Design, Synthesis and Biological Evaluation of New Pyrimidine Derivatives as Anticancer Agents

 ,
,  , ,
, ,  , , ,
, , ,
Abstract
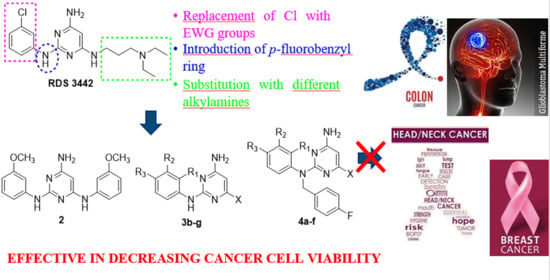
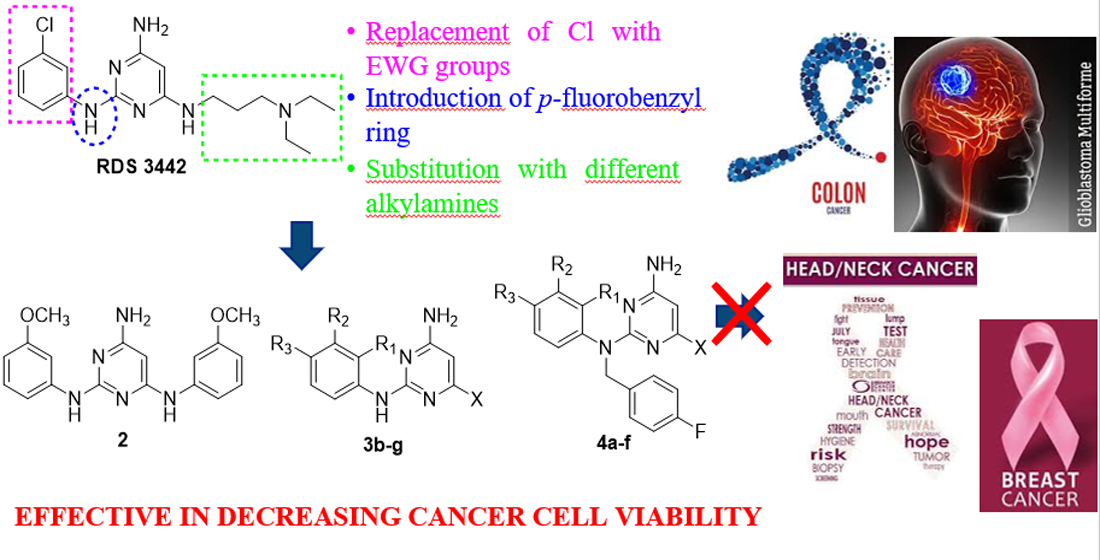
:
1. Introduction
2. Results and Discussion
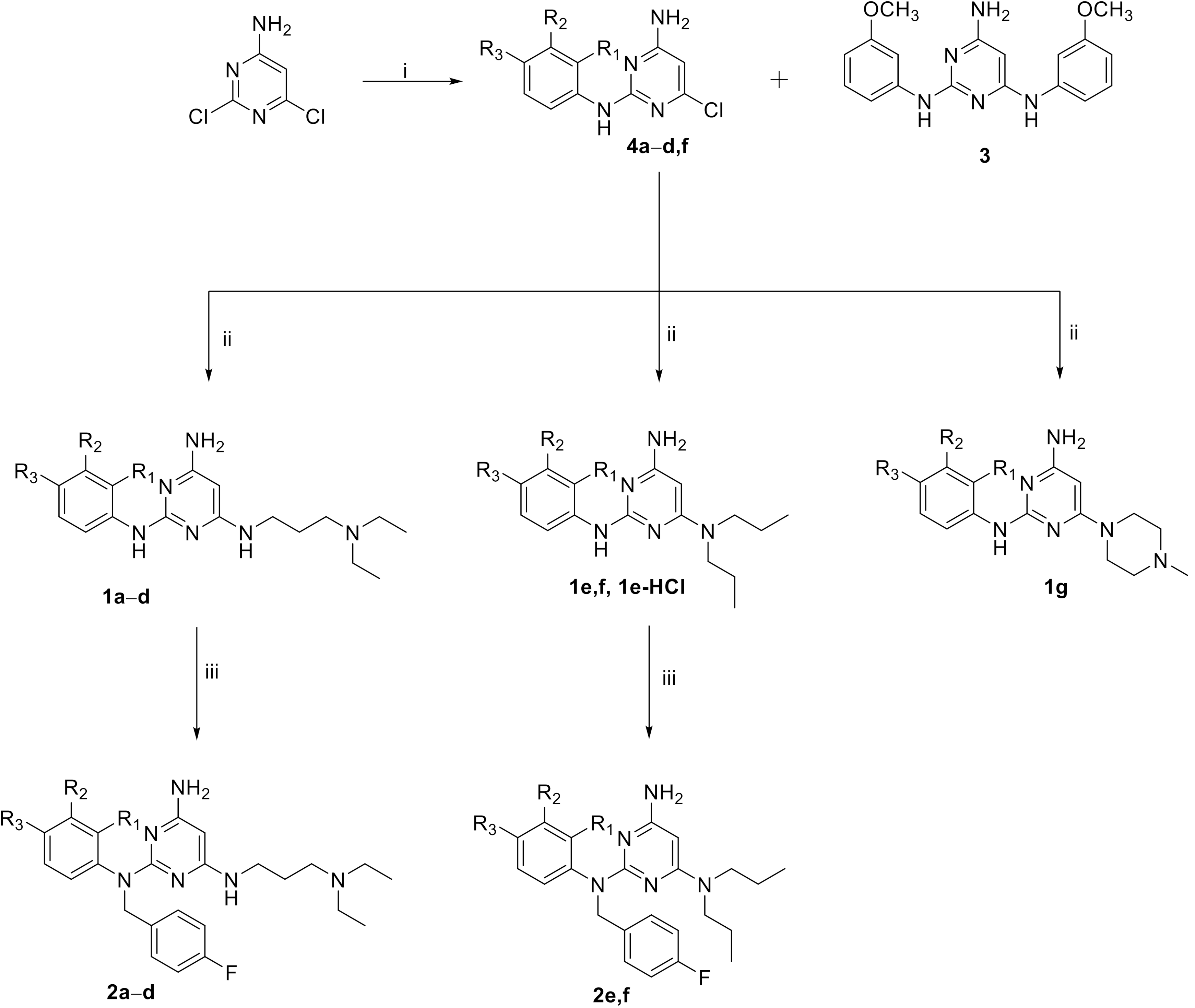
2.1. Chemistry
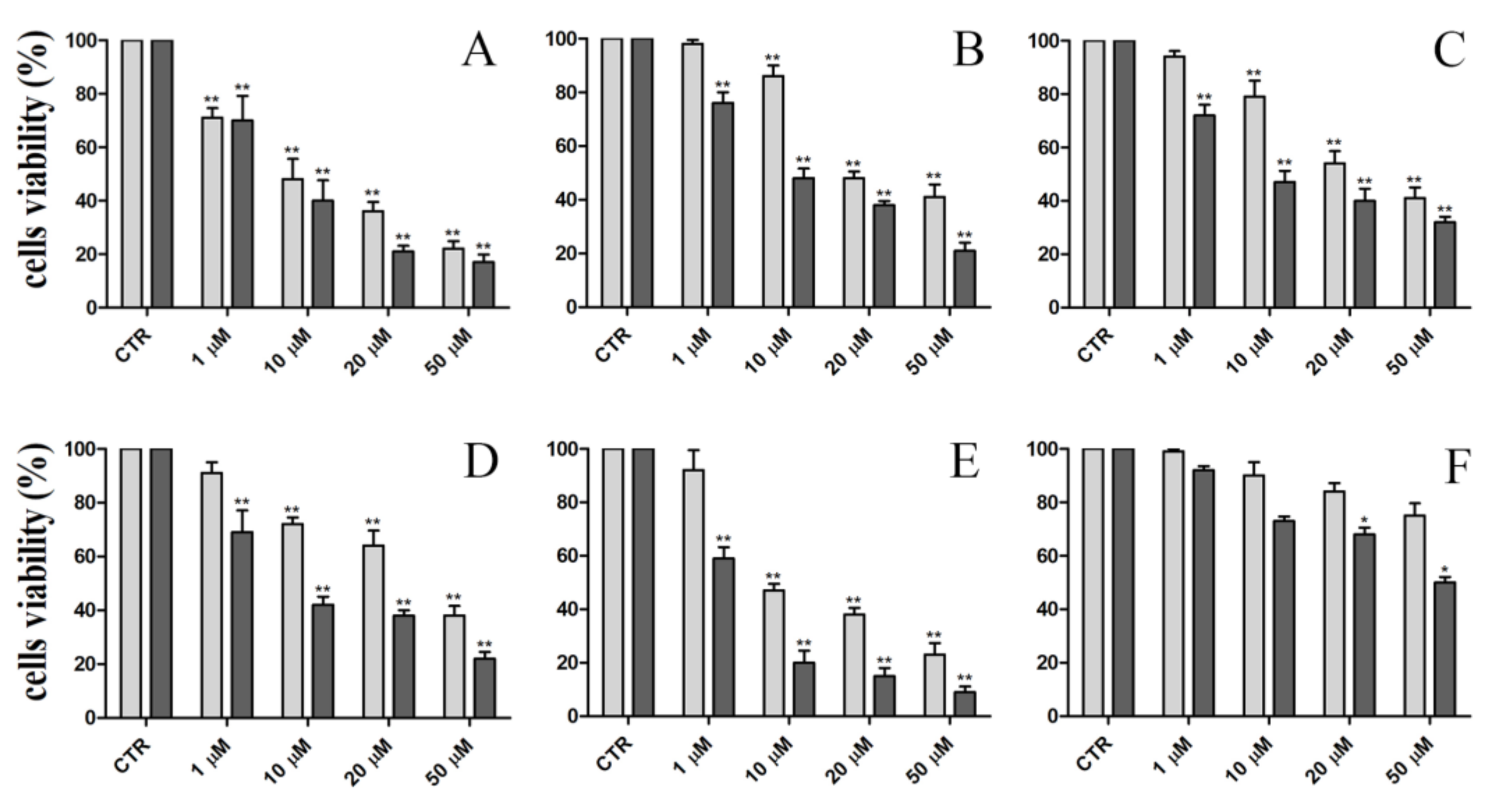
2.2. In Vitro Proliferation Assay
3. Materials and Methods
3.1. Chemistry
3.1.1. General Instrumentation
3.1.2. Microwave Irradiation Experiments
3.1.3. General Experimental Procedures
3.1.4. Specific Procedures and Characterization
3.2. Biological Assays
3.2.1. Cell Cultures and Treatments
3.2.2. Cytotoxicity Assay
3.2.3. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Hanahan, D. Rethinking the war on cancer. Lancet 2014, 383, 558–563. [Google Scholar] [CrossRef]
- Jo, Y.; Choi, N.; Kim, K.; Koo, H.J.; Choi, J.; Kim, H.N. Chemoresistance of cancer cells: Requirements of tumor microenvironment-mimicking in vitro models in anti-cancer drug development. Theranostics 2018, 8, 5259–5275. [Google Scholar] [CrossRef] [PubMed]
