
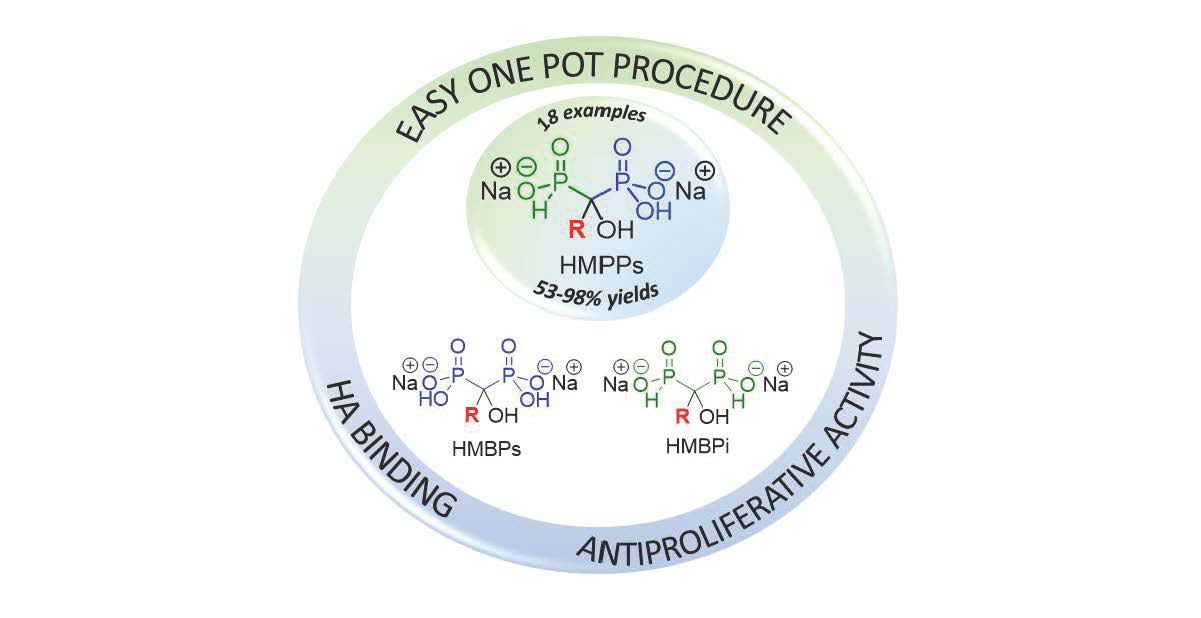
One-Pot Synthesis of Phosphinylphosphonate Derivatives and Their Anti-Tumor Evaluations

 , ,
, , 
Abstract
:
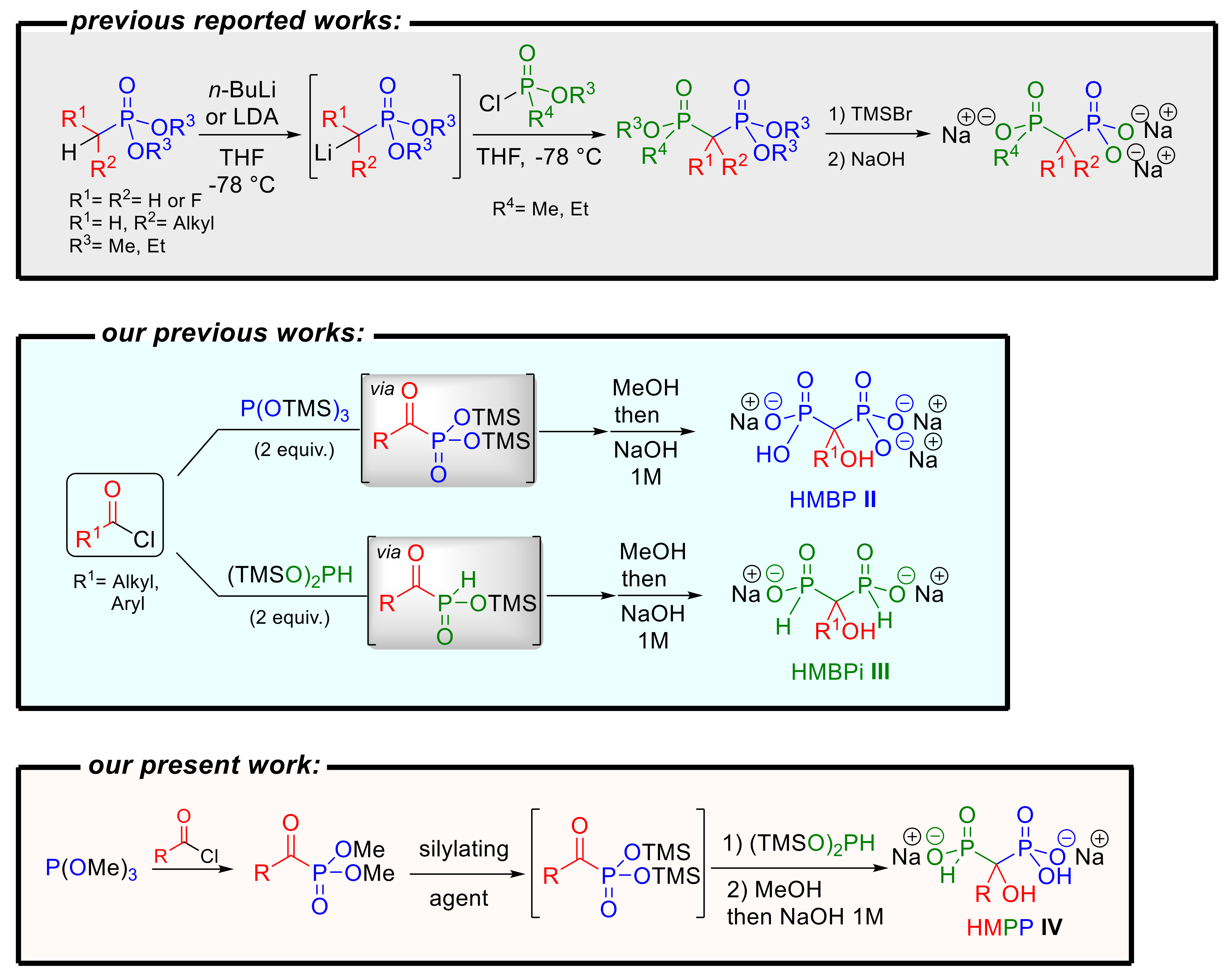
1. Introduction
2. Results and Discussion
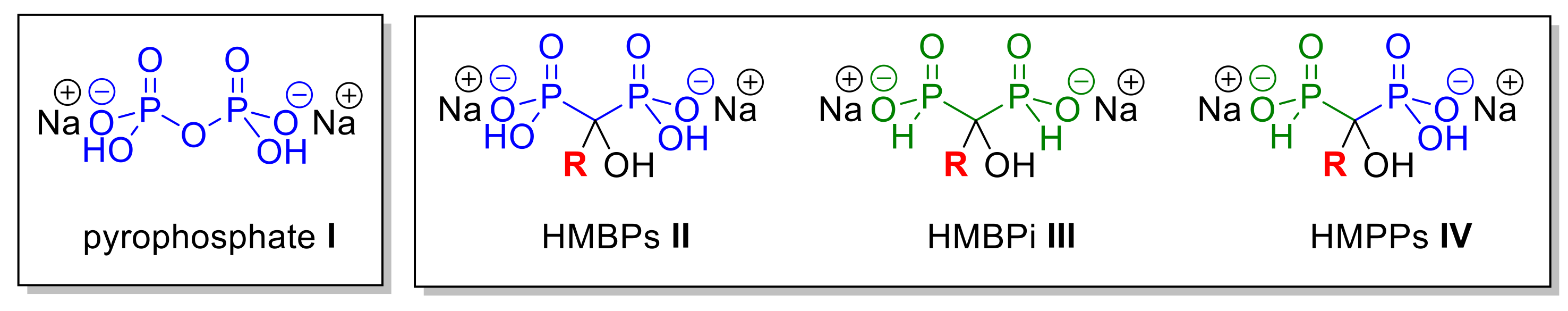
2.1. Chemistry
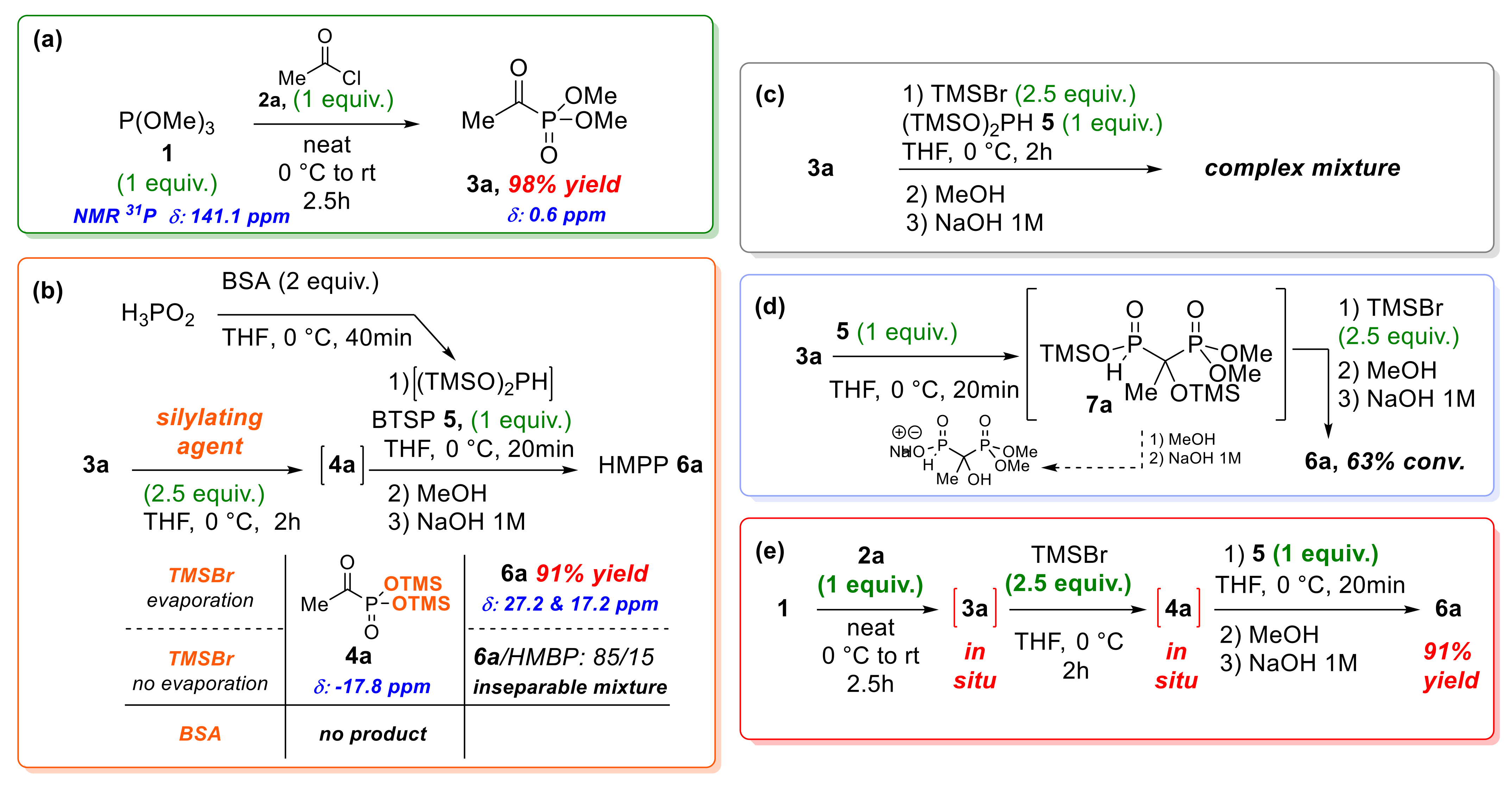
2.1.1. Development of an Optimized One-Pot Sequence
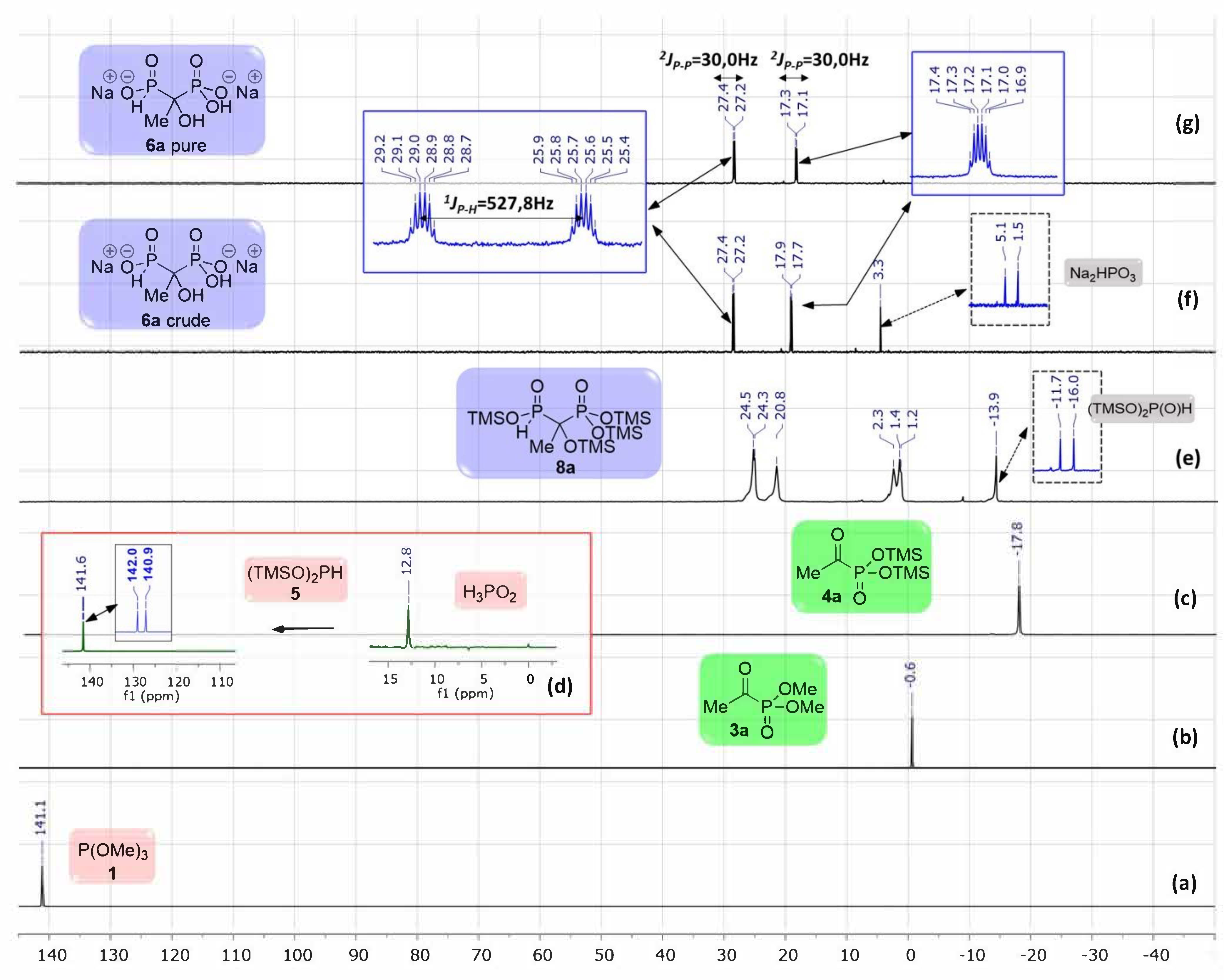
2.1.2. NMR Monitoring of the One-Pot Reaction
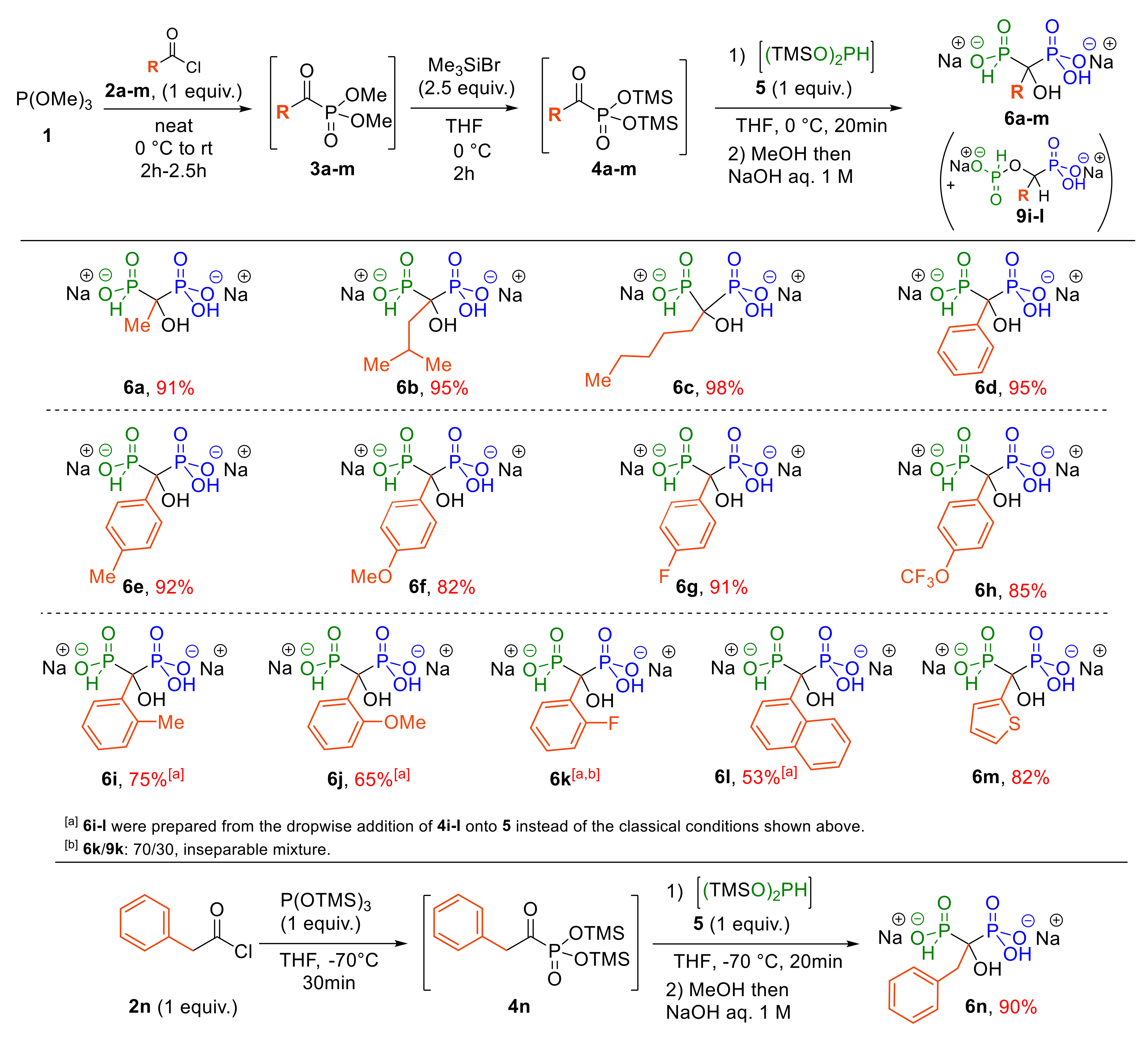
2.1.3. Scope of the Reaction
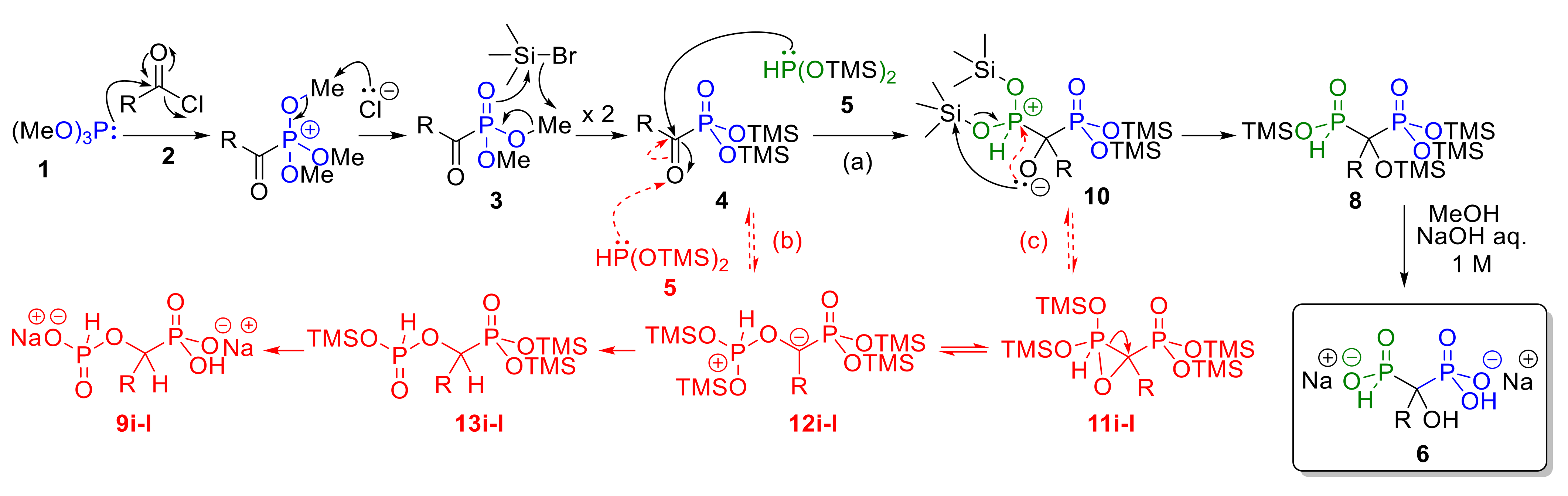
2.1.4. Postulated Mechanism
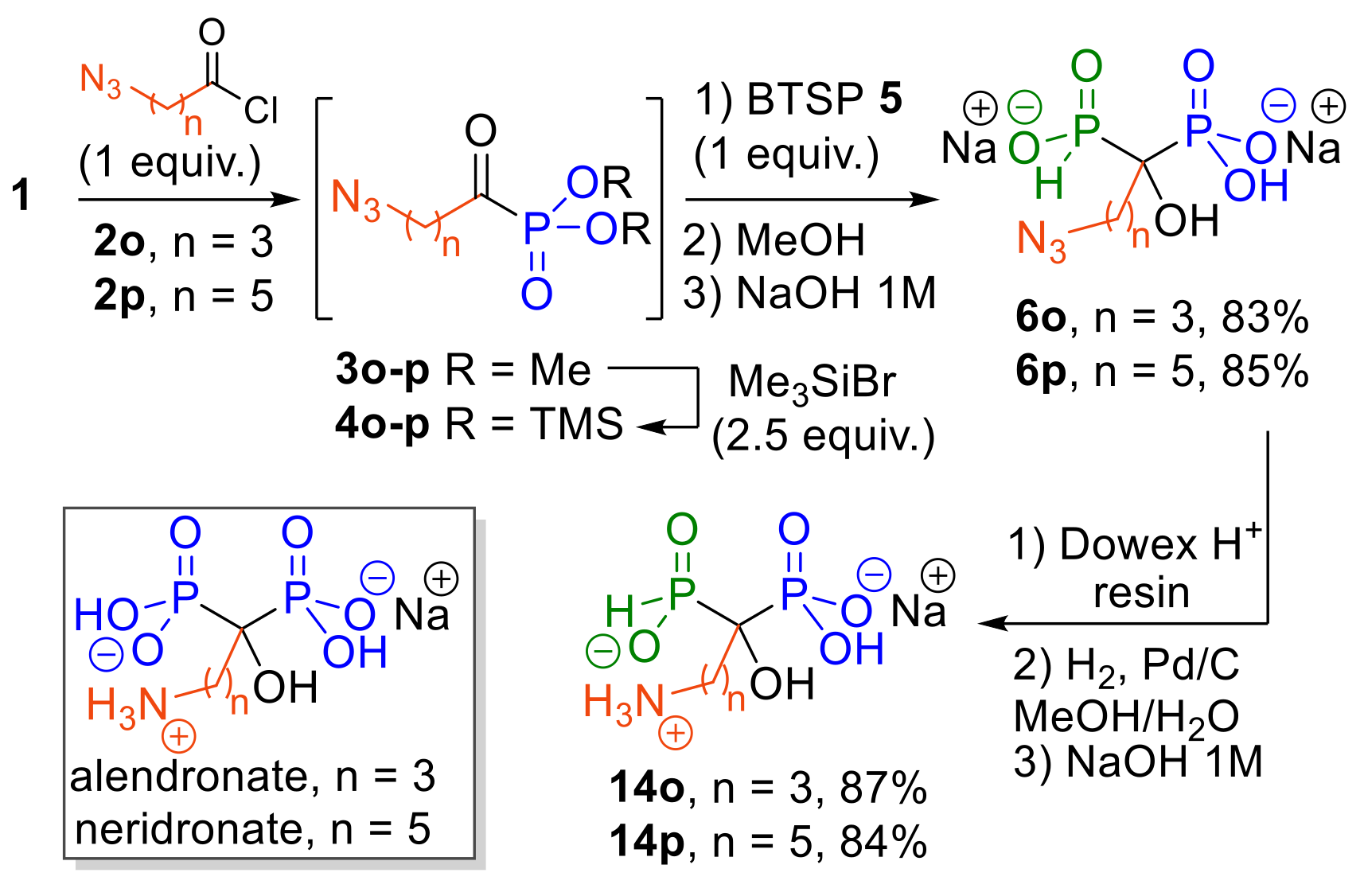
2.1.5. Synthesis of Aminoalkyl-HMPPs
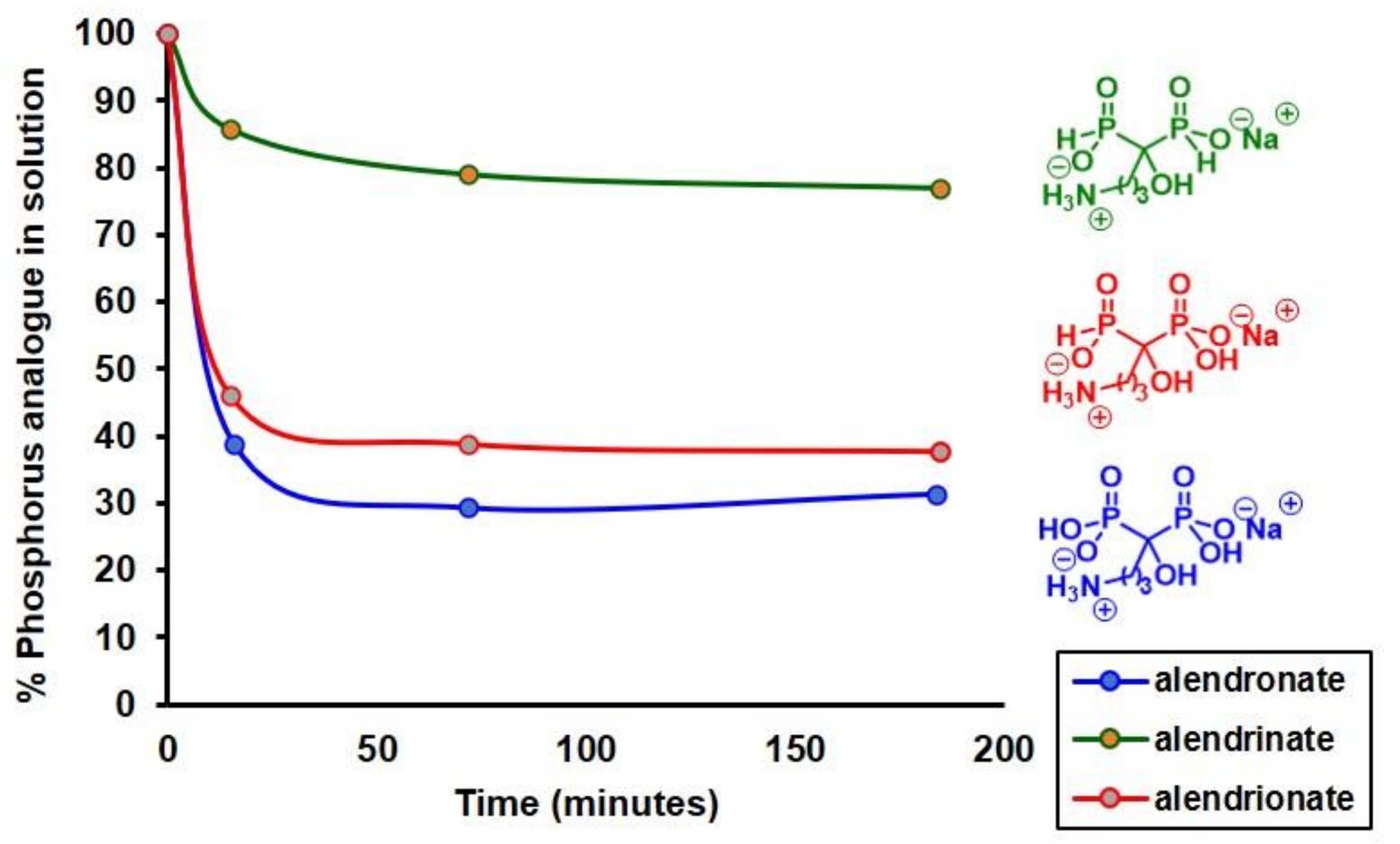
2.2. Complexation Study
2.3. Antiproliferative Evaluation
3. Materials and Methods
3.1. General Information
