
Theoretical Investigation of Carbon Dioxide Adsorption on Li+-Decorated Nanoflakes


Abstract
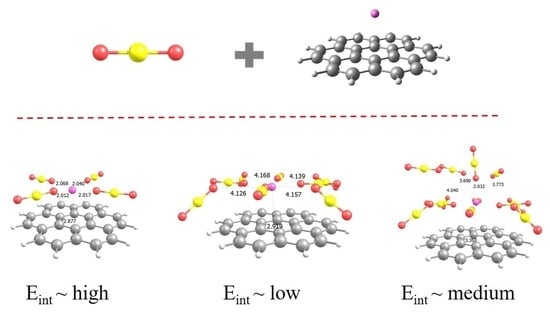
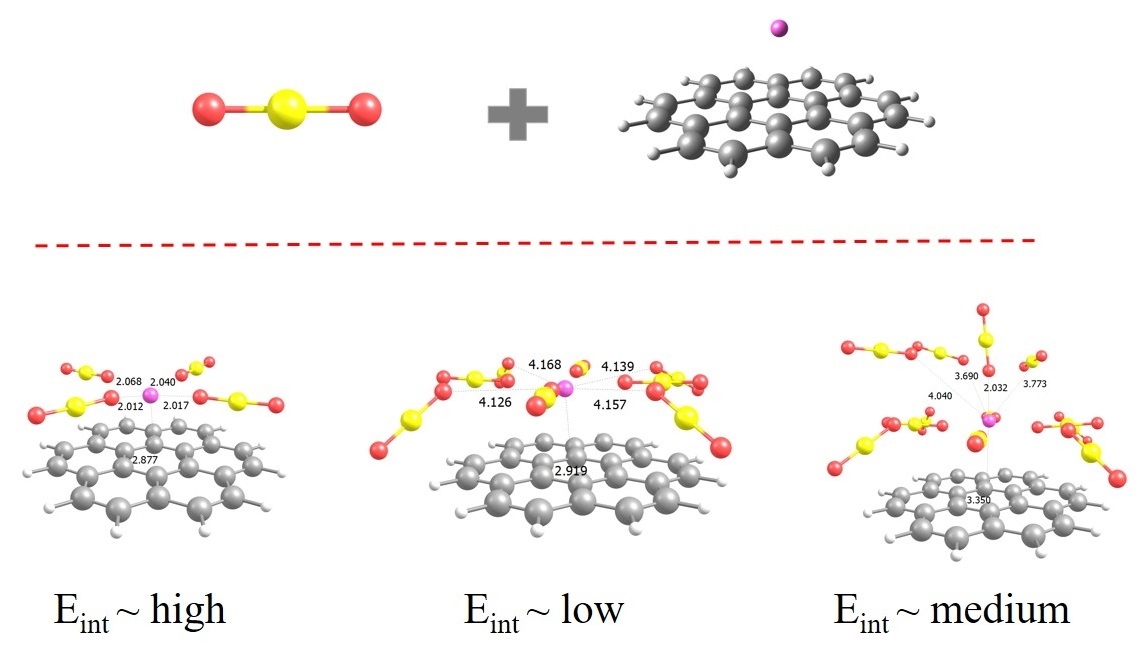
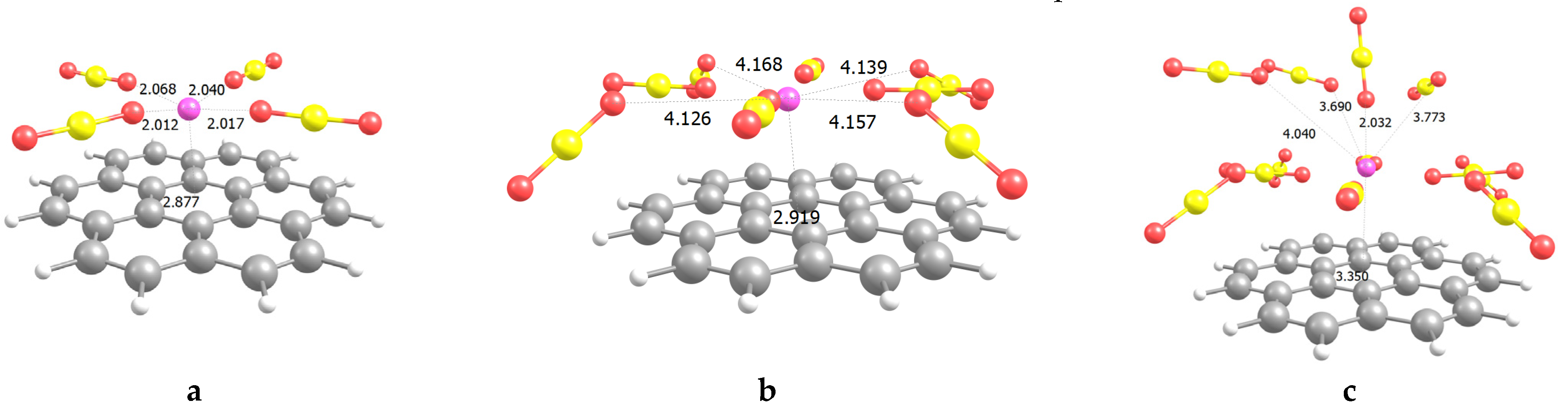
:
1. Introduction
2. Results and Discussions
2.1. CO2 Adsorption on Li+@coronene Complexes
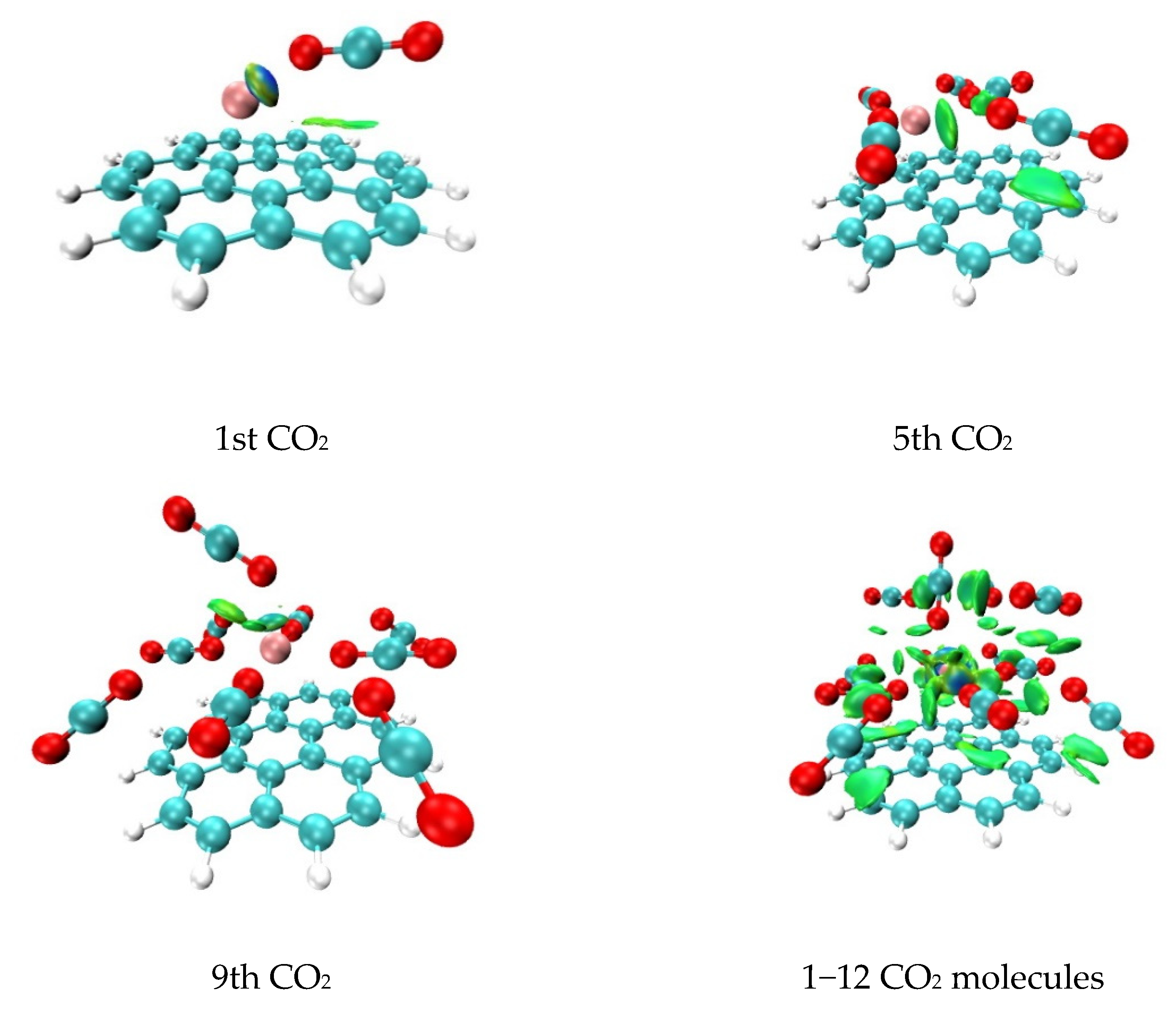
2.2. Analysis of Non-Covalent Interactions between CO2 Molecules and the Li+@coronene Complex by Quantum Theory of Atoms in Molecules (QTAIM)
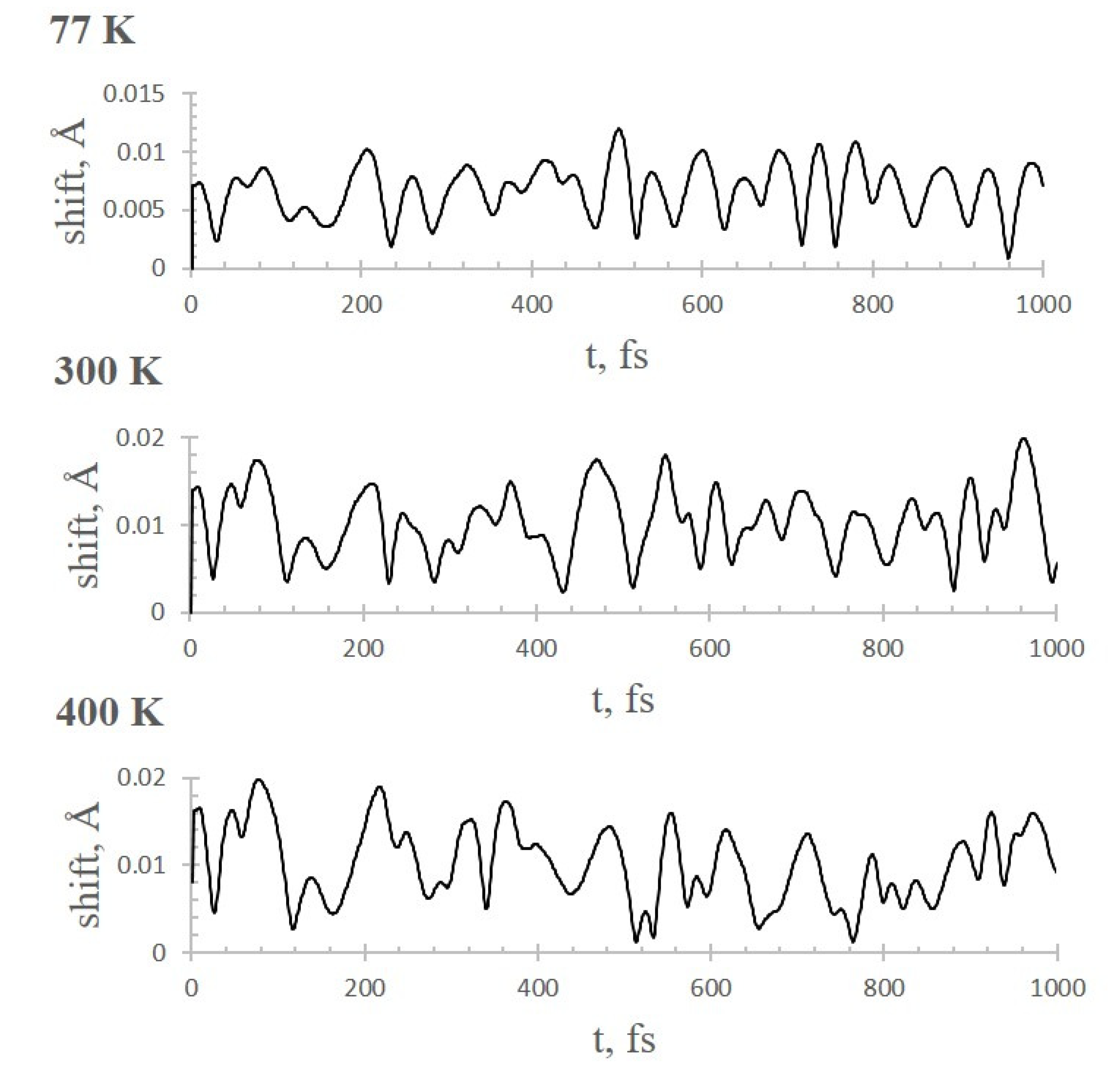
2.3. AIMD Simulations of CO2 Adsorption on Li+@coronene
3. Computational Methods
3.1. Computational Details
3.2. Validation of Methodology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Yu, C.-H.; Huang, C.-H.; Tan, C.-S. A Review of CO2 Capture by Absorption and Adsorption. Aerosol Air Qual. Res. 2012, 12, 745–769. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Shi, X.; Liu, Y.; Xiao, H.; Li, J.; Chen, X. Nanomaterials for adsorption and conversion of CO2 under gentle conditions. Mater. Today 2021. [Google Scholar] [CrossRef]
- Song, X.; Liu, Y.; Yang, Y.; Li, W.; Zhao, M. Strain-tunable CO2 storage by black phosphorene and alpha-PC from combined first principles and molecular dynamics studies. Phys. Chem. Chem. Phys. 2019, 21, 20107–20117. [Google Scholar] [CrossRef] [PubMed]
- Akilan, R.; Ravichandran, D.; Vinnarasi, S.; Shankar, R. Adsorption of H2 and CO2 gas molecules on Li/Na decorated Si2BN nano-sheet for energy harvesting applications—A density functional study. Mater. Lett. 2020, 279, 128487. [Google Scholar] [CrossRef]
- Regufe, M.J.; Ribeiro, A.M.; Ferreira, A.F.P.; Rodrigues, A. CO2 Storage on Zeolites and Other Adsorbents. In Nanoporous Materials for Gas Storage. Green Energy and Technology; Kaneko, K., Rodríguez-Reinoso, F., Eds.; Springer: Singapore, 2019; pp. 359–381. [Google Scholar]
- Yu, Y.; Li, X.; Krishna, R.; Liu, Y.; Cui, Y.; Du, J.; Liang, Z.; Song, X.; Yu, J. Enhancing CO2 Adsorption and Separation Properties of Aluminophosphate Zeolites by Isomorphous Heteroatom Substitutions. ACS Appl. Mater. Interfaces 2018, 10, 43570–43577. [Google Scholar] [CrossRef]
- Al Mesfer, M.K.; Danish, M. Breakthrough adsorption study of activated carbons for CO2 separation from flue gas. J. Environ. Chem. Eng. 2018, 6, 4514–4524. [Google Scholar] [CrossRef]
