; Lin, P.-S.; Schmidt-Ullrich, R.; Mikkelsen, R.B. Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Res. 2001, 61, 3894–3901. [Google Scholar] [PubMed]
- Tulard, A.; Hoffschir, F.; de Boisferon, F.H.; Luccioni, C.; Bravard, A. Persistent oxidative stress after ionizing radiation is involved in inherited radiosensitivity. Free Radic. Biol. Med. 2003, 35, 68–77. [Google Scholar] [CrossRef]
- Rajendran, S.; Harrison, S.H.; Thomas, R.A.; Tucker, J.D. The role of mitochondria in the radiation-induced bystander effect in human lymphoblastoid cells. Radiat. Res. 2011, 175, 159–171. [Google Scholar] [CrossRef] [Green Version]
- Little, J.B. Radiation carcinogenesis. Carcinogenesis 2000, 21, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Morgan, W.F.; Bair, W.J. Issues in low dose radiation biology: The controversy continues. A perspective. Radiat. Res. 2013, 179, 501–510. [Google Scholar] [CrossRef]
- Prise, K.M.; O‘sullivan, J.M. Radiation-induced bystander signalling in cancer therapy. Nat. Rev. Cancer 2009, 9, 351–360. [Google Scholar] [CrossRef]
- Nikjoo, H.; Liamsuwan, T. 9.03-Biophysical Basis of Ionizing Radiation. In Comprehensive Biomedical Physics; Brahme, A., Ed.; Elsevier: Oxford, UK, 2014; pp. 65–104. [Google Scholar] [CrossRef]
- Olivieri, G.; Bodycote, J.; Wolff, S. Adaptive response of human lymphocytes to low concentrations of radioactive thymidine. Science 1984, 223, 594. [Google Scholar] [CrossRef] [PubMed]
- Wolff, S.; Afzal, V.; Wiencke, J.K.; Olivieri, G.; Michaeli, A. Human lymphocytes exposed to low doses of ionizing radiations become refractory to high doses of radiation as well as to chemical mutagens that induce double-strand breaks in DNA. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1988, 53, 39–47. [Google Scholar] [CrossRef]
- Vijayalaxmi; Burkart, W. Resistance and cross-resistance to chromosome damage in human blood lymphocytes adapted to bleomycin. Mutat. Res. 1989, 211, 1–5. [Google Scholar] [CrossRef]
- Wolff, S. The adaptive response in radiobiology: Evolving insights and implications. Environ. Health Perspect 1998, 106 (Suppl. S1), 277–283. [Google Scholar] [CrossRef]
- Sankaranarayanan, K.; Duyn, A.v.; Loos, M.J.; Natarajan, A.T. Adaptive response of human lymphocytes to low-level radiation from radioisotopes or X-rays. Mutat. Res./Fundam. Mol. Mech. Mutagen. 1989, 211, 7–12. [Google Scholar] [CrossRef]
- Siede, W.; Eckardt, F. Indications for an inducible component of error-prone DNA repair in yeast. Br. J. Cancer 1984, 6, 103–106. [Google Scholar]
- Eckardt-Schupp, F.; Klaus, C. Radiation inducible DNA repair processes in eukaryotes. Biochimie 1999, 81, 161–171. [Google Scholar] [CrossRef]
- Ojima, M.; Eto, H.; Ban, N.; Kai, M. Radiation-induced bystander effects induce radioadaptive response by low-dose radiation. Radiat Prot. Dosim. 2011, 146, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Lindhard, J. On the properties of a gas of charged particles. Dan. Vid. Selsk Mat.-Fys. Medd. 1954, 28, 8. [Google Scholar]
- Emfietzoglou, D.; Nikjoo, H. Accurate electron inelastic cross sections and stopping powers for liquid water over the 0.1-10 keV range based on an improved dielectric description of the Bethe surface. Radiat. Res. 2007, 167, 110–120. [Google Scholar] [CrossRef]
- Tan, Z.; Xia, Y.; Zhao, M.; Liu, X. Electron stopping power and inelastic mean free path in amino acids and protein over the energy range of 20–20,000 eV. Radiat. Environ. Biophys. 2006, 45, 135–143. [Google Scholar] [CrossRef]