- López-Verdín, S.; Lavalle-Carrasco, J.; Carreón-Burciaga, R.G.; Serafín-Higuera, N.; Molina-Frechero, N.; González-González, R.; Bologna-Molina, R. Molecular markers of anticancer drug resistance in head and neck squamous cell carcinoma: A literature review. Cancers 2018, 10, 376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luqmani, Y.A. Mechanisms of drug resistance in cancer chemotherapy. Med. Princ. Pract. 2005, 14, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Noch, E.K.; Ramakrishna, R.; Magge, R. Challenges in the treatment of glioblastoma: Multisystem mechanisms of therapeutic resistance. World Neurosurg. 2018, 116, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Nedeljković, M.; Damjanović, A. Mechanisms of chemotherapy resistance in triple-negative breast cancer-How we can rise to the challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurwidya, F.; Takahashi, F.; Murakami, A.; Takahashi, K. Epithelial mesenchymal transition in drug resistance and metastasis of lung cancer. Cancer Res. Treat. 2012, 44, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Van der Jeught, K.; Xu, H.C.; Li, Y.J.; Lu, X.B.; Ji, G. Drug resistance and new therapies in colorectal cancer. World J. Gastroenterol. 2018, 24, 3834–3848. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Kim, J.H. Increasing incidence and improving survival of oral tongue squamous cell carcinoma. Sci. Rep. 2020, 10, 7877. [Google Scholar] [CrossRef] [PubMed]