- Shi, S.; Liu, Y. Nitrogen-doped activated carbons derived from microalgae pyrolysis by-products by microwave/KOH activation for CO2 adsorption. Fuel 2021, 306, 121762. [Google Scholar] [CrossRef]
- Chen, S.-Y.; Hui, Y.; Yang, Y.-B. Monte Carlo simulations of adsorption and separation of binary mixtures of CO2, SO2, and H2S by charged single-walled carbon nanotubes. Soft Mater. 2020, 18, 262–273. [Google Scholar] [CrossRef]
- Lee, K.-J.; Kim, S.-J. Theoretical Investigation of CO2 Adsorption on Graphene. Bull. Korean Chem. Soc. 2013, 34, 3022–3026. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Tang, C.; He, T.; Wijethunge, D.; Yan, C.; Zhu, Z.; Du, A. Computational screening of MN4 (M = Ti-Cu) based metal organic frameworks for CO2 reduction using the d-band centre as a descriptor. Nanoscale 2020, 12, 6188–6194. [Google Scholar] [CrossRef]
- Meconi, G.M.; Zangi, R. Adsorption-induced clustering of CO2 on graphene. Phys. Chem. Chem. Phys. 2020, 22, 21031–21041. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.K.; Ramaprabhu, S. Carbon dioxide adsorption in graphene sheets. AIP Adv. 2011, 1, 032152. [Google Scholar] [CrossRef]
- Xia, K.; Xiong, R.; Chen, Y.; Liu, D.; Tian, Q.; Gao, Q.; Han, B.; Zhou, C. Tuning the pore structure and surface chemistry of porous graphene for CO2 capture and H2 storage. Colloids Surf. A Physicochem. Eng. Asp. 2021, 622, 126640. [Google Scholar] [CrossRef]
- Papageorgiou, D.G.; Kinloch, I.A.; Young, R.J. Mechanical properties of graphene and graphene-based nanocomposites. Prog. Mater. Sci. 2017, 90, 75–127. [Google Scholar] [CrossRef]
- Kumar, A.; Sharma, K.; Dixit, A.R. A review of the mechanical and thermal properties of graphene and its hybrid polymer nanocomposites for structural applications. J. Mater. Sci. 2018, 54, 5992–6026. [Google Scholar] [CrossRef]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef]
- Gadipelli, S.; Guo, Z.X. Graphene-based materials: Synthesis and gas sorption, storage and separation. Prog. Mater. Sci. 2015, 69, 1–60. [Google Scholar] [CrossRef] [Green Version]
- Singla, M.; Jaggi, N. Theoretical investigations of hydrogen gas sensing and storage capacity of graphene-based materials: A review. Sens. Actuators A Phys. 2021, 332, 113118. [Google Scholar] [CrossRef]
