1. Introduction
Anthocyanins are ubiquitous in plants, and the total number of anthocyanin structures identified after isolation from plant extracts is about 800. These pigments are mainly divided into two groups, anthocyanins and 3-deoxyanthocyanins, with the latter group lacking an oxygen-function on the anthocyanidin 3-position on the C-ring [
1]. The vast majority of the common anthocyanins are based on just 6 different aglycones (anthocyanidins) varying with the substitutions on the B-ring, and range in colour from salmon red to dark purple and blue [
2]. In addition, a third heterogenous group (pyranoanthocyanins) [
3,
4] formed from anthocyanins during storage and processing in plant-derived foods, has gained much attention mostly because of their influence on colour evolution in wine during maturation [
5]. A couple of pyranoanthocyanins have also been reported to occur in minor amounts in intact plant materials, for instance strawberries [
6]. The various pyranoanthocyanins have in common an extra pyranic ring formed by cyclic addition onto both carbon 4 and the hydroxyl group at carbon 5 of the anthocyanidin skeleton. In solution, each anthocyanin exists in a network of reactions between the flavylium cation, hemiketals, chalcones and quinoidal bases [
7,
8,
9]. The vast majority of structure elucidations of anthocyanins have been undertaken in acidic environments on the flavylium cation form, which is considered to be the most stable form. In recent years it has been shown that synthetic flavylium compounds analogous to anthocyanidins also possess the same general network of chemical reactions [
10].
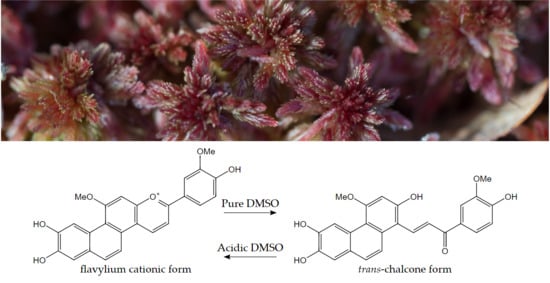
In this context, pigment 1, Sphagnorubin C isolated from sphagnum moss, is an interesting molecule belonging to the 3-deoxyanthocyanidin group. However, due to the two extraordinary aromatic rings connected to the A-ring, this pigment possesses properties not previously highlighted for any natural anthocyanidin/anthocyanin before. The purpose of this work was to present the first full structural characterization of a naturally occurring anthocyanidin/anthocyanin chalcone form. Other aims were to focus on how the extra aromatic rings of 1 extend the type of network of equilibrium forms displayed by natural anthocyanins, and to reveal the colours of the different forms of 1 in this network and their potential use.
3. Discussion
The structure of Sphagnorubin C (
1) in its flavylium cation form has previously been elucidated using derivatisation techniques in combination with ultraviolet–visible (UV–Vis), infrared (IR) and
1H spectroscopy and mass spectrometry [
12,
13]. In a comparison of the assignments of
1H shift values verified for
1 in its flavylium cation form in the same solvent (DMSO), complete assignments of H2’ and H6’ and a switch between H12 and H7 were the only differences, which in the present study were determined by 2D NMR techniques.
Pigment
1 has previously not been supplied with
13C-NMR data. However, highly relevant for the present paper, the 3’-OH analogue (Sphagnorubin B) of
1 has previously been made into a pentaacetate-chalcone derivative in the slightly alkaline solvent when reacted with ethanoic anhydride and pyridine [
13]. This derivative was supplied with
13C-NMR data, although many of the signals were not completely assigned.