- Robert, B.M.; Dakshinamoorthy, M.; Ganapathyagraharam Ramamoorthy, B.; Dhandapani, M.; Thangaiyan, R.; Muthusamy, G.; Madhavan Nirmal, R.; Rajendra Prasad, N. Predicting tumor sensitivity to chemotherapeutic drugs in oral squamous cell carcinoma patients. Sci. Rep. 2018, 8, 15545. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, S.D.; Hier, M.; Mlynarek, A.; Kowalski, L.P.; Alaoui-Jamali, M.A. Recurrent oral cancer: Current and emerging therapeutic approaches. Front. Pharmacol. 2012, 3, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taglieri, L.; Saccoliti, F.; Nicolai, A.; Peruzzi, G.; Madia, V.N.; Tudino, V.; Messore, A.; Di Santo, R.; Artico, M.; Taurone, S.; et al. Discovery of a pyrimidine compound endowed with antitumor activity. Invest. New Drugs 2020, 38, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.K. A critical overview of targeted therapies for glioblastoma. Front. Oncol. 2018, 8, 419. [Google Scholar] [CrossRef] [PubMed]
- Colotti, G.; Saccoliti, F.; Gramiccia, M.; Di Muccio, T.; Prakash, J.; Yadav, S.; Dubey, V.K.; Vistoli, G.; Battista, T.; Mocci, S.; et al. Structure-guided approach to identify a novel class of anti-leishmaniasis diaryl sulfide compounds targeting the trypanothione metabolism. Amino acids 2020, 52, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Saccoliti, F.; Angiulli, G.; Pupo, G.; Pescatori, L.; Madia, V.N.; Messore, A.; Colotti, G.; Fiorillo, A.; Scipione, L.; Gramiccia, M.; et al. Inhibition of Leishmania infantum trypanothione reductase by diaryl sulfide derivatives. J. Enzyme Inhib. Med. Chem. 2017, 32, 304–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taglieri, L.; Nardo, T.; Vicinanza, R.; Ross, J.M.; Scarpa, S.; Coppotelli, G. Thyroid hormone regulates fibronectin expression through the activation of hypoxia inducible factor 1. Biochem. Biophys. Res. Commun. 2017, 493, 1304–1310. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

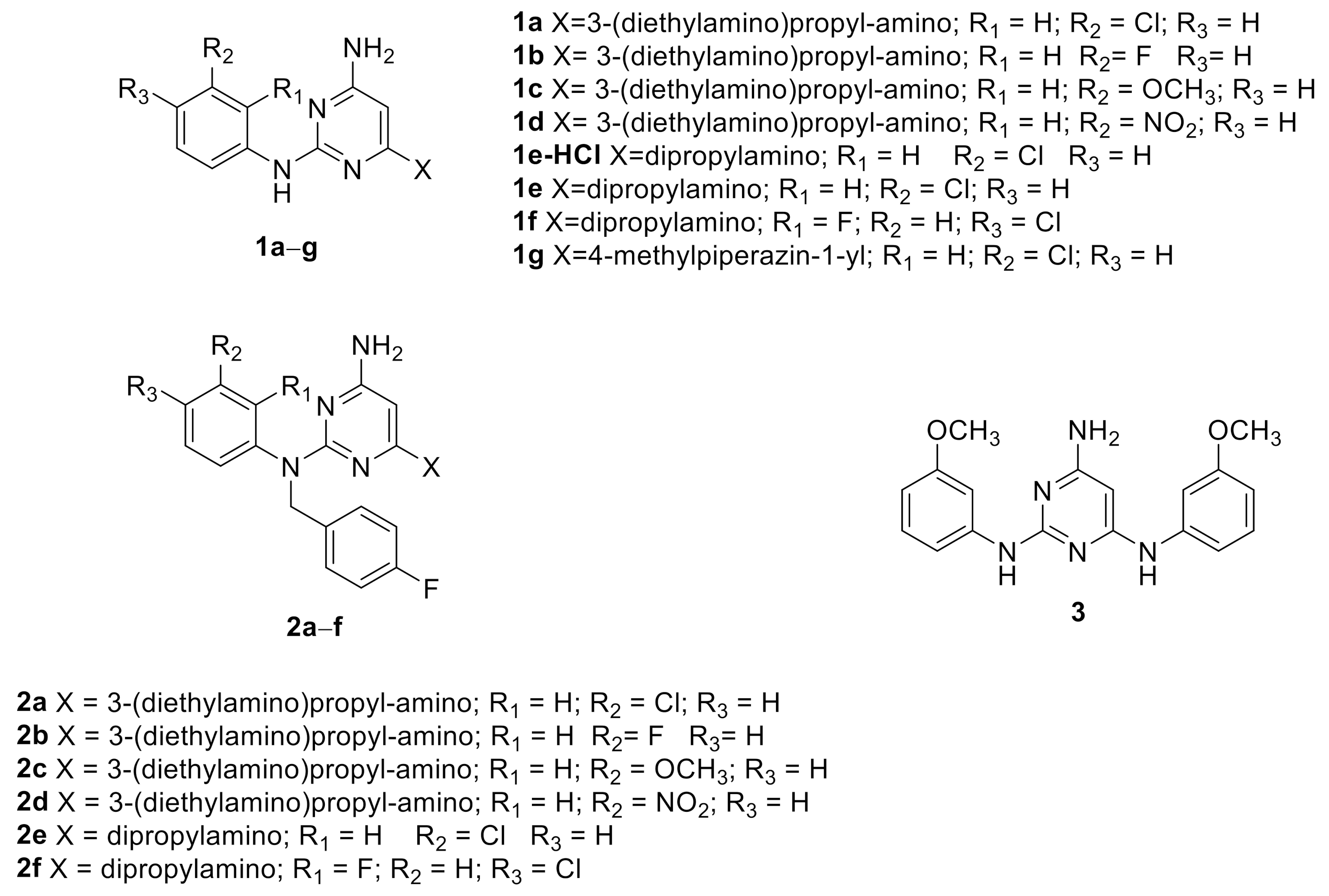
| Cpd | R1 | R2 | R3 | X |
|---|---|---|---|---|
| 1b | H | F | H | 3-(diethylamino)propyl-amino |
| 1c | H | OCH3 | H | 3-(diethylamino)propyl-amino |
| 1d | H | NO2 | H | 3-(diethylamino)propyl-amino |
| 1e-HCl | H | Cl | H | Dipropylamino |
| 1e | H | Cl | H | Dipropylamino |
| 1f | F | H | Cl | Dipropylamino |
| 1g | H | Cl | H | 4-methylpiperazin-1-yl |
| 2a | H | Cl | H | 3-(diethylamino)propyl-amino |