- D’Alessandro, D.M.; Smit, B.; Long, J.R. Carbon dioxide capture: Prospects for new materials. Angew. Chem. 2010, 49, 6058–6082. [Google Scholar] [CrossRef] [Green Version]
- Politakos, N.; Barbarin, I.; Cordero-Lanzac, T.; Gonzalez, A.; Zangi, R.; Tomovska, R. Reduced Graphene Oxide/Polymer Monolithic Materials for Selective CO2 Capture. Polymers 2020, 12, 936. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Lin, Y.; Hagio, T.; Hu, Y.H. Surface-microporous graphene for CO2 adsorption. Catal. Today 2020, 356, 514–518. [Google Scholar] [CrossRef]
- dos Santos, T.C.; Mancera, R.C.; Rocha, M.V.J.; da Silva, A.F.M.; Furtado, I.O.; Barreto, J.; Stavale, F.; Archanjo, B.S.; de M. Carneiro, J.W.; Costa, L.T.; et al. CO2 and H2 adsorption on 3D nitrogen-doped porous graphene: Experimental and theoretical studies. J. CO2 Util. 2021, 48, 101517. [Google Scholar] [CrossRef]
- Ghosh, K.; Mridha, N.K.; Khan, A.A.; Baildya, N.; Dutta, T.; Biswas, K.; Ghosh, N.N. CO2 activation on transition metal decorated graphene quantum dots: An insight from first principles. Phys. E Low-Dimens. Syst. Nanostructures 2022, 135, 114993. [Google Scholar] [CrossRef]
- Liu, A.; Long, J.; Yuan, S.; Cen, W.; Li, J. Synergetic promotion by oxygen doping and Ca decoration on graphene for CO2 selective adsorption. Phys. Chem. Chem. Phys. 2019, 21, 5133–5141. [Google Scholar] [CrossRef]
- Tawfik, S.A.; Cui, X.Y.; Ringer, S.P.; Stampfl, C. Multiple CO2 capture in stable metal-doped graphene: A theoretical trend study. RSC Adv. 2015, 5, 50975–50982. [Google Scholar] [CrossRef] [Green Version]
- To, J.W.; He, J.; Mei, J.; Haghpanah, R.; Chen, Z.; Kurosawa, T.; Chen, S.; Bae, W.G.; Pan, L.; Tok, J.B.; et al. Hierarchical N-Doped Carbon as CO2 Adsorbent with High CO2 Selectivity from Rationally Designed Polypyrrole Precursor. J. Am. Chem. Soc. 2016, 138, 1001–1009. [Google Scholar] [CrossRef]
- Re Fiorentin, M.; Gaspari, R.; Quaglio, M.; Massaglia, G.; Saracco, G. Nitrogen doping and CO2 adsorption on graphene: A thermodynamical study. Phys. Rev. B 2018, 97. [Google Scholar] [CrossRef]
- Öztürk, Z. Lithium decoration characteristics for hydrogen storage enhancement in novel periodic porous graphene frameworks. Int. J. Hydrog. Energy 2021, 46, 11804–11814. [Google Scholar] [CrossRef]
