
A Valence-Bond-Based Multiconfigurational Density Functional Theory: The λ-DFVB Method Revisited


Abstract
:1. Introduction
2. Methodology
- (1)
- Perform a normal VBSCF calculation to obtain the VBSCF density matrix;
- (2)
- Determine the λ value with Equations (9)–(13);
- (3)
- Compute the complement Hartree–exchange–correlation functional, with Equation (8);
- (4)
- Build potential in terms of one-electron integrals with Equation (15);
- (5)
- Optimize {CK} and {ϕi} by a modified VBSCF route, Equation (14);
- (6)
- Compute the λ-DFVB energy by Equation (16) with the optimized {CK} and {ϕi}.
- (1)
- Perform a normal VBSCF calculation to obtain the VBSCF density matrix;
- (2)
- Compute NOONs {ni} from the VBSCF wave function, and get the λ value using Equation (19);
- (3)
- Compute the complement Hartree–exchange–correlation functional, with Equation (8);
- (4)
- Compute the λ-DFVB(IS) energy by Equation (20).
3. Computational Details
4. Results and Discussion
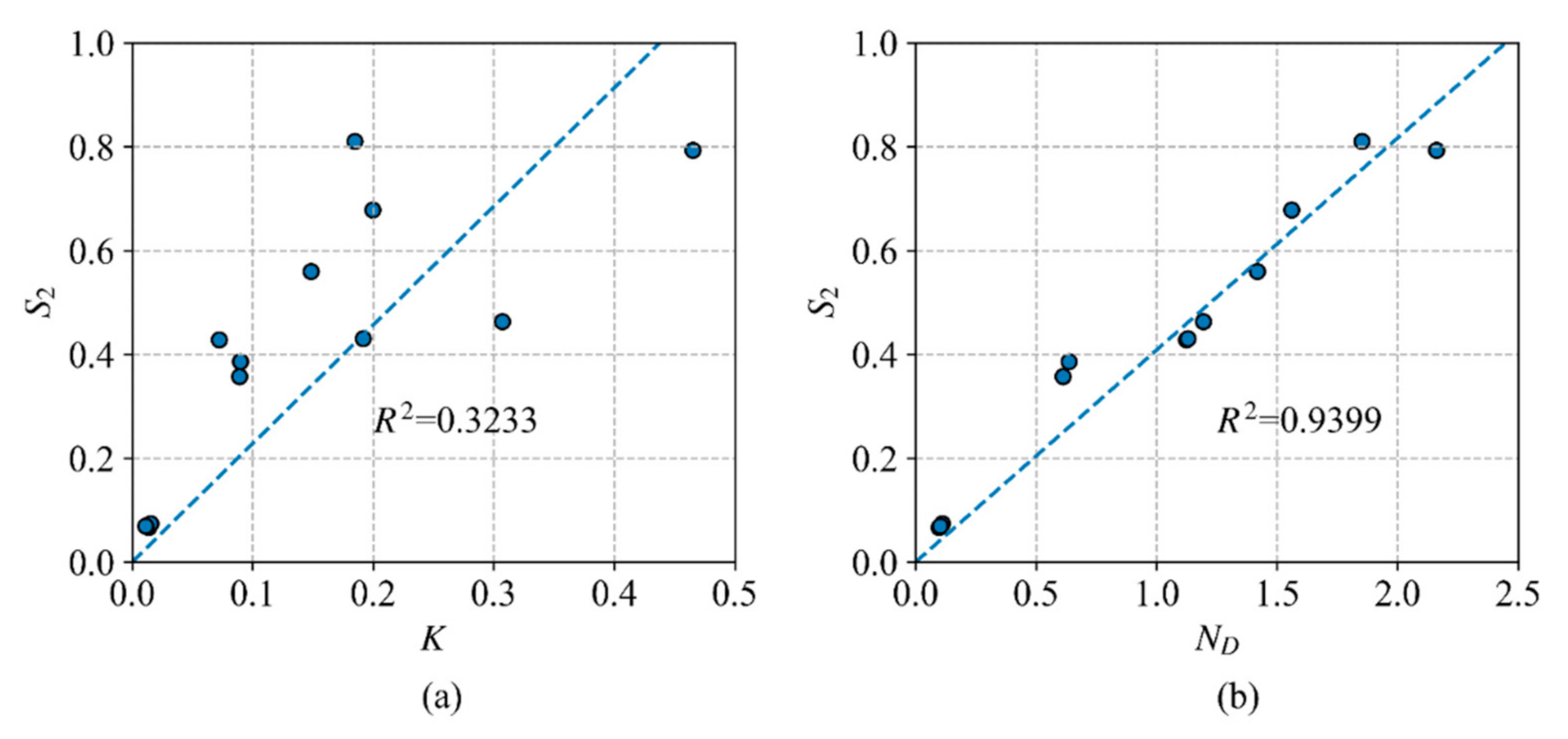
4.1. The Validity of the ND Index
4.2. Dissociation of Diatomic Molecules
4.3. Atomization Energies of Six Molecules
4.4. Atomic Excitation Energies
4.5. Chemical Reaction Barriers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A
References
- Roos, B.O.; Taylor, P.R.; Sigbahn, P.E. A complete active space SCF method (CASSCF) using a density matrix formulated super-CI approach. Chem. Phys. 1980, 48, 157–173. [Google Scholar] [CrossRef]
- Van Lenthe, J.; Balint-Kurti, G. The valence-bond scf (VB SCF) method: Synopsis of theory and test calculation of oh potential energy curve. Chem. Phys. Lett. 1980, 76, 138–142. [Google Scholar] [CrossRef]
- van Lenthe, J.H.; Balint-Kurti, G.G. The valence-bond self-consistent field method (VB–SCF): Theory and test calculations. J. Chem. Phys. 1983, 78, 5699–5713. [Google Scholar] [CrossRef]
- Andersson, K.; Malmqvist, P.Å.; Roos, B.O. Second-order perturbation theory with a complete active space self-consistent field reference function. J. Chem. Phys. 1992, 96, 1218–1226. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M.; Almlöf, J.; Heiberg, A.; Roos, B.O. The complete active space SCF (CASSCF) method in a Newton–Raphson formulation with application to the HNO molecule. J. Chem. Phys. 1981, 74, 2384–2396. [Google Scholar] [CrossRef]
- Wu, W.; Song, L.; Cao, Z.; Zhang, Q.; Shaik, S. Valence Bond Configuration Interaction: A Practical ab Initio Valence Bond Method That Incorporates Dynamic Correlation. J. Phys. Chem. A 2002, 106, 2721–2726. [Google Scholar] [CrossRef]