| 2b | H | F | H | 3-(diethylamino)propyl-amino |
| 2c | H | OCH3 | H | 3-(diethylamino)propyl-amino |
| 2d | H | NO2 | H | 3-(diethylamino)propyl-amino |
| 2e | H | Cl | H | Dipropylamino |
| 2f | F | H | Cl | Dipropylamino |
| 3 | - | - | - | - |
| Cpd | EC50 (μM) 1 | CC50 (μM) 2 | ||||
|---|---|---|---|---|---|---|
| HT-29 | U-87 MG | MDA-MB231 | CAL27 | FaDu | HF | |
| 1b | 29.8 (48 h) ± 3.2 | 47.3 (48 h) ± 3.6 | 35.7 (48 h) ± 3.2 | 20.3 (48 h) ± 1.8 | 43.4 (48 h) ± 3.9 | 88 (48 h) ± 9.0 |
| 1c | 57 (48 h) ± 2.4 | 68 (48 h) ± 5.0 | 70.5 (48 h) ± 6.0 | 57 (48 h) ± 4.0 | 71 (48 h) ± 5.6 | nd 3 |
| 1d | 40.4 (48 h) ± 4.8 | 54.9 (48 h) ± 4.8 | 49.2 (48 h) ± 3.5 | 35.3 (48 h) ± 3.9 | 46.8 (48 h) ± 5.0 | 97 (48 h) ± 13.0 |
| 1e-HCl | 29.3 (48 h) ± 1.7 | 36.6 (48 h) ± 2.9 | 50.2 (48 h) ± 4.5 | 30.3 (48 h) ± 2.4 | 39.8 (48 h) ± 4.0 | 76 (48 h) ± 5.5 |
| 1e | 28.5 (48 h) ± 1.9 | 45.3 (48 h) ± 3.8 | 34.2 (48 h) ± 2.5 | 24.7 (48 h) ± 1.8 | 29.7 (48 h) ± 3.2 | 68 (48 h) ± 5.2 |
| 1f | 50 (48 h) ± 3.3 | 75 (48 h) ± 4.9 | 58 (48 h) ± 6.1 | 55 (48 h) ± 5.7 | 67 (48 h) ± 4.9 | 98 (48 h) ± 8.0 |
| 1g | 60 (48 h) ± 5.8 | 82 (48 h) ± 7.0 | 55 (48 h) ± 3.7 | 60 (48 h) ± 6.2 | 87 (48 h) ± 7.0 | nd 3 |