- Ni, J.; Quintana, M.; Song, S. Adsorption of small gas molecules on transition metal (Fe, Ni and Co, Cu) doped graphene: A systematic DFT study. Phys. E Low-Dimens. Syst. Nanostructures 2020, 116, 113768. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Sharifi, F.; Nematollahi, P. A comparative theoretical study of CO oxidation reaction by O2 molecule over Al- or Si-decorated graphene oxide. J. Mol. Graph. Model. 2016, 69, 8–16. [Google Scholar] [CrossRef]
- Kheirabadi, R.; Vakili, M. Computational modeling in enhanced CO2 and C2H2 capture on chalcogen atom (Se, Te)-decorated graphene: Structural and mechanistic aspects. J. Iran. Chem. Soc. 2021. [Google Scholar] [CrossRef]
- Kumar, R.; Suresh, V.M.; Maji, T.K.; Rao, C.N. Porous graphene frameworks pillared by organic linkers with tunable surface area and gas storage properties. Chem. Commun. 2014, 50, 2015–2017. [Google Scholar] [CrossRef]
- Salih, E.; Ayesh, A.I. Pt-doped armchair graphene nanoribbon as a promising gas sensor for CO and CO2: DFT study. Phys. E Low-Dimens. Syst. Nanostructures 2021, 125, 114418. [Google Scholar] [CrossRef]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Lefebvre, C.; Rubez, G.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Hénon, E. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 2017, 19, 17928–17936. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, C.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Piquemal, J.-P.; Hénon, E. The Independent Gradient Model: A New Approach for Probing Strong and Weak Interactions in Molecules from Wave Function Calculations. ChemPhysChem 2018, 19, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Umadevi, D.; Sastry, G.N. Molecular and Ionic Interaction with Graphene Nanoflakes: A Computational Investigation of CO2, H2O, Li, Mg, Li+, and Mg2+ Interaction with Polycyclic Aromatic Hydrocarbons. J. Phys. Chem. C 2011, 115, 9656–9667. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, Z.; Gong, Y.; Zhou, S.; Wang, J.; Wang, Z.; Wei, S.; Guo, W.; Lu, X. Penta-graphene as a promising controllable CO2 capture and separation material in an electric field. Appl. Surf. Sci. 2020, 502, 144067. [Google Scholar] [CrossRef]