- Song, L.; Wu, W.; Zhang, Q.; Shaik, S. A practical valence bond method: A configuration interaction method approach with perturbation theoretic facility. J. Comput. Chem. 2004, 25, 472–478. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, X.; Ying, F.; Gu, J.; Zhang, H.; Wu, W. Nonorthogonal orbital based n-body reduced density matrices and their applications to valence bond theory. III. Second-order perturbation theory using valence bond self-consistent field function as reference. J. Chem. Phys. 2014, 141, 134118. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Song, J.; Shaik, S.; Hiberty, P.C.; Wu, W. Valence Bond Perturbation Theory. A Valence Bond Method That Incorporates Perturbation Theory. J. Phys. Chem. A 2009, 113, 11560–11569. [Google Scholar] [CrossRef] [PubMed]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef] [Green Version]
- Miehlich, B.; Stoll, H.; Savin, A. A correlation-energy density functional for multideterminantal wavefunctions. Mol. Phys. 1997, 91, 527–536. [Google Scholar] [CrossRef]
- Filatov, M.; Shaik, S. Spin-restricted density functional approach to the open-shell problem. Chem. Phys. Lett. 1998, 288, 689–697. [Google Scholar] [CrossRef]
- Filatov, M.; Shaik, S. Application of spin-restricted open-shell Kohn-Sham method to atomic and molecular multiplet states. J. Chem. Phys. 1999, 110, 116–125. [Google Scholar] [CrossRef]
- Grafenstein, J.; Cremer, D. The combination of density functional theory with multi-configuration methods—CAS-DFT. Chem. Phys. Lett. 2000, 316, 569–577. [Google Scholar] [CrossRef]
- Perez-Jimenez, A.J.; Perez-Jorda, J.M.; Illas, F. Density functional theory with alternative spin densities: Application to magnetic systems with localized spins. J. Chem. Phys. 2004, 120, 18–25. [Google Scholar] [CrossRef]
- Grafenstein, J.; Cremer, D. Development of a CAS-DFT method covering non-dynamical and dynamical electron correlation in a balanced way. Mol. Phys. 2005, 103, 279–308. [Google Scholar] [CrossRef]
- Fromager, E.; Toulouse, J.; Jensen, H.J.A. On the universality of the long-/short-range separation in multiconfigurational density-functional theory. J. Chem. Phys. 2007, 126, 074111. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Cheng, C.-L.; Van Voorhis, T. Configuration interaction based on constrained density functional theory: A multireference method. J. Chem. Phys. 2007, 127. [Google Scholar] [CrossRef]
- Cembran, A.; Song, L.; Mo, Y.; Gao, J. Block-Localized Density Functional Theory (BLDFT), Diabatic Coupling, and Their Use in Valence Bond Theory for Representing Reactive Potential Energy Surfaces. J. Chem. Theory Comput. 2009, 5, 2702–2716. [Google Scholar] [CrossRef] [Green Version]
- Fromager, E.; Real, F.; Wahlin, P.; Wahlgren, U.; Jensen, H.J.A. On the universality of the long-/short-range separation in multiconfigurational density-functional theory. II. Investigating f(0) actinide species. J. Chem. Phys. 2009, 131, 054107. [Google Scholar] [CrossRef] [PubMed]
- Sharkas, K.; Savin, A.; Jensen, H.J.A.; Toulouse, J. A multiconfigurational hybrid density-functional theory. J. Chem. Phys. 2012, 137, 044104. [Google Scholar] [CrossRef] [PubMed]
- Fromager, E.; Knecht, S.; Jensen, H.J.A. Multi-configuration time-dependent density-functional theory based on range separation. J. Chem. Phys. 2013, 138, 084101. [Google Scholar] [CrossRef] [PubMed]
