
Entrained Flow Gasification of Polypropylene Pyrolysis Oil

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
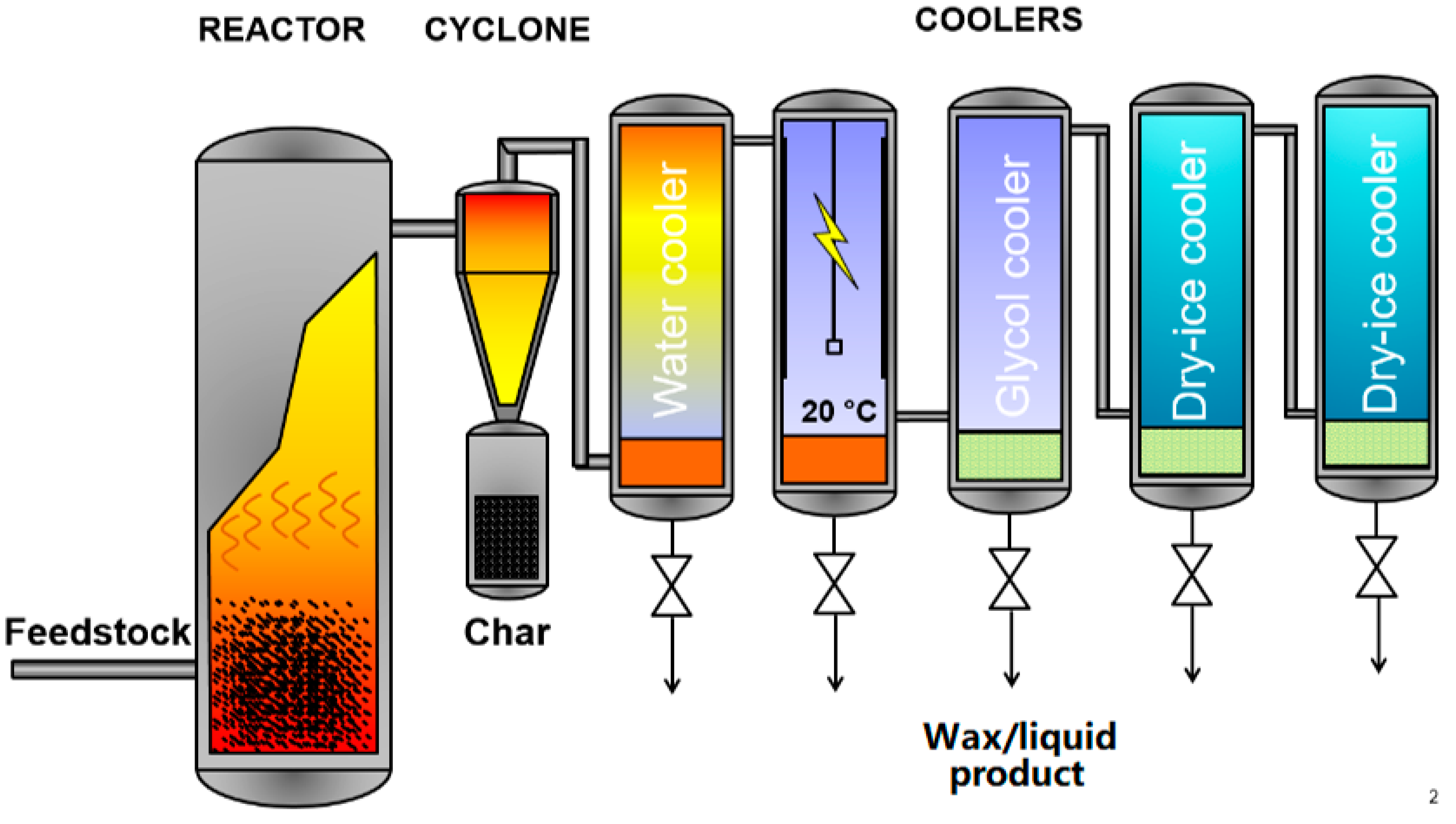
2.1. Pyrolysis
2.1.1. Non-Condensable Gases
2.1.2. Liquid and Waxy Products
2.2. Gasification
2.2.1. Syngas Yields and Elemental Mass Balances
2.2.2. Gasification Performance Parameters
2.2.3. Filter Deposits
2.3. Overall Process Performance
3. Materials and Methods
3.1. Pyrolysis
3.2. Analytical Methods
3.3. Gasification
3.4. Gasification Performance Parameters
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- PlasticsEurope. Plastics—The Facts 2019; Plastics Europe: Brussels, Belgium, 2019. [Google Scholar]
- De-la-Torre, G.; Dioses-Salinas, D.; Pizarro-Ortega, C.; Santillán, L. New plastic formulations in the Anthropocene. Sci. Total Environ. 2021, 745, 142216. [Google Scholar] [CrossRef]
- Pabortsava, K.; Lampitt, R.S. High concentrations of plastic hidden beneath the surface of the Atlantic Ocean. Nat. Commun. 2020, 11, 4073. [Google Scholar] [CrossRef]
- Rai, P.K.; Lee, J.; Brown, R.J.; Kim, K.-H. Environmental fate, ecotoxicity biomarkers, and potential health effects of micro- and nano-scale plastic contamination. J. Hazard. Mater. 2020, 403, 123910. [Google Scholar] [CrossRef]
- Gray, R. What’s the Real Price of Getting Rid of Plastic Packaging? BBC Work-Life: London, UK, 2018. [Google Scholar]
- BPF. Understanding the Debate about Plastic; British Plastic Federation: London, UK, 2019. [Google Scholar]
- Accorsi, R.; Versari, L.; Manzini, R. Glass vs. plastic: Life cycle assessment of extra-virgin olive oil bottles accross global supply chains. Sustainability 2015, 7, 2718–2840. [Google Scholar] [CrossRef] [Green Version]
- Humbert, S.; Rossi, V.; Margni, M.; Jolliet, O.; Loerincik, Y. Life cycle assessment of two baby food packaging alternatives: Glass jars vs. plastic pots. Int. J. Life Cycle Assess. 2009, 14, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Kouloumpis, V.; Pell, R.; Correa-Cano, M.; Yan, X. Potential trade-offs between eliminating plastics and mitigating climate change: An LCA perspective on Polyethylene Terephthalate (PET) bottles in Cornwall. Sci. Total Environ. 2020, 727, 138681. [Google Scholar] [CrossRef]
- Stefanini, R.; Borghesi, G.; Ronazano, A.; Viganali, G. Plastic of glass: A new environmental assessment with a marine litter indicator for the comparison of pasteurized milk bottles. Int. J. Life Cucle Assess. 2021, 26, 767–784. [Google Scholar] [CrossRef]
- Tenhunen, A.; Pöhler, H. A Circular Economy of Plastics; VTT Technical Research Centre of Finland: Espoo, Finland, 2020. [Google Scholar]