- Wang, C.; Fang, Y.; Duan, H.; Liang, G.; Li, W.; Chen, D.; Long, M. DFT study of CO2 adsorption properties on pristine, vacancy and doped graphenes. Solid State Commun. 2021, 337, 114436. [Google Scholar] [CrossRef]
- Cabrera-Sanfelix, P. Adsorption and reactivity of CO(2) on defective graphene sheets. J. Phys. Chemistry. A 2009, 113, 493–498. [Google Scholar] [CrossRef]
- Liu, Y.; Wilcox, J. CO2 adsorption on carbon models of organic constituents of gas shale and coal. Environ. Sci. Technol. 2011, 45, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Cinke, M.; Li, J.; Bauschlicher, C.W.; Ricca, A.; Meyyappan, M. CO2 adsorption in single-walled carbon nanotubes. Chem. Phys. Lett. 2003, 376, 761–766. [Google Scholar] [CrossRef]
- Vidali, G.; Ihm, G.; Kim, H.-Y.; Cole, M.W. Potentials of physical adsorption. Surf. Sci. Rep. 1991, 12, 135–181. [Google Scholar] [CrossRef]
- Montoya, A.; Mondragón, F.; Truong, T.N. CO2 adsorption on carbonaceous surfaces: A combined experimental and theoretical study. Carbon 2003, 41, 29–39. [Google Scholar] [CrossRef]
- Okamoto, Y.; Miyamoto, Y. Ab Initio Investigation of Physisorption of Molecular Hydrogen on Planar and Curved Graphenes. J. Phys. Chem. B 2001, 105, 3470–3474. [Google Scholar] [CrossRef]
- Petrushenko, I.K.; Tikhonov, N.I.; Petrushenko, K.B. Hydrogen adsorption on pillar[6]arene: A computational study. Phys. E Low-Dimens. Syst. Nanostructures 2021, 130, 114719. [Google Scholar] [CrossRef]
- Petrushenko, I.K.; Bettinger, H.F. Hydrogen adsorption on inorganic benzenes decorated with alkali metal cations: Theoretical study. Phys. Chem. Chem. Phys. 2021, 23, 5315–5324. [Google Scholar] [CrossRef] [PubMed]
- Petrushenko, I.K.; Tsar’kova, A.I.; Petrushenko, K.B. Hydrogen adsorption on BN-embedded tetrabenzopentacene as a promising nanoflake for energy storage: Theoretical insights. Diam. Relat. Mater. 2020, 108, 107968. [Google Scholar] [CrossRef]
- Darvishnejad, M.H.; Reisi-Vanani, A. Multiple CO2 capture in pristine and Sr-decorated graphyne: A DFT-D3 and AIMD study. Comput. Mater. Sci. 2020, 176, 109539. [Google Scholar] [CrossRef]
- Venkataramanan, N.S.; Suvitha, A.; Kawazoe, Y. Intermolecular interaction in nucleobases and dimethyl sulfoxide/water molecules: A DFT, NBO, AIM and NCI analysis. J. Mol. Graph. Model. 2017, 78, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Sun, X.; Zhang, L.; Zhang, C.; Jiang, G. Hydrogen storage of Li4&B36 cluster. Sci. Rep. 2018, 8, 1940. [Google Scholar] [CrossRef] [PubMed]
