
Molecular Modelling Studies on Pyrazole Derivatives for the Design of Potent Rearranged during Transfection Kinase Inhibitors

Abstract
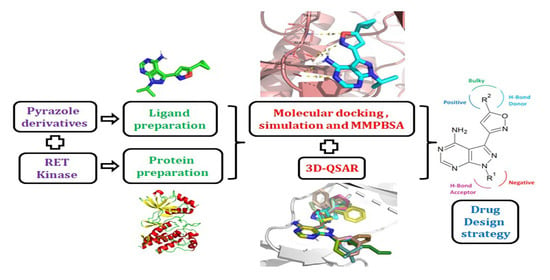
:
1. Introduction
2. Results
2.1. Molecular Docking
2.2. Molecular Dynamics Simulation
2.3. MM/PBSA Binding Free Energy Calculation
2.4. 3D-QSAR
Validation of CoMFA and CoMSIA Models
2.5. Contour Map Analysis
2.5.1. CoMFA Contour Maps
2.5.2. CoMSIA Contour Maps
2.6. Designing of RET Kinase Inhibitors
3. Discussion
4. Materials and Methods
4.1. Test Set/Training Set Selection for 3D-QSAR Analyses
4.2. Modeling of the Missing Residues
4.3. Preparation of the Protein and Molecular Docking
4.4. Molecular Dynamics Simulations
4.5. MM/PBSA Binding Free Energy Calculations
4.6. Receptor-Based CoMFA and CoMSIA Models
3D-QSAR Model Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Trenker, R.; Jura, N. Receptor tyrosine kinase activation: From the ligand perspective. Curr. Opin. Cell Biol. 2020, 63, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shen, T.; Mooers, B.H.M.; Hilberg, F.; Wu, J. Drug resistance profiles of mutations in the RET kinase domain. Br. J. Pharmacol. 2018, 175, 3504–3515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, A.Y.; McCusker, M.G.; Russo, A.; Scilla, K.A.; Gittens, A.; Arensmeyer, K.; Mehra, R.; Adamo, V.; Rolfo, C.D. RET fusions in solid tumors. Cancer Treat. Rev. 2019, 81, 101911. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr.; Sadeghi-Nejad, A. Role of RET protein-tyrosine kinase inhibitors in the treatment RET-driven thyroid and lung cancers. Pharmacol. Res. 2018, 128, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Elisei, R.; Schlumberger, M.J.; Müller, S.P.; Schöffski, P.; Brose, M.S.; Shah, M.H.; Licitra, L.; Jarzab, B.; Medvedev, V.; Kreissl, M.C.; et al. Cabozantinib in Progressive Medullary Thyroid Cancer. J. Clin. Oncol. 2013, 31, 3639–3646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, H.; Kwak, Y.; Choi, S.; Cho, H.; Kim, N.D.; Sim, T. A Pyrazolo[3,4-d]pyrimidin-4-amine Derivative Containing an Isoxazole Moiety Is a Selective and Potent Inhibitor of RET Gatekeeper Mutants. J. Med. Chem. 2015, 59, 358–373. [Google Scholar] [CrossRef] [PubMed]
- De Falco, V.; Carlomagno, F.; Li, H.-Y.; Santoro, M. The molecular basis for RET tyrosine-kinase inhibitors in thyroid cancer. Best Pr. Res. Clin. Endocrinol. Metab. 2017, 31, 307–318. [Google Scholar] [CrossRef]
- Yoon, H.; Shin, I.; Nam, Y.; Kim, N.D.; Lee, K.-B.; Sim, T. Identification of a novel 5-amino-3-(5-cyclopropylisoxazol-3-yl)-1-isopropyl-1H-pyrazole-4-carboxamide as a specific RET kinase inhibitor. Eur. J. Med. Chem. 2017, 125, 1145–1155. [Google Scholar] [CrossRef] [PubMed]
- Mologni, L.; Gambacorti-Passerini, C.; Goekjian, P.; Scapozza, L. RET kinase inhibitors: A review of recent patents (2012–2015). Expert Opin. Ther. Patents 2016, 27, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Matsui, J.; Yamamoto, Y.; Funahashi, Y.; Tsuruoka, A.; Watanabe, T.; Wakabayashi, T.; Uenaka, T.; Asada, M. E7080, a novel inhibitor that targets multiple kinases, has potent antitumor activities against stem cell factor producing human small cell lung cancer H146, based on angiogenesis inhibition. Int. J. Cancer 2007, 122, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Killock, D. SELECT—Lenvatinib in thyroid cancer. Nat. Rev. Clin. Oncol. 2015, 12, 189. [Google Scholar] [CrossRef] [PubMed]