- Stoyanova, A.; Teale, A.M.; Toulouse, J.; Helgaker, T.; Fromager, E. Alternative separation of exchange and correlation energies in multi-configuration range-separated density-functional theory. J. Chem. Phys. 2013, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li Manni, G.; Carlson, R.K.; Luo, S.; Ma, D.; Olsen, J.; Truhlar, D.G.; Gagliardi, L. Multiconfiguration Pair-Density Functional Theory. J. Chem. Theory Comput. 2014, 10, 3669–3680. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Grofe, A.; Ren, H.; Bao, P. Beyond Kohn-Sham Approximation: Hybrid Multistate Wave Function and Density Functional Theory. J. Phys. Chem Lett 2016, 7, 5143–5149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Verma, P.; Cramer, C.J.; Gagliardi, L.; Truhlar, D.G. Combining Wave Function Methods with Density Functional Theory for Excited States. Chem. Rev. 2018, 118, 7249–7292. [Google Scholar] [CrossRef]
- Mostafanejad, M.; Liebenthal, M.D.; DePrince, A.E. Global Hybrid Multiconfiguration Pair-Density Functional Theory. J. Chem. Theory Comput. 2020, 16, 2274–2283. [Google Scholar] [CrossRef] [Green Version]
- Qu, Z.; Ma, Y. Variational Multistate Density Functional Theory for a Balanced Treatment of Static and Dynamic Correlations. J. Chem. Theory Comput. 2020, 16, 4912–4922. [Google Scholar] [CrossRef]
- Savin, A. A Combined Density Functional and Configuration Interaction Method. Int. J. Quantum Chem. 1988, 34, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D.; Savin, A.; Stoll, H. Extension of the Local-Spin-Density Exchange-Correlation Approximation to Multiplet States. Theor. Chim. Acta 1995, 91, 147–156. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ernzerhof, M.; Burke, K.; Savin, A. On-Top Pair-Density Interpretation of Spin Density Functional Theory, with Applications to Magnetism. Int. J. Quantum Chem. 1997, 61, 197–205. [Google Scholar] [CrossRef]
- McDouall, J.J.W. Combining two-body density functionals with multiconfigurational wavefunctions: Diatomic molecules. Mol. Phys. 2003, 101, 361–371. [Google Scholar] [CrossRef]
- Gusarov, S.; Malmqvist, P.A.; Lindh, R. Using on-top pair density for construction of correlation functionals for multideterminant wave functions. Mol. Phys. 2004, 102, 2207–2216. [Google Scholar] [CrossRef]
- Wu, W.; Zhong, S.-j.; Shaik, S. VBDFT(s): A Hückel-type semi-empirical valence bond method scaled to density functional energies. Application to linear polyenes. Chem. Phys. Lett. 1998, 292, 7–14. [Google Scholar] [CrossRef]
- Wu, W.; Danovich, D.; Shurki, A.; Shaik, S. Using Valence Bond Theory to Understand Electronic Excited States: Application to the Hidden Excited State (21Ag) of C2nH2n+2(n = 2−14) Polyenes. J. Phys. Chem. A 2000, 104, 8744–8758. [Google Scholar] [CrossRef]