3.2. Chemistry
3.2.1. General Procedure Pathway (b) with Evaporation
3.2.2. General Procedure for Pathway (e)
3.2.3. General Synthesis Procedure for 6i–j,l
3.2.4. General Synthesis Procedure for 6n
- 1-Hydroxyethane-1,1-(H-phosphinylphosphonate) disodium salt 6a. White powder (1.06 g, 91% yield (from pathway (b) with evaporation), 1.06 g, 91% yield (from pathway (e)); IR (neat, cm−1) ν = 3415 br, 2955 w, 2884 w, 2853 w, 1150 m, 1040 s, 848 s, 733 m 31P{1H} NMR (162 MHz, D2O) δ 27.3 (d, 2JP-P = 31.7 Hz), 17.2 (d, 2JP-P = 31.7 Hz); 31P NMR (162 MHz, D2O) δ 27.3 (ddq, 1JP-H = 526.8 Hz, 2JP-P = 31.7 Hz, 3JP-H = 14.8 Hz), 17.2 (dq, 2JP-P = 31.7 Hz, 3JP-H = 14.8 Hz); 1H NMR (400 MHz, D2O) δ 6.83 (d, 1JP-H = 526.8 Hz, 1H, P-H), 1.36 (t, 3JP-H = 14.3 Hz, 3H); 13C NMR (101 MHz, D2O) δ 71.8 (dd, 1JP-C = 140.3 Hz, 94.3 Hz), 17.2; MS (ESI−) m/z: 