- Tannir, N.M.; Pal, S.K.; Atkins, M.B. Second-line treatment landscape for renal cell carcinoma: A comprehensive review. Oncologist 2018, 23, 540. [Google Scholar] [CrossRef] [Green Version]
- Wells, S.A., Jr.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, R.; Jarzab, B.; Vasselli, J.R. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: A randomized, double-blind phase III trial. J. Clin. Oncol. 2012, 30, 134. [Google Scholar] [CrossRef] [Green Version]
- Hoy, S.M. Ponatinib: A review of its use in adults with chronic myeloid leukaemia or Philadelphia chromosome-positive acute lymphoblastic leukaemia. Drugs 2014, 74, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Mologni, L.; Redaelli, S.; Morandi, A.; Plaza-Menacho, I.; Gambacorti-Passerini, C. Ponatinib is a potent inhibitor of wild-type and drug-resistant gatekeeper mutant RET kinase. Mol. Cell. Endocrinol. 2013, 377, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ettrich, T.; Seufferlein, T. Regorafenib. In Toxicity Assessment; Springer Nature: Berlin, Germany, 2014; Volume 201, pp. 185–196. [Google Scholar]
- Anders, J.; Kjær, S.; Ibáñez, C.F. Molecular Modeling of the Extracellular Domain of the RET Receptor Tyrosine Kinase Reveals Multiple Cadherin-like Domains and a Calcium-binding Site. J. Biol. Chem. 2001, 276, 35808–35817. [Google Scholar] [CrossRef] [Green Version]
- Pathak, P.; Naumovich, V.; Grishina, M.; Shukla, P.K.; Verma, A.; Potemkin, V. Quinazoline based 1, 3, 5-triazine derivatives as cancer inhibitors by impeding the phosphorylated RET tyrosine kinase pathway: Design, synthesis, docking, and QSAR study. Archiv der Pharmazie 2019, 352, 1900053. [Google Scholar] [CrossRef]
- Eisenberg, D.; Schwarz, E.; Komaromy, M.; Wall, R. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J. Mol. Biol. 1984, 179, 125–142. [Google Scholar] [CrossRef]
- Terzyan, S.S.; Shen, T.; Liu, X.; Huang, Q.; Teng, P.; Zhou, M.; Hilberg, F.; Cai, J.; Mooers, B.H.M.; Wu, J. Structural basis of resistance of mutant RET protein-tyrosine kinase to its inhibitors nintedanib and vandetanib. J. Biol. Chem. 2019, 294, 10428–10437. [Google Scholar] [CrossRef]
- Lemkul, J.A. From Proteins to Perturbed Hamiltonians: A Suite of Tutorials for the GROMACS-2018 Molecular Simulation Package [Article v1.0]. Living J. Comput. Mol. Sci. 2019, 1, 5068. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Bang, S.J.; Cho, S.J. Comparative molecular field analysis (CoMFA) and comparative molecular similarity index analysis (CoMSIA) study of mutagen X. Bull. Korean Chem. Soc. 2004, 25, 1525–1530. [Google Scholar]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef]
- Klebe, G.; Abraham, U.; Mietzner, T. Molecular Similarity Indices in a Comparative Analysis (CoMSIA) of Drug Molecules to Correlate and Predict Their Biological Activity. J. Med. Chem. 1994, 37, 4130–4146. [Google Scholar] [CrossRef]
- San Juan, A.A.; Cho, S.J. 3D-QSAR study of microsomal prostaglandin E 2 synthase (mPGES-1) inhibitors. J. Mol. Model. 2007, 13, 601–610. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Predictive QSAR modeling based on diversity sampling of experimental datasets for the training and test set selection. J. Comput. Mol. Des. 2002, 16, 357–369. [Google Scholar] [CrossRef]