- Wu, W.; Luo, Y.; Song, L.; Shaik, S. VBDFT(s)—a semi-empirical valence bond method: Application to linear polyenes containing oxygen and nitrogen heteroatoms. Phys. Chem. Chem. Phys. 2001, 3, 5459–5465. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Song, L.; Wu, W.; Danovich, D.; Shaik, S. The Ground and Excited States of Polyenyl Radicals C2n−1H2n+1 (n = 2–13): A Valence Bond Study. ChemPhysChem 2004, 5, 515–528. [Google Scholar] [CrossRef]
- Wu, W.; Shaik, S. VB-DFT: A nonempirical hybrid method combining valence bond theory and density functional energies. Chem. Phys. Lett. 1999, 301, 37–42. [Google Scholar] [CrossRef]
- Ying, F.; Su, P.; Chen, Z.; Shaik, S.; Wu, W. DFVB: A Density-Functional-Based Valence Bond Method. J. Chem. Theory Comput. 2012, 8, 1608–1615. [Google Scholar] [CrossRef]
- Zhou, C.; Zhang, Y.; Gong, X.; Ying, F.; Su, P.; Wu, W. Hamiltonian Matrix Correction Based Density Functional Valence Bond Method. J. Chem. Theory Comput. 2017, 13, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Ying, F.; Zhou, C.; Zheng, P.; Luan, J.; Su, P.; Wu, W. λ-Density Functional Valence Bond: A Valence Bond-Based Multiconfigurational Density Functional Theory with a Single Variable Hybrid Parameter. Front. Chem 2019, 7, 225. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Kais, S. Entanglement as measure of electron–electron correlation in quantum chemistry calculations. Chem. Phys. Lett. 2005, 413, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Ziesche, P.; Gunnarsson, O.; John, W.; Beck, H. Two-site Hubbard model, the Bardeen-Cooper-Schrieffer model, and the concept of correlation entropy. Phys. Rev. B 1997, 55, 10270–10277. [Google Scholar] [CrossRef]
- Tishchenko, O.; Zheng, J.; Truhlar, D.G. Multireference Model Chemistries for Thermochemical Kinetics. J. Chem. Theory Comput. 2008, 4, 1208–1219. [Google Scholar] [CrossRef]
- Ramos-Cordoba, E.; Salvador, P.; Matito, E. Separation of dynamic and nondynamic correlation. Phys. Chem. Chem. Phys. 2016, 18, 24015–24023. [Google Scholar] [CrossRef] [Green Version]
- Shaik, S.; Hiberty, P.C. A Chemist’s Guide to Valence Bond. Theory; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007. [Google Scholar] [CrossRef]
- Wu, W.; Su, P.; Shaik, S.; Hiberty, P.C. Classical Valence Bond Approach by Modern Methods. Chem. Rev. 2011, 111, 7557–7593. [Google Scholar] [CrossRef]
- Li, J.; Pauncz, R. Efficient evaluation of the algebrants of VB wave functions using the successive expansion method. I. SpinS = 0. Int. J. Quantum Chem. 1997, 62, 245–259. [Google Scholar] [CrossRef]
- Chirgwin, B.H.; Coulson, C.A.; Randall, J.T. The electronic structure of conjugated systems. VI. Proc. R. Soc. Lond. Ser. A. Math. Phys. Sci. 1950, 201, 196–209. [Google Scholar] [CrossRef]
- Hiberty, P.C.; Flament, J.P.; Noizet, E. Compact and accurate valence bond functions with different orbitals for different configurations: Application to the two-configuration description of F2. Chem. Phys. Lett. 1992, 189, 259–265. [Google Scholar] [CrossRef]
- Hiberty, P.C.; Humbel, S.; Byrman, C.P.; Lenthe, J.H.v. Compact valence bond functions with breathing orbitals: Application to the bond dissociation energies of F2 and FH. J. Chem. Phys. 1994, 101, 5969–5976. [Google Scholar] [CrossRef] [Green Version]