- Gal, J.F.; Maria, P.C.; Decouzon, M.; Mo, O.; Yanez, M.; Abboud, J.L. Lithium-cation/pi complexes of aromatic systems. The effect of increasing the number of fused rings. J. Am. Chem. Soc. 2003, 125, 10394–10401. [Google Scholar] [CrossRef]
- Sevilla, M.; Fuertes, A.B. CO2 adsorption by activated templated carbons. J. Colloid Interface Sci. 2012, 366, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Himeno, S.; Komatsu, T.; Fujita, S. High-Pressure Adsorption Equilibria of Methane and Carbon Dioxide on Several Activated Carbons. J. Chem. Eng. Data 2005, 50, 369–376. [Google Scholar] [CrossRef]
- Zhao, Y.; Ding, H.; Zhong, Q. Preparation and characterization of aminated graphite oxide for CO2 capture. Appl. Surf. Sci. 2012, 258, 4301–4307. [Google Scholar] [CrossRef]
- Thiruvenkatachari, R.; Su, S.; Yu, X.X.; Bae, J.-S. Application of carbon fibre composites to CO2 capture from flue gas. Int. J. Greenh. Gas. Control. 2013, 13, 191–200. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Sun, F.; Liu, X.; Gao, J.; Qie, Z.; Zhao, G. The effect of nitrogen-containing functional groups on SO2 adsorption on carbon surface: Enhanced physical adsorption interactions. Surf. Sci. 2018, 677, 78–82. [Google Scholar] [CrossRef]
- Rafique, M.; Uqaili, M.A.; Mirjat, N.H.; Tunio, M.A.; Shuai, Y. Ab-initio investigations on titanium (Ti) atom-doped divacancy monolayer h-BN system for hydrogen storage systems. Phys. E Low-Dimens. Syst. Nanostructures 2019, 109, 169–178. [Google Scholar] [CrossRef]
- Muhammad, R.; Shuai, Y.; Irfan, A.; He-Ping, T. First-principles investigations of manganese oxide (MnOx) complex-sandwiched bilayer graphene systems. RSC Adv. 2018, 8, 23688–23697. [Google Scholar] [CrossRef] [Green Version]
- Muhammad, R.; Shuai, Y.; Tan, H.-P. First-principles study on hydrogen adsorption on nitrogen doped graphene. Phys. E Low-Dimens. Syst. Nanostructures 2017, 88, 115–124. [Google Scholar] [CrossRef]
- Rafique, M.; Shuai, Y.; Tan, H.-P.; Hassan, M. Manipulating intrinsic behaviors of graphene by substituting alkaline earth metal atoms in its structure. RSC Adv. 2017, 7, 16360–16370. [Google Scholar] [CrossRef] [Green Version]