| 3 | 28.2 (24 h) ± 1.4 11.7 (48 h) ± 0.9 | 22.9 (48 h) ± 2.5 | 10.2 (48 h) ± 1.5 | 20.3 (24 h) ± 2.8 10.4 (48 h) ± 0.8 | 42.1 (24 h) ± 3.6 19.7 (48 h) ± 2.0 | 65 (48 h) ± 4.1 |
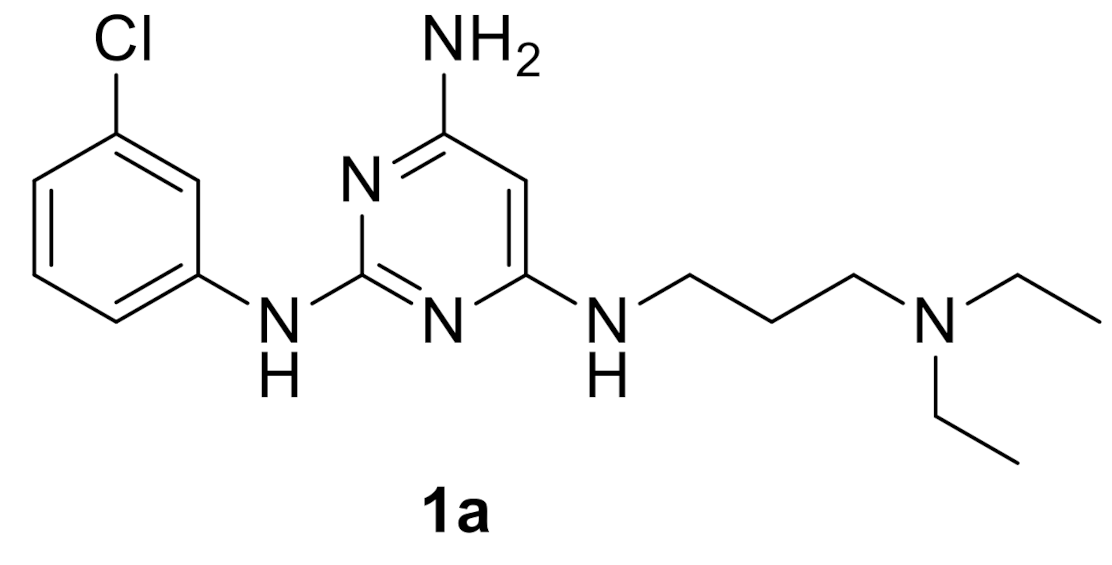
| 1a | 51.8 (48 h) ± 4.2 | 75.2 (48 h) ± 6.2 | 34.8 (48 h) ± 2.8 | 54.2 (48 h) ± 4.8 | 69.3 (48 h) ± 5.8 | 73.6 (48 h) ± 6.0 |
| Cpd | EC50 (µM) 1 | CC50 (µM) 2 | ||||
|---|---|---|---|---|---|---|
| HT-29 | U-87 MG | MDA-MB231 | CAL27 | FaDu | HF | |
| 2a | 10.2 (24 h) ± 0.6 5.4 (48 h) ± 0.4 | 22.0 (24 h) ± 1.3 7.5 (48 h) ± 0.6 | 18.0 (24 h) ± 0.7 7.9 (48 h) ± 0.8 | 9.7 (24 h) ± 0.6 4.3 (48 h) ± 0.5 | 26.2 (24 h) ± 3.2 8.5 (48 h) ± 1.1 | 50.2 (48 h) ± 3.8 |
| 2b | 20.2 (24 h) ± 1.9 10.4 (48 h) ± 0.8 | 17.7 (48 h) ± 1.2 | 7.3 (48 h) ± 0.5 | 5.1 (48 h) ± 0.3 | 23.8 (48 h) ± 2.1 | 54.7 (48 h) ± 4.2 |
| 2c | 34.7 (24 h) ± 2.8 18.3 (48 h) ± 1.2 | 19.8 (48 h) ± 1.4 | 27.2 (48 h) ± 1.8 | 14.9 (48 h) ± 1.1 | 18.2 (48 h) ± 1.3 | 50.3 (48 h) ± 3.7 |
| 2d | 25.2 (24 h) ± 2.0 12.7 (48 h) ± 1.1 | 30.1 (48 h) ± 2.4 | 17.3 (48 h) ± 1.9 | 10.3 (48 h) ± 0.9 | 20.5 (48 h) ± 2.3 | 55.2 (48 h) ± 4.9 |
| 2e | 65.2 (48 h) ± 5.0 | 72.4 (48 h) ± 3.8 | 60.8 (48 h) ± 7.0 | 59.3 (48 h) ± 4.7 | 84.4 (48 h) ± 5.0 | nd 3 |