188.97 [M-H]−, 210.95 [M-2H+Na]−, 170.96 [M-H-H2O]−, 122.99 [M-H-H3PO2−; HRMS (ESI−) m/z: [M-H]− Calcd. for C2H7O6P2 188.9723, found: 188.9726.
- 1-Hydroxy-3-methylbutane-1,1-(H-phosphinylphosphonate) disodium salt 6b. White powder (1.31 g, 95% yield (from pathway (e));); IR (neat, cm−1) ν = 3220br, 2956 w, 2315 w, 1185 s, 1110 m, 1048 s, 745 s; 31P{1H} NMR (162 MHz, D2O) δ 27.8 (d, 2JP-P = 25.9 Hz), 16.6 (d, 2JP-P = 25.9 Hz); 31P NMR (162 MHz, D2O) δ 27.8 (bd, 1JP-H = 531.8 Hz, 2JP-P = 25.9 Hz), 16.9–16.3 (m); 1H NMR (400 MHz, D2O) δ = 6.94 (dm, 1JP-H = 531.8 Hz, 1H), 2.20–2.05 (m, 1H), 1.82–1.67 (m, 2H), 0.95 (d, 3J = 6.6 Hz, 6H); 13C NMR (101 MHz, D2O) δ 75.9 (dd, 1JP-C = 136.2 Hz, 92.1 Hz), 40.0, 24.4 (t, 3JP-C = 8.0 Hz), 24.3, 24.2; MS (ESI−) m/z: 231.02 [M-H]−, 252.96 [M-2H+Na]−, 212.97 [M-H-H2O]−, 165.00 [M-H-H3PO2]−; HRMS (ESI−) m/z: [M-H]− Calcd. for C5H13O6P2 231.0193, found: 231.0195.