- Hohenstein, E.G.; Sherrill, C.D. Density fitting and Cholesky decomposition approximations in symmetry-adapted perturbation theory: Implementation and application to probe the nature of π-π interactions in linear acenes. J. Chem. Phys. 2010, 132, 184111. [Google Scholar] [CrossRef] [Green Version]
- Hohenstein, E.G.; Sherrill, C.D. Wavefunction methods for noncovalent interactions. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 304–326. [Google Scholar] [CrossRef]
- Turney, J.M.; Simmonett, A.C.; Parrish, R.M.; Hohenstein, E.G.; Evangelista, F.A.; Fermann, J.T.; Mintz, B.J.; Burns, L.A.; Wilke, J.J.; Abrams, M.L.; et al. Psi4: An open-source ab initio electronic structure program. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 556–565. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Gryn’ova, G.; Corminboeuf, C. Steric “attraction”: Not by dispersion alone. Beilstein J. Org. Chem. 2018, 14, 1482–1490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, A.; Dinadayalane, T.C.; Leszczynski, J. Effect of ring annelation on Li+–benzene interaction: A computational study. Chem. Phys. Lett. 2007, 443, 205–210. [Google Scholar] [CrossRef]
- Ma, J.C.; Dougherty, D.A. The Cation-pi Interaction. Chem. Rev. 1997, 97, 1303–1324. [Google Scholar] [CrossRef]
- Amicangelo, J.C.; Armentrout, P.B. Absolute Binding Energies of Alkali-Metal Cation Complexes with Benzene Determined by Threshold Collision-Induced Dissociation Experiments and ab Initio Theory. J. Phys. Chem. A 2000, 104, 11420–11432. [Google Scholar] [CrossRef]
- Petrushenko, I.K.; Shipitsin, N.V.; Petrushenko, K.B. Cation-π interactions of inorganic benzenes with Li, Na, and Mg cations: Theoretical insights. Inorg. Chem. Commun. 2020, 118, 108043. [Google Scholar] [CrossRef]
- Vijay, D.; Sakurai, H.; Subramanian, V.; Sastry, G.N. Where to bind in buckybowls? The dilemma of a metal ion. Phys. Chem. Chem. Phys. 2012, 14, 3057–3065. [Google Scholar] [CrossRef]
- Petrushenko, I.K.; Shipitsin, N.V.; Petrushenko, K.B. N-substituted sumanene and cation-π interactions towards Li cations: A theoretical study. Phys. E Low-Dimens. Syst. Nanostructures 2022, 135, 114949. [Google Scholar] [CrossRef]