- Chirico, N.; Gramatica, P. Real External Predictivity of QSAR Models. Part 2. New Intercomparable Thresholds for Different Validation Criteria and the Need for Scatter Plot Inspection. J. Chem. Inf. Model. 2012, 52, 2044–2058. [Google Scholar] [CrossRef]
- Gadhe, C.G.; Madhavan, T.; Kothandan, G.; Cho, S.J. In Silico Quantitative Structure-Activity Relationship Studies on P-gp Modulators of Tetrahydroisoquinoline-Ethyl-Phenylamine Series. BMC Struct. Biol. 2011, 11, 5. [Google Scholar] [CrossRef] [Green Version]
- Roy, K.; Chakraborty, P.; Mitra, I.; Ojha, P.K.; Kar, S.; Das, R.N. Some case studies on application of “rm2” metrics for judging quality of quantitative structure-activity relationship predictions: Emphasis on scaling of response data. J. Comput. Chem. 2013, 34, 1071–1082. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Poso, A.; von Wright, A.; Gynther, J. An empirical and theoretical study on mechanisms of mutagenic activity of hydrazine compounds. Mutat. Res. Fund. Mol. M. 1995, 332, 63–71. [Google Scholar] [CrossRef]
- Thibaut, U.; Folkers, G.; Klebe, G.; Kubinyi, H.; Merz, A.; Rognan, D. Recommendations for CoMFA Studies and 3D QSAR Publications. Quant. Struct. Relatsh. 1994, 13, 1–3. [Google Scholar] [CrossRef]
- Plaza-Menacho, I.; Barnouin, K.; Goodman, K.; Martínez-Torres, R.J.; Borg, A.; Murray-Rust, J.; Mouilleron, S.; Knowles, P.; McDonald, N.Q. Oncogenic RET kinase domain mutations perturb the autophosphorylation trajectory by enhancing substrate presentation in trans. Mol. Cell 2014, 53, 738–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, B.; Sali, A. Protein structure modeling with MODELLER. In Protein Structure Prediction; Springer: New York, NY, USA, 2014; pp. 1–15. [Google Scholar]
- Melo, F.; Sánchez, R.; Sali, A. Statistical potentials for fold assessment. Protein Sci. 2009, 11, 430–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.-Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Da Silva, A.W.S.; Vranken, W.F. ACPYPE-Antechamber python parser interface. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2007, 4, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Gohlke, H.; Kiel, C.; Case, D.A. Insights into Protein–Protein Binding by Binding Free Energy Calculation and Free Energy Decomposition for the Ras–Raf and Ras–RalGDS Complexes. J. Mol. Biol. 2003, 330, 891–913. [Google Scholar] [CrossRef]
- Keretsu, S.; Bhujbal, S.P.; Cho, S.J. Computational study of paroxetine-like inhibitors reveals new molecular insight to inhibit GRK2 with selectivity over ROCK1. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gadhe, C.G.; Kothandan, G.; Cho, S.J. Large variation in electrostatic contours upon addition of steric parameters and the effect of charge calculation schemes in CoMFA on mutagenicity of MX analogues. Mol. Simul. 2012, 38, 861–871. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Full Model | Test Set 11 | ||
|---|---|---|---|---|
| CoMFA | CoMSIA (EHA) | CoMFA | CoMSIA (EHA) | |
| q2 | 0.563 | 0.509 | 0.649 | 0.557 |
| ONC | 6 | 4 | 6 | 5 |
| SDEP | 0.666 | 0.682 | 0.679 | 0.744 |
| r2 | 0.927 | 0.745 | 0.955 | 0.864 |
| SEE | 0.272 | 0.491 | 0.242 | 0.412 |
| F value | 59.187 | 21.918 | 71.390 | 26.749 |
| BS-r2 | - | - | 0.982 | 0.945 |
| BS-SD | - | - | 0.013 | 0.038 |