- Hiberty, P.C.; Shaik, S. Breathing-orbital valence bond method—a modern valence bond method that includes dynamic correlation. Theor. Chem. Acc. 2002, 108, 255–272. [Google Scholar] [CrossRef]
- Mayer, I. On bond orders and valences in the Ab initio quantum chemical theory. Int. J. Quantum Chem. 1986, 29, 73–84. [Google Scholar] [CrossRef]
- Takatsuka, K.; Fueno, T.; Yamaguchi, K. Distribution of odd electrons in ground-state molecules. Theor. Chim. Acta 1978, 48, 175–183. [Google Scholar] [CrossRef]
- Staroverov, V.N.; Davidson, E.R. Distribution of effectively unpaired electrons. Chem. Phys. Lett. 2000, 330, 161–168. [Google Scholar] [CrossRef]
- Bochicchio, R.C.; Torre, A.; Lain, L. Comment on ‘Characterizing unpaired electrons from one-particle density matrix’ [M. Head-Gordon, Chem. Phys. Lett. 372 (2003) 508–511]. Chem. Phys. Lett. 2003, 380, 486–487. [Google Scholar] [CrossRef]
- Head-Gordon, M. Characterizing unpaired electrons from the one-particle density matrix. Chem. Phys. Lett. 2003, 372, 508–511. [Google Scholar] [CrossRef]
- Head-Gordon, M. Reply to comment on ‘characterizing unpaired electrons from the one-particle density matrix’. Chem. Phys. Lett. 2003, 380, 488–489. [Google Scholar] [CrossRef]
- Song, L.; Mo, Y.; Zhang, Q.; Wu, W. XMVB: A program for ab initio nonorthogonal valence bond computations. J. Comput. Chem. 2005, 26, 514–521. [Google Scholar] [CrossRef]
- Chen, Z.; Ying, F.; Chen, X.; Song, J.; Su, P.; Song, L.; Mo, Y.; Zhang, Q.; Wu, W. XMVB 2.0: A new version of Xiamen valence bond program. Int. J. Quantum Chem. 2015, 115, 731–737. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Fdez. Galván, I.; Vacher, M.; Alavi, A.; Angeli, C.; Aquilante, F.; Autschbach, J.; Bao, J.J.; Bokarev, S.I.; Bogdanov, N.A.; Carlson, R.K.; et al. OpenMolcas: From Source Code to Insight. J. Chem. Theory Comput. 2019, 15, 5925–5964. [Google Scholar] [CrossRef]
- Lynch, B.J.; Truhlar, D.G. Small Representative Benchmarks for Thermochemical Calculations. J. Phys. Chem. A 2003, 107, 8996–8999. [Google Scholar] [CrossRef]
- Zheng, J.; Zhao, Y.; Truhlar, D.G. The DBH24/08 Database and Its Use to Assess Electronic Structure Model Chemistries for Chemical Reaction Barrier Heights. J. Chem. Theory Comput. 2009, 5, 808–821. [Google Scholar] [CrossRef] [PubMed]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Papajak, E.; Leverentz, H.R.; Zheng, J.; Truhlar, D.G. Efficient Diffuse Basis Sets: Cc-pVxZ+ and maug-cc-pVxZ. J. Chem. Theory Comput. 2009, 5, 1197–1202. [Google Scholar] [CrossRef]
- Verma, P.; Truhlar, D. Geometries for Minnesota Database 2019. 2019. [Google Scholar] [CrossRef]
- Johnson, R.D., III. NIST Computational Chemistry Comparison and Benchmark Database, NIST Standard Reference Database Number 101, Release 21, August 2020. Available online: http://cccbdb.nist.gov/ (accessed on 4 January 2021).