- Reisi-Vanani, A.; Shabani, Z. Evaluation of the hydrogen adsorption onto Li and Li+ decorated circumtrindene (C36H12): A theoretical study. Int. J. Hydrogen Energy 2017, 42, 22973–22986. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. CO2 | Eel | Eex | Eind | Edisp | Eint |
|---|---|---|---|---|---|
| 1 | −14.34 (50) | 11.98 | −8.99 (31) | −5.52 (19) | −16.87 |
| 2 | −13.83 (47) | 13.06 | −8.48 (29) | −7.32 (25) | −16.57 |
| 3 | −13.4 (49) | 12.79 | −7.11 (26) | −6.57 (24) | −14.29 |
| 4 | −11.08 (45) | 13.15 | −5.44 (23) | −7.89 (32) | −11.26 |
| 5 | −6.44 (44) | 9.03 | −1.58 (10) | −6.75 (46) | −5.73 |
| 6 | −7.52 (46) | 9.36 | −1.74 (11) | −6.95 (43) | −6.85 |
| 7 | −7.02 (46) | 9.04 | −1.62 (11) | −6.65 (43) | −6.24 |
| 8 | −5.96 (44) | 7.9 | −1.37 (10) | −6.13 (46) | −5.55 |
| 9 | −6.88 (48) | 7.29 | −3.29 (22) | −4.28 (30) | −7.16 |
| 10 | −8.65 (47) | 9.92 | −3.71 (20) | −5.96 (33) | −8.94 |
| 11 | −10.11 (48) | 11.47 | −4.24 (20) | −6.85 (32) | −9.73 |
| 12 | −5.94 (48) | 6.31 | −1.48 (12) | −5.03 (40) | −6.14 |
| Complex | ρ(r) | ∇2ρ(r) | H(r) | V(r) | G(r) |
|---|---|---|---|---|---|
| 1st CO2 | 0.0279 | 0.2117 | 0.0121 | −0.0287 | 0.0408 |
| 2nd CO2 | 0.0271 | 0.2084 | 0.0121 | −0.0279 | 0.0400 |
| 3rd CO2 | 0.0246 | 0.1913 | 0.0113 | −0.0252 | 0.0365 |
| 4th CO2 | 0.0160 | 0.1145 | 0.0065 | −0.0156 | 0.0221 |
| 9th CO2 | 0.0144 | 0.0969 | 0.0050 | −0.0143 | 0.0193 |
| 10th CO2 | 0.0068 | 0.0238 | 0.0005 | −0.0049 | 0.0054 |
| 11th CO2 | 0.0094 | 0.0415 | 0.0019 | −0.0067 | 0.0085 |
| 12th CO2 | 0.0077 | 0.0332 | 0.0016 | −0.0052 | 0.0067 |
| Li+@coronene | |||
|---|---|---|---|
| GD, DFT | GD, AIMD | ||
| - | 77 K | 300 K | 400 K |
| 5.0 (9.3) 1 | 5.0 (9.3) | 3.2 (6.0) | 2.6 (4.9) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrushenko, I.K.; Ivanov, N.A.; Petrushenko, K.B. Theoretical Investigation of Carbon Dioxide Adsorption on Li+-Decorated Nanoflakes. Molecules 2021, 26, 7688. https://doi.org/10.3390/molecules26247688
Petrushenko IK, Ivanov NA, Petrushenko KB. Theoretical Investigation of Carbon Dioxide Adsorption on Li+-Decorated Nanoflakes. Molecules. 2021; 26(24):7688. https://doi.org/10.3390/molecules26247688
Chicago/Turabian StylePetrushenko, Igor K., Nikolay A. Ivanov, and Konstantin B. Petrushenko. 2021. "Theoretical Investigation of Carbon Dioxide Adsorption on Li+-Decorated Nanoflakes" Molecules 26, no. 24: 7688. https://doi.org/10.3390/molecules26247688