- EU. A European Strategy for Plastics in a Circular Economy; European Commission: Strasbourg, France, 2018. [Google Scholar]
- Ragaert, K.; Delva, L.; Van Geem, K. Mechanical and chemical recycling of solid plastic waste. Waste Manag. 2017, 69, 24–58. [Google Scholar] [CrossRef]
- Eriksen, M.; Christiansen, J.; Daugaard, A.; Astrup, T. Closing the loop for PET, PE and PP waste from housholds: Influence of material properties and product design for plastic recycling. Waste Manag. 2019, 96, 75–85. [Google Scholar] [CrossRef]
- Schyns, Z.O.G.; Shaver, M.P. Mechanical Recycling of Packaging Plastics: A Review. Macromol. Rapid Commun. 2020, 42, e2000415. [Google Scholar] [CrossRef]
- Thiounn, T.; Smith, R. Advances and approaches for chemical recyling of plastic waste. J. Polym. Sci. 2020, 58, 1347–1364. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Panda, A.; Sinch, R. A review on thertiary recycling of high-density polyethylene to fuel. Resour. Conserv. Recycl. 2011, 55, 893–910. [Google Scholar] [CrossRef]
- Al-Salem, S.M.; Lettieri, P.; Baeyens, J. Recycling and recovery routes of plastic solid waste (PSW): A review. Waste Manag. 2009, 29, 2625–2643. [Google Scholar] [CrossRef] [PubMed]
- Solis, M.; Silveira, S. Technologies for chemical recycling of household plastics—A technical reveiw and TRL assessment. Waste Manag. 2020, 105, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.S.; Czernik, S.R.; Piskorz, J.; Radlein, D.S.A.G. Fast pyrolysis of plastic wastes. Energy Fuels 1990, 4, 407–411. [Google Scholar] [CrossRef]
- Miandad, R.; Barakat, M.; Aburiazaiza, A.S.; Rehan, M.; Ismail, I.; Nizami, A. Effect of plastic waste types on pyrolysis liquid oil. Int. Biodeterior. Biodegrad. 2016, 119, 239–252. [Google Scholar] [CrossRef]
- Butler, E.; Devlin, G.; McDonnell, K. Waste Polyolefins to Liquid Fuels via Pyrolysis: Review of Commercial State-of-the-Art and Recent Laboratory Research. Waste Biomass Valorization 2011, 2, 227–255. [Google Scholar] [CrossRef] [Green Version]
- Miandad, R.; Barakat, M.; Aburiazaiza, A.S.; Rehan, M.; Nizami, A. Catalytic pyrolysis of plastic waste: A review. Process. Saf. Environ. Prot. 2016, 102, 822–838. [Google Scholar] [CrossRef]
- Arena, U.; Mastellone, M. Fluidized bed pyrolysis of plastic wastes. In Feedstock Recycling and Pyrolysis of Waste Plastics: Converting Waste Plastics into Diesel and Other Fuels; Wiley: Chichester, West Sussex, UK, 2006; pp. 435–474. [Google Scholar]
- Butler, E.; Devlin, G.; Meier, D.; McDonnell, K. A review of recent laboratory research and commercial developments in fast pyrolysis and upgrading. Renew. Sustain. Energy Rev. 2011, 15, 4171–4186. [Google Scholar] [CrossRef] [Green Version]