- 1-Hydroxyhexane-1,1-(H-phosphinylphosphonate) disodium salt 6c. White powder (1.42 g, 98% yield (from pathway (e)); IR (neat, cm−1) ν = 3350 br, 2960 w, 2300 w, 1185 s, 1105 m, 961 s, 747 s; 31P{1H} NMR (162 MHz, D2O) δ 28.7 (d, 2JP-P = 27.0 Hz), 15.2 (d, 2JP-P = 27.1 Hz); 31P NMR (162 MHz, D2O) δ 28.7 (dm, 1JP-H = 527.1 Hz, 2JP-P = 27.0 Hz), 15.4–15.2 (m); 1H NMR (400 MHz, D2O) δ 7.01 (bd, 1JP-H = 527.2 Hz, 1H, P-H), 1.94–1.65 (m, 2H), 1.59–1.44 (m, 2H), 1.35–1.19 (m, 4H), 0.85 (t, 3J = 6.9 Hz, 3H); 13C NMR (101 MHz, D2O) δ 75.7 (dd, 1JP-C = 130.9 Hz, 93.7 Hz), 32.6, 32.4, 23.4 (t, 3JP-C = 6.5 Hz), 22.1, 13.5; MS (ESI−) m/z: 245.04 [M-H]−, 267.02 [M-2H+Na]−, 227.02 [M-H-H2O]−, 179.05 [M-H-H3PO2]−;HRMS (ESI−) m/z: [M-H]− Calcd. for C6H15O6P2 245.0349, found: 245.0351.