- Lemmon, E.; McLinden, M.; Friend, D. Thermophysical Properties of Fluid Systems in NIST Chemistry WebBook, NIST Standard Reference Database Number 69; Linstrom, P.J., Mallard, W.G., Eds.; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2019.
- Leininger, M.L.; Sherrill, C.D.; Allen, W.D.; III, H.F.S. Benchmark configuration interaction spectroscopic constants for X 1Σg+ C2 and X 1Σ+ CN+. J. Chem. Phys. 1998, 108, 6717–6721. [Google Scholar] [CrossRef]
- Carlson, R.K.; Li Manni, G.; Sonnenberger, A.L.; Truhlar, D.G.; Gagliardi, L. Multiconfiguration Pair-Density Functional Theory: Barrier Heights and Main Group and Transition Metal Energetics. J. Chem. Theory Comput. 2015, 11, 82–90. [Google Scholar] [CrossRef]
- Verma, P.; Wang, Y.; Ghosh, S.; He, X.; Truhlar, D.G. Revised M11 Exchange-Correlation Functional for Electronic Excitation Energies and Ground-State Properties. J. Phys. Chem. A 2019, 123, 2966–2990. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.E. CRC Series in Evaluated Data in Atomic Physics; Gallagher, J., Ed.; CRC Press: Boca Raton, FL, USA, 1993. [Google Scholar]
- Kramida, A.; Martin, W.C. A Compilation of Energy Levels and Wavelengths for the Spectrum of Neutral Beryllium (Be I). J. Phys. Chem. Ref. Data 1997, 26, 1185–1194. [Google Scholar] [CrossRef]
- Martin, W.C.; Kaufman, V.; Musgrove, A. A Compilation of Energy Levels and Wavelengths for the Spectrum of Singly-Ionized Oxygen (O II). J. Phys. Chem. Ref. Data 1993, 22, 1179–1212. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Active Space | CASPT2 | VBSCF | λ-DFVB | BLYP | B3LYP | Ref. 1 | ||
|---|---|---|---|---|---|---|---|---|
| K | IS | |||||||
| H2 | (2, 2) | 0.004 | 0.014 | 0.003 | 0.003 | 0.005 | 0.002 | 0.741 |
| F2 | (2, 2) | 0.007 | 0.055 | −0.040 | −0.035 | 0.020 | −0.015 | 1.412 |
| HF | (2, 2) | −0.001 | −0.001 | −0.009 | −0.008 | 0.016 | 0.005 | 0.917 |
| N2 | (6, 6) | 0.005 | 0.005 | 0.000 | −0.003 | 0.005 | −0.007 | 1.098 |
| C2 | (8, 8) | 0.007 | 0.012 | 0.008 | 0.008 | 0.013 | 0.004 | 1.243 |
| MUE | 0.005 | 0.017 | 0.012 | 0.011 | 0.012 | 0.007 | ||
| Active Space | CASPT2 | VBSCF | λ-DFVB | BLYP | B3LYP | Ref. 1 | ||