- Encinar, J.; González, J. Pyrolysis of synthetic polymers and plastic wastes. Kinetic study. Fuel Process. Technol. 2008, 89, 678–686. [Google Scholar] [CrossRef]
- López, A.; de Marco, I.; Caballero, B.; Laresgoiti, M.; Adrados, A. Influence of time and temperature on pyrolysis of plastic wastes in a semi-batch reactor. Chem. Eng. J. 2011, 173, 62–71. [Google Scholar] [CrossRef]
- Qureshi, M.S.; Oasmaa, A.; Pihkola, H.; Deviatkin, I.; Tenhunen, A.; Mannila, J.; Minkkinen, H.; Pohjakallio, M.; Laine-Ylijoki, J. Pyrolysis of plastic waste: Opportunities and challenges. J. Anal. Appl. Pyrolysis 2020, 152, 104804. [Google Scholar] [CrossRef]
- Singh, R.K.; Ruj, B.; Sadhukhan, A.; Gupta, P. Impact of fast and slow pyrolysis on the degradation of mixed plastic waste: Product yield analysis and their characterization. J. Energy Inst. 2019, 92, 1647–1657. [Google Scholar] [CrossRef]
- Elordi, G.; Olazar, M.; Lopez, G.; Artetxe, M.; Bilbao, J. Product Yields and Compositions in the Continuous Pyrolysis of High-Density Polyethylene in a Conical Spouted Bed Reactor. Ind. Eng. Chem. Res. 2011, 50, 6650–6659. [Google Scholar] [CrossRef]
- Mastral, F.J.; Esperanza, E.; Garcia, P.; Juste, M. Pyrolysis of high-density polyethylene in a fluidised bed reactor. Influence of the temperature and residence time. J. Anal. Appl. Pyrolysis 2002, 63, 1–15. [Google Scholar] [CrossRef]
- Onwudili, J.; Insura, N.; Williams, P. Composition of products from the pyrolysis of polyethylene and polystyrene in a closed batch reactor: Effects of temperature and residence time. J. Anal. Appl. Pyrolysis 2009, 86, 293–303. [Google Scholar] [CrossRef]
- Al-Salem, S.M.; Antelava, A.; Constantinou, A.; Manos, G.; Dutta, A. A review on thermal and catalytic pyrolysis of plastic solid waste (PSW). J. Environ. Manag. 2017, 197, 177–198. [Google Scholar] [CrossRef] [PubMed]
- Nanda, S.; Berruti, F. Thermochemical conversion of plastic waste to fuels: A review. Environ. Chem. Lett. 2020, 19, 123–148. [Google Scholar] [CrossRef]
- Angyal, A.; Miskolczi, N.; Bartha, L. Petrochemical feedstock by thermal cracking of plastic waste. J. Anal. Appl. Pyrolysis 2007, 79, 409–414. [Google Scholar] [CrossRef]
- Donaj, P.J.; Kaminsky, W.; Buzeto, F.; Yang, W. Pyrolysis of polyolefins for increasing the yield of monomers’ recovery. Waste Manag. 2011, 32, 840–846. [Google Scholar] [CrossRef]
- Lopez, G.; Artetxe, M.; Amutio, M.; Alvarez, J.; Bilbao, J.; Olazar, M. Recent advances in the gasification of waste plastics. A critical overview. Renew. Sustain. Energy Rev. 2018, 82, 576–596. [Google Scholar] [CrossRef]