- 1-Hydroxy-1-phenylmethane-1,1-(H-phosphinylphosphonate) disodium salt 6d. White powder (1.41 g, 95% yield (from pathway (b) with evaporation), 1.41 g, 95% yield (from pathway (e)); IR (neat, cm−1) ν = 3265 br, 2972 w, 2318 w, 1600 w, 1490 w, 1445 w, 1395 w, 1195 s, 1120 m, 975 m, 730 m; 31P{1H} NMR (162 MHz, D2O) δ 25.5 (d, 2JP-P = 24.4 Hz), 14.2 (d, 2JP-P = 24.4 Hz); 31P NMR (162 MHz, D2O) δ 25.5 (dd, 1JP-H = 541.3 Hz, 2JP-P = 24.4 Hz), 14.3 (d, 2JP-P = 24.4 Hz); 1H NMR (400 MHz, D2O) δ 7.64 (d, 3J = 7.6 Hz, 2H), 7.37 (t, 3J = 7.5 Hz, 2H), 7.32–7.26 (m, 1H), 7.03 (d, 1JP-H = 541.4 Hz, 1H, P-H); 13C NMR (101 MHz, D2O) δ 137.6, 127.9 (2C), 126.6 (2C), 125.8 (t, 3JP-C = 4.1 Hz), 77.4 (dd, 1JP-C = 135.9 Hz, 89.7 Hz); MS (ESI−) m/z: 250.99 [M-H]−, 272.97 [M-2H+Na]−, 232.98 [M-H-H2O]−, 185.00 [M-H-H3PO2]−; HRMS (ESI−) m/z: [M-H]− Calcd. for C7H9O6P2 250.9880, found: 250.9882.
- 1-Hydroxy-1-(4-tolyl)methane-1,1-(H-phosphinylphosphonate) disodium salt 6e. White powder (1.43 g, 92% yield (from pathway (e)); IR (neat, cm−1) ν = 3235 br, 2975 w, 2900 w, 2310 w, 1649 w, 1510 w, 1336 w, 1188 s, 1095 s, 980 m, 754 m; 31P{1H} NMR (162 MHz, D2O) δ 26.0 (d, 2JP-P = 24.8 Hz), 14.2 (d, 2JP-P = 24.8 Hz); 31P NMR (162 MHz, D2O) δ 26.0 (dd, 1JP-H = 538.8 Hz, 2JP-P = 24.8 Hz), 14.2 (d, 2JP-P= 24.8 Hz); 1H NMR (400 MHz, D2O) δ 7.56 (d, 3J = 8.0 Hz, 2H), 7.24 (d, 3J = 8.0 Hz, 2H), 7.05 (d, 1JP-H = 539.9 Hz, 1H, P-H), 2.34 (s, 3H); 13C NMR (101 MHz, D2O) δ 136.4, 134.9, 128.3 (2C), 125.8 (2C), 77.6 (dd, 1JP-C = 134.4 Hz, 91.0 Hz), 20.0; MS (ESI−) m/z: 265.00 [M-H]−, 286.99 [M-2H+Na]−, 246.99 [M-H-H2O]−199.02 [M-H-H3PO2]−; HRMS (ESI−) m/z: [M-H]− Calcd. for C8H11O6P2 265.0036, found: 265.0040.
- 1-Hydroxy-1-(4-methoxyphenyl)methane-1,1-(H-phosphinylphosphonate) disodium salt 6f. White powder (1.34 g, 82% yield (from pathway (e)); IR (neat, cm−1) ν = 3250 br, 3000 w, 29020 w, 2315 w, 1611 w, 1511 m, 1464 w, 1192 s, 1118 m, 1034 m, 975 w, 740 w; 31P{1H} NMR (162 MHz, D2O) δ 25.7 (d, 2JP-P = 26.4 Hz), 14.4 (d, 2JP-P = 26.4 Hz); 31P NMR (162 MHz, D2O) δ 25.7 (dd, 1JP-H = 539.2 Hz, 2JP-P = 26.4 Hz), 14.4 (d, 2JP-P = 26.4 Hz); 1H NMR (400 MHz, D2O) δ 7.59 (d, 3J = 7.4 Hz, 2H), 7.01 (d, 1JP-H = 539.2 Hz, 1H, P-H), 6.99 (d, 3J = 7.8 Hz, 2H), 3.82 (s, 3H); 13C NMR (101 MHz, D2O) δ 157.5, 130.2, 127.2 (t, 3JP-C = 4.2 Hz, 2C), 113.4 (2C), 77.1 (dd, 1JP-C = 136.3 Hz, 90.3 Hz), 55.3; MS (ESI−) m/z: 281.00 [M-H]−, 302.98 [M-2H+Na]−, 262.99 [M-H-H2O]−, 215.01 [M-H-H3PO2]−; HRMS (ESI−) m/z: [M-H]− Calcd. for C8H11O7P2 280.9986, found: 280.9989.
- 1-Hydroxy-1-(4-fluorophenyl)methane-1,1-(H-phosphinylphosphonate) disodium salt 6g. White powder (1.43 g, 91% yield (from pathway (e)); IR (neat, cm−1) ν = 3224 br, 2988 m, 2308 w, 1605 w, 1506 m, 1195 s, 1092 s, 1054 s, 973 w, 749 m; 31P{1H} NMR (162 MHz, D2O) δ 25.4 (d, 2JP-P = 25.4 Hz), 14.0 (d, 2JP-P = 24.7 Hz); 31P NMR (162 MHz, D2O) δ 25.4 (dd, 1JP-H = 540.5 Hz, 2JP-P = 26.4 Hz), 14.0 (d, 2JP-P = 24.7 Hz); 19F{1H} NMR (377 MHz, D2O) δ −116.7–−116.8 (m); 19F NMR (377 MHz, D2O) δ −116.6–−116.8 (m); 1H NMR (400 MHz, D2O) δ 7.60–7.48 (m, 2H), 7.02 (t, 3J = 8.6 Hz 2H), 6.94 (d, 1JP-H = 540.6 Hz, 1H, P-H); 13C NMR (101 MHz, D2O) δ 161.6 (d, 1JC-F = 242.8 Hz), 133.6, 127.8–127.4 (m, 2C), 114.4 (d, 2JC-F = 21.2 Hz, 2C), 77.0 (dd, 1JP-C = 135.2 Hz, 90.1 Hz); MS (ESI−) m/z: 268.98 [M-H]−, 290.96 [M-2H+Na]−, 250.97 [M-H-H2O]−, 202.99 [M-H-H3PO2]−; HRMS (ESI−) m/z: [M-H]− Calcd. for C7H8FO6P2 268.9786, found: 268.9787.
- 1-Hydroxy-1-(4-trifluoromethoxyphenyl)methane-1,1-(H-phosphinylphosphonate) disodium salt 6h. White powder (1.62 g, 85% yield (from pathway (e)); IR (neat, cm-1) ν = 3320 br, 2319 w, 1650 w, 1610 w, 1192 s, 1114 s, 1070 s, 978 w, 761 m; 31P{1H} NMR (162 MHz, D2O) δ 25.2 (d, 2JP-P = 23.1 Hz), 13.5 (d, 2JP-P = 23.1 Hz); 31P NMR (162 MHz, D2O) δ 25.2 (dd, 1JP-H = 541.9 Hz, 2JP-P = 23.1 Hz), 13.5 (d, 2JP-P = 23.1 Hz); 19F {1H} NMR (377 MHz, D2O) δ −57.7 (s); 19F NMR (377 MHz, D2O) δ −57.7 (bs); 1H NMR (400 MHz, D2O) δ 7.73–7.67 (m, 2H), 7.27 (d, 3J = 8.5 Hz, 2H), 7.03 (bd, 1JP-H = 541.9 Hz, 1H, P-H); 13C NMR (101 MHz, D2O) δ 147.5, 137.2, 127.3 (2C), 120.2 (2C), 120.3 (q, 1JC-F = 256.8 Hz), 77.5 (dd, 1JP-C = 133.5 Hz, 88.7 Hz); MS (ESI−) m/z: 334.97 [M-H]−, 356.95 [M-2H+Na]−, 316.96 [M-H-H2O]−, 268.98 [M-H-H3PO2]−; HRMS (ESI−) m/z: [M-H]− Calcd. for C8H8F3O7P2 334.9703, found: 334.9705.