|---|---|---|---|---|---|---|---|---|
| K | IS | |||||||
| H2 | (2, 2) | −3.6 | −14.2 | −1.0 | −1.1 | 0.0 | 0.7 | 109.5 |
| F2 | (2, 2) | −3.2 | −21.4 | 1.0 | 0.2 | 12.4 | −0.1 | 38.2 |
| HF | (2, 2) | −4.1 | −27.7 | 1.6 | −0.1 | −4.0 | −4.2 | 141.3 |
| N2 | (6, 6) | −11.7 | −24.4 | −3.1 | −6.5 | 11.3 | 0.5 | 228.5 |
| C2 | (8, 8) | −1.9 | −5.4 | −15.9 | −1.6 | −12.7 | −28.5 | 148.0 |
| MUE | 4.9 | 18.6 | 4.5 | 1.9 | 8.1 | 6.8 | ||
| Active Space | CASPT2 | VBSCF | λ-DFVB | BLYP | B3LYP | Ref. 1 | ||
|---|---|---|---|---|---|---|---|---|
| K | IS | |||||||
| SiH4 | (8, 8) | 79.3 | 73.4 | 79.7 | 79.1 | 79.1 | 80.5 | 81.1 |
| S2 | (8, 6) | 98.0 | 76.0 | 92.8 | 93.8 | 104.8 | 100.7 | 103.1 |
| SiO | (6, 6) | 184.6 | 190.3 | 193.3 | 190.1 | 192.7 | 184.8 | 192.4 |
| C3H4 | (8, 8) | 116.4 | 101.5 | 115.0 | 114.5 | 116.9 | 117.0 | 117.5 |
| C2H2O2 | (10, 10) | 124.7 | 108.0 | 125.3 | 123.9 | 128.2 | 126.1 | 126.7 |
| C4H8 | (8, 8) | 94.4 | 77.7 | 93.2 | 92.8 | 94.3 | 95.2 | 95.8 |
| MUE | 3.2 | 14.9 | 3.1 | 3.7 | 1.2 | 2.0 | ||
| Excitation | Active Space | CASPT2 | VBSCF | λ-DFVB | BLYP | B3LYP | Ref. 1 | ||
|---|---|---|---|---|---|---|---|---|---|
| K | IS | ||||||||
| Be | 1S→3P | (2, 4) | 2.78 | 2.81 | 3.66 | 3.08 | 2.48 | 2.46 | 2.73 |
| C | 3P→1D | (4, 4) | 1.26 | 1.53 | 1.14 | 1.19 | 0.33 | 0.38 | 1.26 |
| N+ | 3P→1D | (4, 4) | 1.87 | 2.12 | 1.72 | 1.79 | 0.56 | 0.62 | 1.89 |
| N | 4S→2D | (5, 4) | 2.47 | 2.79 | 2.13 | 2.14 | 0.94 | 1.04 | 2.38 |
| O+ | 4S→2D | (5, 4) | 3.40 | 3.70 | 3.03 | 3.03 | 1.4 | 1.53 | 3.32 |
| O | 3P→1D | (6, 4) | 1.91 | 2.18 | 1.78 | 1.81 | 0.65 | 0.70 | 1.96 |
| MUE | 0.05 | 0.27 | 0.32 | 0.20 | 1.20 | 1.14 | |||
| Active Space | CASPT2 | VBSCF | λ-DFVB | BLYP | B3LYP | Ref. 1 | ||
|---|---|---|---|---|---|---|---|---|
| K | IS | |||||||
| OH + CH4 → CH3 + H2O | (3, 3) | 5.9 | 23.9 | 3.8 | 2.2 | −2.3 | 2.3 | 6.3 |
| reverse | 19.6 | 29.6 | 23.1 | 25.8 | 10.4 | 13.8 | 19.5 | |
| H + OH → O + H2 | (4, 4) | 11.7 | 17.9 | 11.0 | 11.4 | 1.3 | 3.9 | 10.9 |
| reverse | 14.2 | 29.4 | 14.8 | 15.4 | 1.5 | 6.3 | 13.2 | |
| H + H2S → HS + H2 | (3, 3) | 5.1 | 11.8 | 4.2 | 3.1 | −2.3 | −0.7 | 3.9 |