Anthocyanidins and anthocyanins (anthocyanidin with glycosidic moieties) are outstanding compounds by virtue of the way they are involved in a series of equilibrium reactions giving rise to several forms (secondary structures). However, proper
1H and
13C-NMR structural assignments of other forms than the flavylium cationic form, have with just a couple of exceptions not been addressed for anthocyanins and anthocyanidins. As far as we know the first proper spectroscopic data (UV–visible absorption spectrum) for any natural anthocyanin in its chalcone form was published by Brouillard, et al. [
14]. In 1993, Santos, et al. [
15] studied various equilibrium forms of malvidin 3,5-
O-diglucoside (malvin), which coexisted in slightly acidic aqueous conditions. Two-dimensional nuclear Overhauser effect spectroscopy (NOESY) exchange correlation NMR spectra provided valuable information about the structural transformations, including evidence for the relationship between the various equilibrium forms of malvin. For the first time the chalcone forms of a natural anthocyanin were characterized by assignments of the chemical shifts of the four protons of the anthocyanidin. Later the chemical proton shifts of the chalcone forms of malvidin 3-
O-glucoside [
16] and cyanidin 3-
O-glucoside [
17] have been assigned. Several forms of the pyranoanthocyanin vitisin A., including the chalcone form, have previously been reported to be distinguished by NMR [
3]. However, the authors reported that complete unambiguous assignment of the
13C-NMR signals of the chalcone form was not possible. More recently it has clearly been demonstrated that pyranoanthocyanins like vitisins are not able to form hemiketals [
18,
19], and as a consequence unable to form chalcone forms. Thus, the assignments of all
1H and
13C-NMR signals of the
trans-chalcone structure of
1 is as far as we know, the first full structural characterization of a naturally occurring anthocyanin chalcone form.
The difference in structure between anthocyanidins and 3-deoxyanthocyanidins is the lack of oxygen function in the 3-position in the C-ring of the latter group. The impact of a reduced chromophore gives the common 3-deoxyanthocyanins (apigeninidin, luteolinidin and tricetinidin) yellow colours under acidic conditions compared to more scarlet to blue colours of analogous flavylium cationic forms of the common anthocyanins. Another even more striking effect of this hypsochromic shift in the absorption spectra under less acidic/neutral conditions is displayed by the colourless chalcone forms of the common 3-deoxyanthocyanins contrasting the yellow chalcone forms of the analogous anthocyanins. However, the colours of 3-deoxyanthocyanidins are positively reckoned to be much more stable than the colours of the analogous anthocyanins under slightly acidic conditions [
20]. The network consisting of the flavylium cationic and
trans-chalcone forms of pigment
1 in the present paper, proved the best of both of these attributes. The additional D- and E-rings extended the
π-conjugation within the molecule and provided bathochromic shifts in the absorption spectra of the various forms of
1 compared to 3-deoxyanthocyanidins comprising only the normal A–C rings. Thus, the red flavylium cationic form of
1 had its absorption maximum (
λmax) at 540 nm, while the
trans-chalcone form revealed a yellow colour and had its
λmax at 420 nm in pure DMSO.
How was it verified that pigment
1 had higher colour stability than the common anthocyanins (in this paper represented by peonidin 3-
O-glucoside,
2) under slightly acidic to neutral conditions? The flavylium cation and the two hemiketal enantiomers of
2 were in the
1H NMR spectra in
Figure 4 (right) represented by their anomeric protons (see
Supplementary Table S1 for aromatic and anomeric
1H-NMR data). In pure
d-DMSO the flavylium cationic form of
2 was transferred within hours into two hemiketal forms (64% within 1.4 h or 93% after 4.9 h) (
Figure 4, right, a–d), according to
Scheme 2. Few traces of the probable yellow chalcone forms were observed (
Figure 4, right). This is in accordance with previous papers reporting the chalcone forms to constitute typically less than 10% of the various equilibrium forms of anthocyanins (not 3-deoxyanthocyanidins) in slightly acidic or neutral aqueous solutions [
8]. The important factor in this context was that the hemiketal forms of
2 (or other anthocyanins) were colourless, making the colour of the solution fade proportionally with the increase of the hemiketal forms. However, opposite to this pattern the red flavylium cation and the yellow
trans-chalcone forms occurred in various proportions when
1 was examined under similar conditions as described for
2 (
Figure 4, left, a–d). The
trans-chalcone and flavylium cationic forms in the
1H-NMR spectra are in
Figure 4 (left) represented by their H3 and H6′ protons, respectively. Since no colourless forms of
1 were observed in the NMR spectra, a gradual change from reddish flavylium cation colour towards yellow
trans-chalcone colours were observed in the NMR spectra of this sample taken during storage (
Figure 4, left, a–d). To characterize the reversibility of the two-components system described above between the flavylium cation and
trans-chalcone forms of
1, acid (5%
d-TFA,
v/
v) was added to the equilibrated neutral sample (
Figure 4, left, e–f). After 5–6 days of equilibration, the sample had turned back to its red flavylium cationic form, leaving only traces of the
trans-chalcone form (
Figure 4, left, g).