- Ciuffi, B.; Chiaramonti, D.; Rizzo, A.M.; Frediani, M.; Rosi, L. A Critical Review of SCWG in the Context of Available Gasification Technologies for Plastic Waste. Appl. Sci. 2020, 10, 6307. [Google Scholar] [CrossRef]
- Higman, C.; Van der Burgt, M. Gasification; Gulf Professional Publishing: Oxford, UK, 2008; ISBN 978-0-7506-8528-3. [Google Scholar]
- Weiland, F.; Wiinikka, H.; Hedman, H.; Wennebro, J.; Pettersson, E.; Gebart, R. Influence of process parameters on the performance of an oxygen blown entrained flow biomass gasifier. Fuel 2015, 153, 510–519. [Google Scholar] [CrossRef]
- Wiinikka, H.; Johansson, A.-C.; Wennebro, J.; Carlsson, P.; Öhrman, O. Evaluation of black liquor gasification intended for synthetic fuel or power production. Fuel Process. Technol. 2015, 139, 216–225. [Google Scholar] [CrossRef]
- KIT. The Bioliq Process. 2018. Available online: www.bioliq.de (accessed on 1 April 2021).
- Eberhard, M.; Santo, U.; Michelfelder, B.; Günther, A.; Weigand, P.; Matthes, J.; Waibel, P.; Hagenmeyer, V.; Kolb, T. The bioliq entrained-flow gasifier—A model for the German energiwende. ChemBioEng Rev. 2020, 7, 106–118. [Google Scholar] [CrossRef]
- Leijenhorst, E.J.; Assink, D.; van de Beld, L.; Weiland, F.; Wiinikka, H.; Carlsson, P.; Öhrman, O.G.W. Entrained flow gasification of straw- and wood-derived pyrolysis oil in a pressurized oxygen blown gasifier. Biomass Bioenergy 2015, 79, 166–176. [Google Scholar] [CrossRef]
- Bach-Oller, A.; Kirtania, K.; Furusjö, E.; Umeki, K. Co-gasification of black liquor and pyrolysis oil at high temperature: Part 1. Fate of alkali elements. Fuel 2017, 202, 46–55. [Google Scholar] [CrossRef]
- Bach-Oller, A.; Kirtania, K.; Furusjö, E.; Umeki, K. Co-gasification of black liquor and pyrolysis oil at high temperature: Part 2. Fuel conversion. Fuel 2017, 197, 240–247. [Google Scholar] [CrossRef]
- Jafri, Y.; Furusjö, E.; Kirtania, K.; Gebart, R.; Granberg, F. A study of black liquor and pyrolysis oil co-gasification in pilot scale. Biomass Convers. Biorefin. 2017, 8, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Predel, M.; Kaminsky, W. Pyrolysis of mixed polyolefins in a fluidized-bed reactor and on a pyro-GC/MS to yield aliphatic waxes. Polym. Degrad. Stab. 2000, 70, 373–385. [Google Scholar] [CrossRef]
- Westerhout, R.W.J.; Kuipers, J.A.M.P.M.; van Swaaij, W. Experimental determination of the yield of pyrolysis products of polyethene and polypropene. Influence of reaction conditions. Ind. Eng. Chem. Res. 1998, 37, 841–847. [Google Scholar] [CrossRef]
- Arabiourrutia, M.; Elordi, G.; Lopez, G.; Borsella, E.; Bilbao, J.; Olazar, M. Characterization of the waxes obtained by the pyrolysis of polyolefin plastics in a conical spouted bed reactor. J. Anal. Appl. Pyrolysis 2012, 94, 230–237. [Google Scholar] [CrossRef]