- 1-Hydroxy-1-(2-tolyl)methane-1,1-bis(H-phosphonate) disodium salt 6i. The crude product was purified by simple washes (ethanol and methanol) to give a white powder (1.17 g, 75% yield (from pathway (e)); IR (neat, cm−1) ν = 3216 br, 2897 m, 2902 m, 2326 w, 1451 w, 1392 w, 1177 m, 1081 s, 976 w, 752 w; 31P{1H} NMR (162 MHz, D2O) δ 26.9–25.5 (m), 14.9 (d, 2JP-P = 26.9 Hz); 31P NMR (162 MHz, D2O) δ 26.2 (dm, 1JP-H = 547.8 Hz, 2JP-P = 26.9 Hz), 14.9 (d, 2JP-P = 26.9 Hz); 1H NMR (400 MHz, D2O) δ 7.64 (s, 1H), 7.10–7.02 (m, 3H), 7.06 (bd, 1JP-H = 547.8 Hz, 1H, P-H), 2.46 (s, 3H, H8); 13C NMR (101 MHz, D2O) δ 137.7, 136.0, 132.6, 127.7, 126.7, 124.9, 80.2 (dd, 1JP-C = 133.3 Hz, 88.0 Hz), 22.3; MS (ESI−) m/z: 265.00 [M-H]−, 286.99 [M-2H+Na]−, 246.99 [M-H-H2O]−, 199.02 [M-H-H3PO2]−; HRMS (ESI−) m/z: [M-H]− Calcd. for C8H11O6P2 265.0036, found: 265.0036.
- 1-Hydroxy-1-(2-methoxyphenyl)methane-1,1-(H-phosphinylphosphonate) disodium salt 6j. White powder (1.06 g, 65% yield (from pathway (e)); IR (neat, cm−1) ν = 3675 w, 2989 w, 2901 w, 2310 w, 1581 w, 1488 w, 1465 w, 1205 s, 1118 m, 1058 w, 968 w, 758 w; 31P{1H} NMR (162 MHz, D2O) δ 24.7 (d, 2JP-P = 28.7 Hz), 14.8 (d, 2JP-P = 28.7 Hz); 31P NMR (162 MHz, D2O) δ 24.7 (dd, 1JP-H = 561.5 Hz, 2JP-P = 28.7 Hz), 14.8 (d, 2JP-P = 28.7 Hz); 1H NMR (400 MHz, D2O) δ 7.57 (d, 3J = 7.6 Hz, 1H), 7.22 (t, 3J = 7.7 Hz, 1H), 7.14 (d, 1JP-H = 561.5 Hz, 1H, P-H), 7.00–6.88 (m, 2H), 3.75 (s, 3H); 13C NMR (101 MHz, D2O) δ 157.0 (t, 3JP-C = 4.4 Hz, 1C), 128.4, 127.7 (t, 3JP-C = 4.6 Hz, 1C), 125.9, 120.7, 112.1, 78.3 (dd, 1JP-C = 137.9 Hz, 89.6 Hz), 55.5; MS (ESI−) m/z: 281.00 [M-H]−, 302.98 [M-2H+Na]−, 262.99 [M-H-H2O]−, 215.01 [M-H-H3PO2]−; HRMS (ESI−) m/z: [M-H]− Calcd. for C8H11O7P2 280.9986, found: 280.9988.
- 1-Hydroxy-1-(1-napthyl)methane-1,1-(H-phosphinylphosphonate) disodium salt 6l. White powder (1.83 g, 53% yield (from pathway (e)); IR (neat, cm−1) ν = 3312 br, 2372 w, 2995 w, 1181 s, 1095 m, 967 m, 785 s; 31P{1H} NMR (162 MHz, D2O) δ 28.1–23.2 (m), 14.6 (d, 2JP-P = 25.6 Hz); 31P NMR (162 MHz, D2O) δ 25.7 (dm, 1JP-H = 544.5 Hz), 14.6 (d, 2JP-P = 25.6 Hz); 1H NMR (400 MHz, D2O) δ 8.92–8.83 (m, 1H), 7.87 (s, 1H), 7.80–7.73 (m, 1H), 7.69 (d, 3J = 7.9 Hz, 1H), 7.43–7.30 (m, 3H), 7.24 (d, 1JP-H = 545.1 Hz, 1H, P-H); 13C NMR (101 MHz, D2O) δ 134.1, 132.1–131.3 (m, 2C), 128.4, 128.3 (2C), 127.8–127.4 (m), 125.5–125.1 (m, 2C), 124.8, 80.6 (dd, 1JP-C = 132.1 Hz, 86.9 Hz); MS (ESI−) m/z: 301.00 [M-H]−, 322.99 [M-2H+Na]−, 282.99 [M-H-H2O]−, 235.02 [M-H-H3PO2]−; HRMS (ESI−) m/z: [M-H]− Calcd. for C11H11O6P2 301.0036, found: 301.0038.
- 1-Hydroxy-1-(2-thienyl)methane-1,1-(H-phosphinylphosphonate) disodium salt 6m. White powder (1.24 g, 82% yield (from pathway (e)); IR (neat, cm−1) ν = 3305 br, 2961 w, 2308 w, 1666 w, 1423 w, 1434 w, 1369 w, 1190 s, 1095 m, 972 w, 725 w; 31P{1H} NMR (162 MHz, D2O) δ 24.6 (d, 2JP-P = 24.0 Hz), 12.7 (d, 2JP-P = 24.0 Hz); 31P NMR (162 MHz, D2O) δ 24.6 (dd, 1JP-H = 542.5 Hz, 2JP-P = 24.0 Hz), 12.7 (d, 2JP-P = 24.0 Hz); 1H NMR (400 MHz, D2O) δ 7.37–7.30 (m, 1H), 7.21–7.11 (m, 1H), 7.08–7.03 (m, 1H), 7.01 (bd, 1JP-H = 542.5 Hz, 1H, P-H); 13C NMR (101 MHz, D2O) δ 142.8, 127.0 (t, 4JP-C = 2.6 Hz), 123.8 (t, 5JP-C = 2.5 Hz), 123.7 (t, 3JP-C = 5.4 Hz), 77.2 (dd, 1JP-C = 133.1 Hz, 90.6 Hz); MS (ESI−) m/z: 256.94 [M-H]−, 278.93 [M-2H+Na]−, 238.93 [M-H-H2O]−, 190.96 [M-H-H3PO2]−; HRMS (ESI−) m/z: [M-H]− Calcd. for C5H7O6P2S 256.9444, found: 256.9447.
- 1-Hydroxy-2-phenylethane-1,1-(H-phosphinylphosphonate) disodium salt 6n. White powder (1.40 g, 90% yield; IR (neat, cm−1) ν = 3289 br, 2989 m, 2901 m, 2315 w, 1187 s, 1121 m, 1056 m, 756 m; 31P{1H} NMR (162 MHz, D2O) δ 26.7 (d, 2JP-P = 22.4 Hz), 15.8 (d, 2JP-P = 21.9 Hz); 31P NMR (162 MHz, D2O) δ 27.8 (bd, 1JP-H = 533.8 Hz, 2JP-P = 20.3 Hz), 16.0–15.7 (m); 1H NMR (400 MHz, D2O) δ 7.48–7.40 (m, 2H), 7.36–7.18 (m, 3H), 6.88 (dm, 1JP-H = 534.7 Hz, 2H, P-H), 3.38–3.07 (m, 2H); 13C NMR (101 MHz, D2O) δ 137.1–136.2 (m), 131.4 (2C), 127.9 (2C), 126.4, 74.5 (dd, 1JP-C = 134.4 Hz, 94.0 Hz), 37.2; MS (ESI−) m/z: 265.00 [M-H]−, 286.99 [M-2H+Na]−, 246.99 [M-H-H2O]−; HRMS (ESI−) m/z: [M-H]− Calcd. for C8H11O6P2 265.0036, found: 265.0045.
- 1-Hydroxy-1-(3-azidopropyl)methane-1,1-(H-phosphinylphosphonate) disodium salt 6o. White powder (1.26 g, 83% yield (from pathway (e)); IR (neat, cm−1) ν = 3305 br, 2964 w, 2310 w, 1665 w, 1421 w, 1432 w, 1369 w, 1192 s, 1097 m, 972 w, 724 w; 31P{1H} NMR (162 MHz, D2O) δ 27.4 (d, 2JP-P = 27.3 Hz), 16.0 (d, 2JP-P = 27.3 Hz); 31P NMR (162 MHz, D2O) δ 27.4 (dm, 1JP-H = 532.4 Hz, 2JP-P = 27.3 Hz), 16.7–15.8 (m); 1H NMR (400 MHz, D2O) δ 6.95 (bd, 1JP-H = 532.4 Hz, 1H, P-H), 3.32 (t, 3J = 6.0 Hz, 2H), 2.01–1.74 (m, 4H); 13C NMR (101 MHz, D2O) δ 74.4 (dd, 1JP-C = 135.7 Hz, 92.4 Hz), 51.8, 29.3, 23.1 (t, 3JP-C = 6.7 Hz); MS (ESI−) m/z: 258.01 [M-H]−, 279.99 [M-2H+Na]−, 239.99 [M-H-H2O]−, 214.99 [M-H-N3]−, 192.02 [M-H-H3PO2]−; HRMS (ESI−) m/z: [M-H]− Calcd. for C4H10N3O6P2 258.0050, found: 258.0053.