- Kyriakou, I.; Incerti, S.; Francis, Z. Technical Note: Improvements in geant4 energy-loss model and the effect on low-energy electron transport in liquid water. Med. Phys. 2015, 42, 3870–3876. [Google Scholar] [CrossRef]
- Ritchie, R.H.; Howie, A. Electron excitation and the optical potential in electron microscopy. Philos. Mag. A J. Theor. Exp. Appl. Phys. 1977, 36, 463–481. [Google Scholar] [CrossRef]
- Emfietzoglou, D.; Kyriakou, I.; Garcia-Molina, R.; Abril, I.; Nikjoo, H. Inelastic Cross Sections for Low-Energy Electrons in Liquid Water: Exchange and Correlation Effects. Radiat. Res. 2013, 180, 499–513. [Google Scholar] [CrossRef]
- Barkas, W.H. Techniques and theory. In Nuclear Research Emulsions; Academic Press: New York, NY, USA, 1963; Volume 1. [Google Scholar]
- Tung, C.J.; Chao, T.C.; Hsieh, H.W.; Chan, W.T. Low-energy electron interactions with liquid water and energy depositions in nanometric volumes. Nucl. Instrum. Methods Phys. Res. B 2007, 262, 231–239. [Google Scholar] [CrossRef]
- Nikjoo, H.; Uehara, S.; Wilson, W.E.; Hoshi, M.; Goodhead, D.T. Track structure in radiation biology: Theory and applications. Int. J. Radiat. Biol. 1998, 73, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, A.G.; O‘Neill, P.; Stewart, R.D. Induction and repair of clustered DNA lesions: What do we know so far? Radiat. Res. 2013, 180, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.A. Radiation Track Structure: How the Spatial Distribution of Energy Deposition Drives Biological Response. Clin. Oncol. (R. Coll. Radiol.) 2020, 32, 75–83. [Google Scholar] [CrossRef] [PubMed]
- International Commission on Radiation Units and Measurements. Key Data For Ionizing-Radiation Dosimetry: Measurement Standards And Applications (Report 90). J. Int. Comm. Radiat. Units Meas. 2014, 14, 79–80. [Google Scholar]
- Bimbot, R.; Geissel, H.; Paul, H.; Shinner, A.; Sigmund, P. Stopping of Ions Heavier than Helium ICRU Report 73; Oxford University Press: Oxford, UK, 2005. [Google Scholar]
- Zeigler, J.; Ziegler, M.; Biersack, J. SRIM 2008. 04 Software Package. 2008. Available online: http://www.srim.org (accessed on 9 September 2020).
- Cooper, G.M.; Hausman, R.E. The Cell: A Molecular Approach, 7th ed.; Oxford University Press: Oxford, UK, 2018. [Google Scholar]
- Von Sonntag, C. The Chemical Basis of Radiation Biology; Taylor & Francis: London, UK, 1987. [Google Scholar]
- Roots, R.; Smith, K.C. On the Nature of the Oxygen Effect on X-ray-induced DNA Single-strand Breaks in Mammalian Cells. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1974, 26, 467–480. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalospyros, S.A.; Nikitaki, Z.; Kyriakou, I.; Kokkoris, M.; Emfietzoglou, D.; Georgakilas, A.G. A Mathematical Radiobiological Model (MRM) to Predict Complex DNA Damage and Cell Survival for Ionizing Particle Radiations of Varying Quality. Molecules 2021, 26, 840. https://doi.org/10.3390/molecules26040840
Kalospyros SA, Nikitaki Z, Kyriakou I, Kokkoris M, Emfietzoglou D, Georgakilas AG. A Mathematical Radiobiological Model (MRM) to Predict Complex DNA Damage and Cell Survival for Ionizing Particle Radiations of Varying Quality. Molecules. 2021; 26(4):840. https://doi.org/10.3390/molecules26040840
Chicago/Turabian StyleKalospyros, Spyridon A., Zacharenia Nikitaki, Ioanna Kyriakou, Michael Kokkoris, Dimitris Emfietzoglou, and Alexandros G. Georgakilas. 2021. "A Mathematical Radiobiological Model (MRM) to Predict Complex DNA Damage and Cell Survival for Ionizing Particle Radiations of Varying Quality" Molecules 26, no. 4: 840. https://doi.org/10.3390/molecules26040840