- Billaud, J.; Valin, S.; Peyrot, M.; Salvador, S. Influence of H2O, CO2 and O2 addition on biomass gasification in entrained flow reactor conditions: Experiments and modelling. Fuel 2016, 166, 166–178. [Google Scholar] [CrossRef] [Green Version]
- Qin, K.; Lin, W.; Jensen, P.A.; Jensen, A.D. High-temperature entrained flow gasification of biomass. Fuel 2011, 93, 589–600. [Google Scholar] [CrossRef]
- Dufour, A.; Valin, S.; Castelli, P.; Thiery, S.; Boissonnet, G.; Zoulalian, A.; Glaude, P.-A. Mechanisms and Kinetics of Methane Thermal Conversion in a Syngas. Ind. Eng. Chem. Res. 2009, 48, 6564–6572. [Google Scholar] [CrossRef]
- Dupont, C.; Boissonnet, G.; Seiler, J.-M.; Gauthier-Maradei, P.; Schweich, D. Study about the kinetic processes of biomass steam gasification. Fuel 2007, 86, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Jand, N.; Brandani, V.; Foscolo, P. Thermodynamic limits and actual product yields and compositions in biomass gasification processes. Ind. Eng. Chem. Res. 2006, 45, 834–843. [Google Scholar] [CrossRef]
- Weiland, F.; Nilsson, P.; Gebart, R.; Wiinikka, H.; Gudmundsson, A.; Sanati, M. Online characterization of syngas particulates using aerosol mass spectrometry in entrained-flow biomass gasification. Aerosol Sci. Technol. 2014, 48, 1145–1155. [Google Scholar] [CrossRef]
- Weiland, F.; Sweeney, D.J.; Wiinikka, H. Extractive Sampling of Gas and Particulates from the Reactor Core of an Entrained Flow Biomass Gasifier. Energy Fuels 2016, 30, 6405–6412. [Google Scholar] [CrossRef]
- Kremling, M.; Briesemeister, L.; Gaderer, M.; Fendt, S.; Spliethoff, H. Oxygen-blown entrained flow gasification of bio-mass: Impact of fuel parameters and oxygen stoichiometric ratio. Energy Fuels 2017, 31, 2349–2359. [Google Scholar] [CrossRef]
- Chen, H.; Dobbins, R. Crystallogenesis of Particles Formed in Hydrocarbon Combustion. Combust. Sci. Technol. 2000, 159, 109–128. [Google Scholar] [CrossRef]
- Green, G.J.; Takahashi, F.; Walsh, D.E.; Dryer, F.L. Aerodynamic device for generating mono-disperse fuel droplets. Rev. Sci. Instrum. 1989, 60, 646–652. [Google Scholar] [CrossRef]
- Toth, P.; Vikström, T.; Molinder, R.; Wiinikka, H. Structure of carbon black continuously produced from biomass pyrolysis oil. Green Chem. 2018, 20, 3981–3992. [Google Scholar] [CrossRef]
- Wiinikka, H.; Carlsson, P.; Marklund, M.; Grönberg, C.; Pettersson, E.; Lidman, M.; Gebart, R. Experimental investigation of an industrial scale black liquor gasifier. Part 2: Influence of quench operation on product gas composition. Fuel 2012, 93, 117–129. [Google Scholar] [CrossRef]