- 1-Hydroxy-1-(5-azidopentyl)methane-1,1-(H-phosphinylphosphonate) disodium salt 6p. White powder (1.34 g, 85% yield (from pathway (e)); IR (neat, cm−1): ν = 3328 br, 2956 w, 2303 w, 2121 w, 1182 s, 1105 m, 959 m, 746 w. 31P{1H} NMR (162 MHz, D2O) δ 27.4 (d, 2JP-P = 27.5 Hz), 16.0 (d, 2JP-P = 27.5 Hz); 31P NMR (162 MHz, D2O) δ 27.4 (dm, 1JP-H = 530.9 Hz, 2JP-P = 27.5 Hz), 17.1–16.0 (m); 1H NMR (400 MHz, D2O) δ 6.95 (bd, 1JP-H = 530.9 Hz, 1H, P-H), 3.30 (t, 3J = 7.0 Hz, 2H), 1.92–1.75 (m, 2H), 1.67–1.48 (m, 4H), 1.41–1.28 (m, 2H); 13C NMR (101 MHz, D2O) δ 74.8 (dd, 1JP-C = 135.8 Hz, 91.9 Hz), 51.2, 32.8, 27.8, 26.9, 22.9 (t, 3JP-C = 6.1 Hz); MS (ESI−) m/z: 286.04 [M-H]−, 308.02 [M-2H+Na]−, 268.03 [M-H-H2O]−, 243.0194 [M-H-N3]−, 220.05 [M-H-H3PO2]−; HRMS (ESI−) m/z: [M-H]− Calcd. for C6H14N3O6P2 286.0363, found: 286.0364.
3.2.5. Synthesis of Alendrionate 14o and Neridrionate 14p by Hydrogenolysis
- 1-Hydroxy-1-(3-aminopropyl)methane-1,1-(H-phosphinylphosphonate) disodium salt 14o.White powder (0.047 g, 87% yield); IR (neat, cm−1): ν = 3319 br, 2951 w, 2309 w, 1204 w, 1178 s, 1102 m, 975 m, 737 w; 31P{1H} NMR (162 MHz, D2O) δ 26.8 (d, 2JP-P = 27.3 Hz), 15.8 (d, 2JP-P = 27.3 Hz); 31P NMR (162 MHz, D2O) δ 26.8 (dm, 1JP-H = 535.6 Hz), 16.7–16.5 (m); 1H NMR (400 MHz, D2O) δ 6.95 (bd, 1JP-H= 535.6 Hz, 1H, P-H), 3.13–2.82 (m, 2H), 2.06–1.70 (m, 4H); 13C NMR (101 MHz, D2O) δ 73.9 (dd, 1JP-C = 140.7 Hz, 94.4 Hz), 39.9, 28.7, 21.8 (m); MS (ESI−) m/z: 232.01 [M-2H]−; HRMS (ESI−) m/z: [M-2H]− Calcd. for C4H12NO6P2 232.0145, found: 232.0148.
- 1-Hydroxy-1-(5-aminopentyl)methane-1,1-(H-phosphinylphosphonate) disodium salt 14p. White powder (0.050 g, 84% yield); IR (neat, cm−1): ν = 3320 br, 2951 w, 2309 w, 1205 w, 1179 s, 1101 m, 974 m, 738 w; 31P{1H} NMR (162 MHz, D2O) δ 27.0 (d, 2JP-P = 28.6 Hz), 16.8 (d, 2JP-P = 29.4 Hz); 31P NMR (162 MHz, D2O) δ 27.0 (dm, 1JP-H = 532.4 Hz), 17.3–16.4 (m); 1H NMR (400 MHz, D2O) δ 7.00 (bd, 1JP-H = 532.4 Hz, 1H, P-H), 3.05 (bs, 2H), 2.00–1.78 (m, 2H), 1.85–1.59 (m, 4H), 1.56–1.32 (m, 2H); 13C NMR (101 MHz, D2O) δ 74.4 (dd, 1JP-C = 141.0 Hz, 88.1 Hz), 39.2, 31.7, 26.3, 26.2, 22.5 (t, 3JP-C = 6.2 Hz); MS (ESI−) m/z: 260.05 [M-H]−, 282.03 [M-2H+Na]−, 242.04 [M-H-H2O]−, 194.06 [M-H-H3PO2]−; HRMS (ESI−) m/z: [M-H]− Calcd. for C2H16NO6P2 265.0458, found: 260.0459.
3.3. Complexation to Hydroxyapatite (HA)
3.4. Cytotoxicity Assay
3.5. In Vitro Antiproliferative Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Pradere, U.; Garnier-Amblard, E.C.; Coats, S.J.; Amblard, F.; Schinazi, R.F. Synthesis of nucleoside phosphate and phosphonate prodrugs. Chem. Rev. 2014, 114, 9154–9218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virieux, D.; Volle, J.N.; Bakalara, N.; Pirat, J.L. Synthesis and biological applications of phosphinates and derivatives. Top. Curr. Chem. 2015, 360, 39–114. [Google Scholar]
- Horsman, G.P.; Zechel, D.L. Phosphonate Biochemistry. Chem. Rev. 2017, 117, 5704–5783. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, J.S.; Almeida Paz, F.A.; Braga, S.S. Bisphosphonates, Old Friends of Bones and New Trends in Clinics. J. Med. Chem. 2021, 64, 1260–1282. [Google Scholar] [CrossRef]
- van Beek, E.; Löwik, C.; Que, I.; Papapoulos, S. Dissociation of binding and antiresorptive properties of hydroxybisphosphonates by substitution of the hydroxyl with an amino group. J. Bone Miner. Res. 1996, 11, 1492–1497. [Google Scholar] [CrossRef]
- Clézardin, P. Bisphosphonates′ antitumor activity: An unravelled side of a multifaceted drug class. Bone 2011, 48, 71–79. [Google Scholar] [CrossRef]
- Abdelkarim, M.; Guenin, E.; Sainte-Catherine, O.; Vintonenko, N.; Peyri, N.; Perret, G.Y.; Crepin, M.; Khatib, A.M.; Lecouvey, M.; Di Benedetto, M. New symmetrically esterified m-bromobenzyl non-aminobisphosphonates inhibited breast cancer growth and metastases. PLoS ONE 2009, 4, e4685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baranyi, M.; Rittler, D.; Molnar, E.; Shirasawa, S.; Jalsovszky, I.; Varga, I.K.; Hegedus, L.; Nemeth, A.; Dank, M.; Aigner, C.; et al. Next Generation Lipophilic Bisphosphonate Shows Antitumor Effect in Colorectal Cancer In Vitro and In Vivo. Pathol. Oncol. Res. 2020, 26, 1957–1969. [Google Scholar] [CrossRef] [PubMed]
- Hamma-Kourbali, Y.; Di Benedetto, M.; Ledoux, D.; Oudar, O.; Leroux, Y.; Lecouvey, M.; Kraemer, M. A novel non-containing-nitrogen bisphosphonate inhibits both in vitro and in vivo angiogenesis. Biochem. Biophys. Res. Commun. 2003, 310, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H. Bisphosphonates: A review of their pharmacokinetic properties. Bone 1996, 18, 75–85. [Google Scholar] [CrossRef]
- Cremers, S.; Ebetino, F.H.; Phipps, R. On the pharmacological evaluation of bisphosphonates in humans. Bone 2020, 139, 115501. [Google Scholar] [CrossRef] [PubMed]
- Dussart, J.; Guedeney, N.; Deschamp, J.; Monteil, M.; Gager, O.; Legigan, T.; Migianu-Griffoni, E.; Lecouvey, M. A convenient synthetic route towards H-bisphosphinates. Org. Biomol. Chem. 2018, 16, 6969–6979. [Google Scholar] [CrossRef]
- Guedeney, N.; Dussart, J.; Deschamp, J.; Ouechtati, M.; Migianu-Griffoni, E.; Lecouvey, M. A convenient one-pot synthesis of 1-hydroxymethylene-1,1-bisphosphinic acids. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 323–325. [Google Scholar] [CrossRef]
- Dussart, J.; Deschamp, J.; Monteil, M.; Gager, O.; Legigan, T.; Migianu-Griffoni, E.; Lecouvey, M. Formation of 1-Hydroxymethylene-1,1-Bisphosphinates through the Addition of a Silylated Phosphonite on Various Trivalent Derivatives. J. Org. Chem. 2020, 85, 14559–14569. [Google Scholar] [CrossRef] [PubMed]
- Biller, S.A.; Forster, C. The synthesis of isoprenoid (phosphinylmethyl)phosphonates. Tetrahedron 1990, 46, 6645–6658. [Google Scholar] [CrossRef]
- Biller, S.A.; Dickson, J.K.J. Hydroxyphosphinyl phosphonate squalene synthetase inhibitors and their use in pharmaceutical compositions. European Patent EP0589473A1, 19 October 1993. [Google Scholar]