It is well known that all anthocyanins, including 3-deoxyanthocyanidins, are represented by a network of chemical forms, which are reversibly interconverted by external stimuli such as pH variation, light, etc. [
8]. Based on the results described above, the network depicted for
1 is presented in
Scheme 1. It can be viewed as a single acid-base equilibrium involving the flavylium cation and the overall conjugated base. The conjugated base represents both the quinoidal bases, hemiketal forms and the
cis- and
trans-chalcones. The hemiketal and
cis-chalcone forms are short-lived, transient forms, which were not observed in the NMR spectra. In this network there is a low isomerization barrier between the
cis- and
trans-chalcone forms. Thus, at low pH values the reddish flavylium cationic form (
λmax at 540 nm) is the only form occurring in considerable amounts, while at more moderately acidic and at neutral pH values the yellow
trans-chalcone form (
λmax at 420 nm) is increasing with pH to be the most dominant form. The group of Pina [
10] has previously reported a similar network with similar colours for the synthetic compound 2-(4-hydroxystyryl)-1-naphthopyrylium. Here they reported that a photochromic system with good switching colours from yellow to red (or from orange to red depending on pH) based on this network could be designed. Another interesting paper from this group has also reported a similar natural photochromic system based on dracoflavylium [
21]. However, this compound does not have the conjugation of the D- and E-rings of compound
1, which gives less colour (
λmax at 374 nm) and less occurrence of the
trans-chalcone form compared to
1. The occurrence of the
trans-chalcone form was in this paper improved by stabilization of the
trans-chalcone form in the presence of β-cyclodextrin. The common anthocyanins (but the 3-deoxyanthocyanins) have similar colours as
1 for their flavylium cationic and
trans-chalcone forms. However, opposite to the network of
1, these natural anthocyanins have a high isomerization barrier between their
cis- and
trans-chalcone forms and turn into colourless hemiketal forms under moderately acidic to neutral conditions. However, photochemistry of the colourless hemiketal forms of common anthocyanins have been shown to have a potential role in plant protection from UV-B radiation [
22]. The stable, reversible two-component colour system involving the reddish flavylium cation and the yellow
trans-chalcone forms of
1 might thus be of advantage compared to previously reported multistate systems of natural anthocyanins.
4. Materials and Methods
4.1. Isolation of Sphagnorubin C (1)
Sphagnorubins are natural pigments tightly bound to the cell walls of peat moss (
Sphagnum sp.) [
23], which makes them quite difficult to isolate. Additionally, in contrast to most anthocyanins, they are not soluble in water, and poorly soluble in pure methanol.
Brightly red coloured peat moss (8 kg wet wt.) was collected in the western part of Norway (60.2744 N, 5.4974 E), air dried and homogenised using a blender. The dried plant material was extracted 4 times for 24 h in 8 L 0.5% TFA in methanol (v/v). The combined extracts were concentrated to a low volume under reduced pressure. A low volume of 0.5% TFA in water (v/v) was added and the remaining methanol was removed under reduced pressure. The extract was partitioned against ethyl acetate, where three layers formed: water on the bottom, a thick layer of precipitate containing most of the Sphagnorubins in the middle and ethyl acetate on the top. The water and middle layer were removed and partitioned six more times against ethyl acetate. The purified extract was evaporated to dryness, recovered using a minimal volume of 0.5% TFA in methanol (v/v) and applied to a column of Amberlite XAD-7 resin (Supelco, Bellefonte, USA). Most of the pigments were eluted as a precipitate using 0.5% TFA in water (v/v), the eluate was evaporated to dryness under reduced pressure. Sphagnorubin C (1) was isolated from the extract using semi-preparative high-performance liquid chromatography (HPLC), which produced pure pigment suitable for 1D and 2D NMR spectroscopy and visible absorption spectroscopy.