- Lange, J.-P. Methanol synthesis: A short review of technology improvements. Catal. Today 2001, 64, 3–8. [Google Scholar] [CrossRef]
- Dry, M. The Fisher-Tropsch process: 1950–2000. Catal. Today 2002, 71, 227–241. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fraction | wt.% |
|---|---|
| Pyrolytic gases | 17 |
| Organic liquids | 14 |
| Organic waxes | 53 |
| Organic product total | 67 |
| Sum of products | 84 |
| Gas Component | Yield (wt.%) |
|---|---|
| Methane | 1.0 |
| Ethane | 1.3 |
| Ethene | 1.4 |
| Propane | 0.4 |
| Propene | 7.6 |
| n-Butane | 0.1 |
| trans-2-Butane | 0.1 |
| 1-Butene | 0.2 |
| i-Butene | 1.3 |
| 1,3-Butadiene | 0.2 |
| n-Pentane | 0.4 |
| 1-Pentene | 0.2 |
| Benzene | 0.1 |
| Other C3+ | 2.8 |
| C | H | N | H/C | HHV | LHV | Viscosity at 40 °C | Density at 40 °C | Melting Temp. | Hydrocarbon Range | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| wt.% | wt.% | wt.% | - | MJ/kg | MJ/kg | mm2/s | g/cm3 | °C | C7–C20 | C21–C37 | C37+ | End Point | |
| Liquid | 83.9 | 13.8 | <0.1 | 0.16 | 46.3 | 43.3 | 2.3 | 0.77 | n.m. | 70 | 22 | 8 | C77 |
| Wax | 83.7 | 13.8 | <0.1 | 0.16 | 46.2 | 43.2 | n.m. | n.m. | 98 | 47 | 27 | 26 | C102 |
| Gas Specie | Yield (mol/kg Fuel) 1200 °C | Yield (mol/kg Fuel) 1300 °C | ||||
|---|---|---|---|---|---|---|
| λ 0.35 | λ 0.43 | λ 0.50 | λ 0.35 | λ 0.43 | λ 0.50 | |
| H2 | 44.4 | 37.0 | 34.9 | 52.4 | 46.6 | 40.6 |
| CO | 41.6 | 42.4 | 44.4 | 48.3 | 51.2 | 51.1 |
| H2O | 17.3 | 23.3 | 28.7 | 15.5 | 21.2 | 27.3 |
| CO2 | 7.9 | 12.8 | 17.6 | 6.2 | 10.1 | 15.2 |
| CH4 | 2.4 | 2.2 | 2.2 | 1.6 | 1.6 | 1.5 |
| C2H2 | 1.9 | 1.5 | 1.2 | 1.8 | 1.2 | 0.7 |
| C2H4 | 0.13 | 0.09 | 0.07 | 0.12 | 0.08 | 0.05 |
| Massbalance (wt.%) 1200 °C | Massbalance (wt.%) 1300 °C | |||||
|---|---|---|---|---|---|---|
| λ | C 1 | H | O | C 1 | H | O |
| 0.35 | 80 | 101 | 104 | 86 | 105 | 103 |
| 0.43 | 87 | 97 | 103 | 94 | 106 | 103 |
| 0.50 | 96 | 100 | 102 | 99 | 107 | 103 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiland, F.; Qureshi, M.S.; Wennebro, J.; Lindfors, C.; Ohra-aho, T.; Shafaghat, H.; Johansson, A.-C. Entrained Flow Gasification of Polypropylene Pyrolysis Oil. Molecules 2021, 26, 7317. https://doi.org/10.3390/molecules26237317
Weiland F, Qureshi MS, Wennebro J, Lindfors C, Ohra-aho T, Shafaghat H, Johansson A-C. Entrained Flow Gasification of Polypropylene Pyrolysis Oil. Molecules. 2021; 26(23):7317. https://doi.org/10.3390/molecules26237317
Chicago/Turabian StyleWeiland, Fredrik, Muhammad Saad Qureshi, Jonas Wennebro, Christian Lindfors, Taina Ohra-aho, Hoda Shafaghat, and Ann-Christine Johansson. 2021. "Entrained Flow Gasification of Polypropylene Pyrolysis Oil" Molecules 26, no. 23: 7317. https://doi.org/10.3390/molecules26237317