- Magnin, D.R.; Dickson, J.K.; Logan, J.V.; Lawrence, R.M.; Chen, Y.; Sulsky, R.B.; Ciosek, C.P.; Biller, S.A.; Harrity, T.W.; Jolibois, K.G.; et al. 1,1-Bisphosphonate Squalene Synthase Inhibitors: Interplay Between the Isoprenoid Subunit and the Diphosphate Surrogate. J. Med. Chem. 1995, 38, 2596–2605. [Google Scholar] [CrossRef] [PubMed]
- Bisseret, P.; Eustache, J. H-Phosphonylphosphonate triethylester: The first member of a novel family of stable bisphosphorylated compounds; its short synthesis and reactivity with aldehydes. Tetrahedron Lett. 2001, 42, 8451–8453. [Google Scholar] [CrossRef]
- Antczak, M.I.; Montchamp, J.-L. Synthesis of 1,1-bis-phosphorus compounds from organoboranes. Tetrahedron Lett. 2008, 49, 5909–5913. [Google Scholar] [CrossRef]
- Berger, O.; Gavara, L.; Montchamp, J.-L. Chemistry of the Versatile (Hydroxymethyl)phosphinyl P(O)CH2OH Functional Group. Org. Lett. 2012, 14, 3404–3407. [Google Scholar] [CrossRef] [PubMed]
- Dussart, J.; Deschamp, J.; Migianu-Griffoni, E.; Lecouvey, M. From Industrial Method to the Use of Silylated P(III) Reagents for the Synthesis of Relevant Phosphonylated Molecules. Org. Process Res. Dev. 2020, 24, 637–651. [Google Scholar] [CrossRef]
- Migianu, E.; Guénin, E.; Lecouvey, M. New Efficient Synthesis of 1-Hydroxymethylene-1,1-Bisphosphonate Monomethyl Esters. Synlett 2005, 425–428. [Google Scholar] [CrossRef]
- McKenna, C.E.; Higa, M.T.; Cheung, N.H.; McKenna, M.-C. The facile dealkylation of phosphonic acid dialkyl esters by bromotrimethylsilane. Tetrahedron Lett. 1977, 18, 155–158. [Google Scholar] [CrossRef]
- Dussart, J.; Deschamp, J.; Monteil, M.; Gager, O.; Migianu-Griffoni, E.; Lecouvey, M. A General Protocol for the Synthesis of H-α-Hydroxyphosphinates. Synthesis 2019, 51, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Fitch, S.J.; Moedritzer, K. NMR study of the P-C(OH)-P to P-C-O-P Rearrangement: Tetraethyl 1-Hydroxyalkylidenediphosphonates. J. Am. Chem. Soc. 1962, 84, 1876–1879. [Google Scholar] [CrossRef]
- Kumaraswamy, S.; Senthamizh Selvi, R.; Swamy, K.C.K. Synthesis of New α-Hydroxy-, α-Halogeno- and Vinylphosphonates Derived from 5,5-Dimethyl-1,3,2-dioxaphosphinan-2-one. Synthesis 1997, 1997, 207–212. [Google Scholar] [CrossRef]
- El Manouni, D.; Leroux, Y.; Burgada, E.R. Synthese d′α cétophosphonates et d′esters d′hydroxy méthylene diphosphonates III. Phosphorus Sulfur Silicon Relat. Elem. 1989, 42, 73–83. [Google Scholar] [CrossRef]
- Griffiths, D.V.; Jamali, H.A.R.; Tebby, J.C. Reactions of Phosphites with Acid Chlorides. Phosphite Attack at the Carbonyl Oxygen of α-Ketophosphonates. Phosphorus Sulfur Silicon Relat. Elem. 1981, 11, 95–99. [Google Scholar] [CrossRef]
- Kaushik, M.P.; Gupta, A.K.; Lal, B.; Vaidyanathaswamy, R. Unusual Reaction of Triethylphosphite with Pyridoyl Chlorides. Phosphorus Sulfur Silicon Relat. Elem. 1992, 71, 175–177. [Google Scholar] [CrossRef]
- Pajkert, R.; Röschenthaler, G.-V. Organophosphorus Chemistry; Specialist Periodical Reports; Allen, D.W., Tebby, J.C., Loakes, D., Eds.; The Royal Society of Chemistry: London, UK, 2014; Volume 43, p. 348. [Google Scholar]
- van Beek, E.; Hoekstra, M.; van De Ruit, M.; Löwik, C.; Papapoulos, S. Structural requirements for bisphosphonate actions in vitro. J. Bone Miner. Res. 1994, 9, 1875–1882. [Google Scholar] [CrossRef]
- Puljula, E.; Turhanen, P.; Vepsalainen, J.; Monteil, M.; Lecouvey, M.; Weisell, J. Structural requirements for bisphosphonate binding on hydroxyapatite: NMR study of bisphosphonate partial esters. ACS Med. Chem. Lett. 2015, 6, 397–401. [Google Scholar] [CrossRef] [Green Version]
- Legigan, T.; Migianu-Griffoni, E.; Redouane, M.A.; Descamps, A.; Deschamp, J.; Gager, O.; Monteil, M.; Barbault, F.; Lecouvey, M. Synthesis and preliminary anticancer evaluation of new triazole bisphosphonate-based isoprenoid biosynthesis inhibitors. Eur. J. Med. Chem. 2021, 214, 113241. [Google Scholar] [CrossRef] [PubMed]
- Migianu-Griffoni, E.; Chebbi, I.; Kachbi, S.; Monteil, M.; Sainte-Catherine, O.; Chaubet, F.; Oudar, O.; Lecouvey, M. Synthesis and Biological Evaluation of New Bisphosphonate-Dextran Conjugates Targeting Breast Primary Tumor. Bioconjugate Chem. 2014, 25, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Deprèle, S.; Montchamp, J.L. Triethylborane-Initiated Room Temperature Radical Addition of Hypophosphites to Olefins: Synthesis of Monosubstituted Phosphinic Acids and Esters. J. Org. Chem. 2001, 66, 6745–6755. [Google Scholar] [CrossRef] [PubMed]
- Kieczykowski, G.R.; Jobson, R.B.; Melillo, D.G.; Reinhold, D.F.; Grenda, V.J.; Shinkai, I. Preparation of (4-Amino-1-Hydroxybutylidene)bisphosphonic Acid Sodium Salt, MK-217 (Alendronate Sodium). An Improved Procedure for the Preparation of 1-Hydroxy-1,1-bisphosphonic Acids. J. Org. Chem. 1995, 60, 8310–8312. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Molecule | MDA-MB-231(Breast) | MIA PaCa-2(Pancreas) | A549 (Lung) |
|---|---|---|---|---|
| 1 | alendronate | 20.6 ± 4.1 | 17.2 ± 4.8 | 7.9 ± 0.7 |
| 2 | alendrinate | 75 ± 4 | >100 | 84 ± 4 |
| 3 | 14o | >100 | >100 | 53 ± 17 |
| 4 | neridronate | >100 | >100 | 69 ± 3 2 |
| 5 | neridrinate | >100 | >100 | >100 |
| 6 | 14p | 72 ± 5 | >100 | 26.6 ± 3.1 |
| 7 | zoledronate | 10.0 ± 0.3 | 9.0 ± 0.5 | 8.1 ± 0.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dussart-Gautheret, J.; Deschamp, J.; Legigan, T.; Monteil, M.; Migianu-Griffoni, E.; Lecouvey, M. One-Pot Synthesis of Phosphinylphosphonate Derivatives and Their Anti-Tumor Evaluations. Molecules 2021, 26, 7609. https://doi.org/10.3390/molecules26247609
Dussart-Gautheret J, Deschamp J, Legigan T, Monteil M, Migianu-Griffoni E, Lecouvey M. One-Pot Synthesis of Phosphinylphosphonate Derivatives and Their Anti-Tumor Evaluations. Molecules. 2021; 26(24):7609. https://doi.org/10.3390/molecules26247609
Chicago/Turabian StyleDussart-Gautheret, Jade, Julia Deschamp, Thibaut Legigan, Maelle Monteil, Evelyne Migianu-Griffoni, and Marc Lecouvey. 2021. "One-Pot Synthesis of Phosphinylphosphonate Derivatives and Their Anti-Tumor Evaluations" Molecules 26, no. 24: 7609. https://doi.org/10.3390/molecules26247609