| 2f | 30.0 (48 h) ± 1.5 | 67.0 (48 h) ± 5.0 | 58.3 (48 h) ± 6.5 | 45.4 (48 h) ± 1.8 | 75.3 (48 h) ± 6.9 | nd 3 |
| 1a | 51.8 (48 h) ± 4.2 | 75.2 (48 h) ± 6.2 | 34.8 (48 h) ± 2.8 | 54.2 (48 h) ± 4.8 | 69.3 (48 h) ± 5.8 | 73.6 (48 h) ± 6.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madia, V.N.; Nicolai, A.; Messore, A.; De Leo, A.; Ialongo, D.; Tudino, V.; Saccoliti, F.; De Vita, D.; Scipione, L.; Artico, M.; et al. Design, Synthesis and Biological Evaluation of New Pyrimidine Derivatives as Anticancer Agents. Molecules 2021, 26, 771. https://doi.org/10.3390/molecules26030771
Madia VN, Nicolai A, Messore A, De Leo A, Ialongo D, Tudino V, Saccoliti F, De Vita D, Scipione L, Artico M, et al. Design, Synthesis and Biological Evaluation of New Pyrimidine Derivatives as Anticancer Agents. Molecules. 2021; 26(3):771. https://doi.org/10.3390/molecules26030771
Chicago/Turabian StyleMadia, Valentina Noemi, Alice Nicolai, Antonella Messore, Alessandro De Leo, Davide Ialongo, Valeria Tudino, Francesco Saccoliti, Daniela De Vita, Luigi Scipione, Marco Artico, and et al. 2021. "Design, Synthesis and Biological Evaluation of New Pyrimidine Derivatives as Anticancer Agents" Molecules 26, no. 3: 771. https://doi.org/10.3390/molecules26030771