| Q2 | - | - | 0.531 | 0.508 |
| r2pred | - | - | 0.652 | 0.660 |
| rm2 | - | - | 0.532 | 0.601 |
| Delta rm2 | - | - | 0.073 | 0.072 |
 | |||
| Compound | R1 | R2 | pIC50 |
| D1 |  |  | 10.31 |
| D2 |  | | 8.85 |
| D3 |  | | 8.97 |
| D4 |  | | 9.02 |
| D5 |  | | 10.26 |
| D6 |  | | 10.74 |
| D7 |  | | 9.08 |
| D8 |  | | 9.20 |
| D9 |  | | 9.22 |
| D10 |  | | 8.80 |
| Designed Compound | Lipophilicity | Water Solubility | Pharmacokinetics | Synthetic Accessibility | Druglikeness | ||
|---|---|---|---|---|---|---|---|
| Log Po/w | Log S (ESOL) | Class | GI Absorption | Log Kp (Skin Permeation) | Lipinski Rule | ||
| D1 | 1.72 | −3.81 | Moderately soluble | Low | −7.46 cm/s | 3.86 | Yes; 0 violation |
| D2 | 1.97 | −4.10 | Moderately soluble | Low | −6.68 cm/s | 4.28 | Yes; 0 violation |
| D3 | 1.08 | −2.19 | Soluble | Low | −8.54 cm/s | 4.57 | Yes; 0 violation |
| D4 | 0.48 | −2.58 | Soluble | Low | −8.61 cm/s | 3.94 | Yes; 1 violation: NorO > 10 |
| D5 | 1.51 | −2.71 | Soluble | Low | −8.26 cm/s | 4.21 | Yes; 0 violation |
| D6 | 1.99 | −1.96 | Soluble | Low | −9.13 cm/s | 4.13 | Yes; 1 violation: NorO > 10 |
| D7 | 0.86 | −3.35 | Moderately soluble | Low | −8.39 cm/s | 3.96 | Yes; 1 violation: NorO > 10 |
| D8 | 1.26 | −1.98 | Soluble | Low | −8.61 cm/s | 3.99 | Yes; 0 violation |
| D9 | 0.47 | −2.31 | Soluble | Low | −8.96 cm/s | 4.12 | Yes; 1 violation: NorO > 10 |
| D10 | 2.43 | −4.14 | Moderately soluble | Low | −7.75 cm/s | 4.31 | Yes; 0 violation |
 Compounds 2–14 |  Compounds 18–35 | |||
| Compound | R1 | R2 | IC50 (µM) | pIC50 |
| 1 |  | 0.139 | 6.857 | |
| 2 * |  |  | 0.236 | 6.627 |
| 3 |  | | 1.639 | 5.785 |
| 4 |  | | 1.661 | 5.780 |
| 5 |  | | 0.416 | 6.381 |
| 6 |  | | 3.395 | 5.469 |
| 7 |  | | 4.803 | 5.318 |
| 8 |  | | 0.078 | 7.108 |
| 9 * |  | | 0.162 | 6.790 |
| 10 |  | | 0.631 | 6.200 |
| 11 * |  | | 1.259 | 5.900 |
| 12 |  | | 2.224 | 5.653 |
| 13 * |  | | 0.044 | 7.357 |
| 14 |  | | 2.695 | 5.569 |
| 15 |  | 4.90 | 5.310 | |
| 16 |  | 8.33 | 5.079 | |
| 17 |  | 0.583 | 6.234 | |
| 18 * | | | 0.190 | 6.721 |
| 19 | | | 0.241 | 6.618 |
| 20 | | | 0.150 | 6.824 |
| 21 * |  | | 0.910 | 6.041 |
| 22 |  | | 0.041 | 7.387 |
| 23 | | | 0.041 | 7.387 |
| 24 | | | 0.210 | 6.678 |
| 25 | | | 0.0014 | 8.854 |
| 26 |  | | 0.241 | 6.618 |
| 27 | | | 0.910 | 6.041 |
| 28 | H | | 0.213 | 6.672 |
| 29 * | | H | 0.052 | 7.284 |
| 30 | |  | 0.027 | 7.569 |
| 31 | | | 0.074 | 7.131 |
| 32 | |  | 0.096 | 7.018 |
| 33 * | |  | 0.068 | 7.167 |
| 34 | |  | 0.002 | 8.699 |
| 35 | |  | 0.005 | 8.301 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhujbal, S.P.; Keretsu, S.; Cho, S.J. Molecular Modelling Studies on Pyrazole Derivatives for the Design of Potent Rearranged during Transfection Kinase Inhibitors. Molecules 2021, 26, 691. https://doi.org/10.3390/molecules26030691
Bhujbal SP, Keretsu S, Cho SJ. Molecular Modelling Studies on Pyrazole Derivatives for the Design of Potent Rearranged during Transfection Kinase Inhibitors. Molecules. 2021; 26(3):691. https://doi.org/10.3390/molecules26030691
Chicago/Turabian StyleBhujbal, Swapnil P., Seketoulie Keretsu, and Seung Joo Cho. 2021. "Molecular Modelling Studies on Pyrazole Derivatives for the Design of Potent Rearranged during Transfection Kinase Inhibitors" Molecules 26, no. 3: 691. https://doi.org/10.3390/molecules26030691