4.2. Samples for Analysis
Each injection on the semi-preparative HPLC system yielded about 0.15 mg of pure 1. One batch was used to produce sample A, which was dissolved in 5% d-TFA in d-DMSO for analysis on NMR. Another batch was used to produce sample B, which was dissolved in pure d-DMSO for analysis on NMR. A third batch was dissolved in 10 mL 0.5% TFA in methanol and aliquoted into samples appropriate for visible absorption spectroscopy. Each aliquot was evaporated to dryness under a stream of nitrogen. These aliquots were used to produce samples C, D, and F1–F4. Sample C was dissolved in 0.5% TFA in DMSO and sample D was dissolved in pure DMSO. Sample F1 was identical to sample D, while samples F2–F4 was prepared by adding respectively one, two and three purification steps in addition to those used for preparation of F1. Each of these purification steps included adding pure methanol to pigment sample followed by evaporation to dryness under a stream of nitrogen. Samples F3 and F4 gave identical results.
4.3. Semi-Preparative High-Performance Liquid Chromatography (HPLC)
Semi-preparative HPLC were performed using a Gilson 321 pump (Gilson, Inc., Middleton, WI, USA) equipped with an UltiMate 3000 variable wavelength detector (Thermo Fisher Scientific, Waltham, MA, USA) set at 540 nm, a 25 × 2.2 cm (10 µm particle size) Econosphere C18 column (Grace, Columbia, MD, USA). The mobile phases were A, 1% TFA in water (v/v), and B, 1% TFA in methanol (v/v), used at a flow of 15 mL/min. A solvent elution profile consisted of initial conditions of 30% B and the following isocratic and gradient elution: 0–14 min gradient to 40% B, 14–20 min gradient to 50% B, 20–30 min isocratic, 30–32 min gradient to 80% B, 32–38 min isocratic, and a finally 38–41 min gradient to 30% B. Prior to injections, samples were filtered through a 0.45 µm filter.
4.4. Analytical HPLC
Analytical HPLC was used to assess the purity of the extract during each step of the isolation procedure. The instrument used was an HP Agilent 1200 analytical HPLC system (HP/Agilent, Santa Clara, USA) equipped with a diode array detector at 520 nm and 280 nm, and a 250 × 25 mm (4.5 µm particle size) Ascentis RP-Amide column (Supelco, Bellefonte, USA. The mobile phases were A, 1% TFA in water (v/v), and B, 1% TFA in acetonitrile (v/v), used at a flow of 1 mL/min. A solvent elution profile consisted of initial conditions of 30% B and the following isocratic and gradient elution: 0–7 min gradient to 40% B, 7–10 min gradient to 50% B, 10–15 min isocratic, 15–17 min gradient to 80% B, 17–22 min isocratic, and 22–25 min gradient to 30%. Injections were 20 µL aliquots injected by an auto-sampler. Prior to injections, samples were filtered through a 0.45 µm filter.
4.5. Nuclear Magnetic Resonance (NMR) Spectroscopy
The NMR experiments were obtained using a Bruker Ultrashield Plus AV-600 MHz (Bruker, Billerica, MA, USA) at 600.13 and 150.92 MHz for 1H and 13C spectra respectively, recorded at 21 °C. Deuterated dimethyl sulfoxide (d-DMSO) and deuterated trifluoroacetic acid (d-TFA) were used as solvents. 1H and 13C signals of the spectra were calibrated using the d-DMSO residual signal at 2.50 ppm and 39.51 ppm, respectively.
4.6. Mass Spectrometry
Purified samples of both the flavylium cation and the
trans-chalcone of
1 were analysed by high-resolution electron spray ionisation mass spectrometry (HR-ESI-MS). The spectra were recorded using a JEOL AccuTOF instrument (JEOL USA, Inc., Peabody, MA, USA). See
supplementary Figures S7 and S8 for details about the individual MS spectra.
4.7. Visible Absorption Spectroscopy
A HunterLab UltraScan PRO spectrophotometer (Hunter Associates Laboratory, Inc., Reston, VA, USA) was used to record visible absorption spectra. Pure solvents were used as background and extracted from each spectrum. The instrument was calibrated every 24 h. During 24 h of interval sampling, the cuvettes were covered with parafilm. The data from the instrument were imported to Microsoft Excel for data analysis.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}