| reverse | 18.3 | 26.1 | 21.3 | 24.2 | 14.7 | 16.2 | 17.2 | |
| H + N2O → N2 + OH | (11, 9) | 19.7 | 30.8 | 18.7 | 18.8 | 8.7 | 11.5 | 17.7 |
| reverse | 81.2 | 108.7 | 78.7 | 75.9 | 62.4 | 73.5 | 82.6 | |
| H + ClH → HCl + H | (3, 3) | 19.7 | 29.6 | 18.2 | 16.8 | 10.5 | 13.1 | 17.8 |
| reverse | 19.7 | 29.6 | 18.2 | 16.8 | 10.5 | 13.1 | 17.8 | |
| CH3 + FCl → CH3F + Cl | (3, 3) | 6.5 | 15.0 | 8.5 | 14.5 | −7.1 | −1.6 | 7.1 |
| reverse | 63.7 | 71.4 | 76.3 | 73.9 | 42.4 | 51.6 | 59.8 | |
| Cl− ··· CH3Cl → ClCH3 ··· Cl− | (4, 3) | 10.0 | 19.3 | 10.3 | 12.7 | 5.2 | 8.7 | 13.5 |
| reverse | 10.0 | 19.3 | 10.3 | 12.7 | 5.2 | 8.7 | 13.5 | |
| F− ··· CH3Cl → FCH3 ··· Cl− | (4, 3) | 4.2 | 5.6 | 1.5 | 2.4 | −1.8 | 0.2 | 3.5 |
| reverse | 28.2 | 47.9 | 31.2 | 34.6 | 20.6 | 26.3 | 29.6 | |
| OH− + CH3F → HOCH3 + F− | (4, 3) | −0.6 | 8.9 | −3.5 | 0.9 | −7.9 | −4.5 | −2.7 |
| reverse | 19.9 | 28.6 | 18.5 | 20.3 | 11.5 | 15.9 | 17.6 | |
| H + N2 → HN2 | (7, 7) | 17.4 | 29.0 | 16.6 | 16.5 | 5.4 | 7.7 | 14.6 |
| reverse | 11.2 | −2.0 | 7.3 | 6.9 | 8.5 | 10.9 | 10.9 | |
| H + C2H4 → CH3CH2 | (3, 3) | 2.7 | 7.6 | 3.3 | 1.7 | −0.7 | −0.2 | 2.0 |
| reverse | 41.4 | 37.7 | 43.6 | 44.3 | 38.2 | 41.8 | 42.0 | |
| HCN → HNC | (10, 9) | 48.2 | 56.3 | 46.8 | 44.4 | 46.8 | 47.4 | 48.1 |
| reverse | 32.6 | 36.9 | 37.0 | 38.3 | 31.9 | 33.5 | 33.0 | |
| MUE | 1.4 | 10.6 | 2.6 | 3.5 | 7.7 | 4.2 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, P.; Ji, C.; Ying, F.; Su, P.; Wu, W. A Valence-Bond-Based Multiconfigurational Density Functional Theory: The λ-DFVB Method Revisited. Molecules 2021, 26, 521. https://doi.org/10.3390/molecules26030521
Zheng P, Ji C, Ying F, Su P, Wu W. A Valence-Bond-Based Multiconfigurational Density Functional Theory: The λ-DFVB Method Revisited. Molecules. 2021; 26(3):521. https://doi.org/10.3390/molecules26030521
Chicago/Turabian StyleZheng, Peikun, Chenru Ji, Fuming Ying, Peifeng Su, and Wei Wu. 2021. "A Valence-Bond-Based Multiconfigurational Density Functional Theory: The λ-DFVB Method Revisited" Molecules 26, no. 3: 521. https://doi.org/10.3390/molecules26030521