2. Chloro- and Bromotrifluoromethylation
Photoredox catalysis has undergone an exponential growth in the last two decades, probably due to the simplicity of the method, the excellent reducing ability of photocatalysis in the excited state, and in most cases, the low cost of the starting materials [
27]. The use of photoredox catalysis such as the widely-used ruthenium(II) polypyridine complexes (e.g., [Ru(bpy)
3]
2+, bpy = 2,2′-bipyridine, or [Ru(Phen)
3]
2+, Phen = 1,10-phenanthroline), the well-known iridium(III) cyclometalated derivatives (e.g.,
fac-Ir(ppy)
3, ppy = 2-phenylpyridine) has been established as an elegant and powerful tool in synthetic chemistry because they can smoothly catalyze single-electron transfer (SET) processes with visible light irradiation under operationally simple conditions at room temperature (
Figure 2). Concretely, the difunctionalization of unsaturated carbon–carbon bonds using this strategy provides, among others, the formation of carbon–fluoroalkyl bonds, in particular, C–CF
3 bonds, which are of great interest both in the agrochemical and pharmaceutical industry. The trifluoromethylation of alkenes, including halotrifluoromethylation and others, has emerged as an interesting and attractive synthetic tool for the incorporation of CF
3 moieties in bioactive molecules [
28]. Different reagents have been used to this end, with R
fX (in particular, CF
3I gas, b.p. – 22 °C), Togni reagent, Umemoto reagent, CF
3SO
2Cl (corrosive and volatile), Langlois and Baran reagents, and
N-trifylpyridinium salts being the most widely used [
29,
30,
31].
Quite recently, different authors used trifluoromethanesulfonyl chloride (CF
3SO
2Cl, b.p. 29–32 °C) as a useful, effective, and inexpensive reagent for the chlorotrifluoromethylation of terminal and internal alkenes and alkynes as it can be easily reduced to generate a trifluoromethyl radical (∙CF
3) intermediate. In this context, in 2014, Jung, Han, and co-workers developed a mild and simple strategy for the photoredox-catalyzed vicinal difunctionalization of terminal and internal alkenes
1 using CF
3SO
2Cl as CF
3 and Cl sources and releasing SO
2 as a single byproduct (
Scheme 1) [
32].
The authors initially examined the difunctionalization of terminal olefins such as
1 [R
1 = TsNH(CH
2)
3] using 1.5 equiv of CF
3SO
2Cl as CF
3 and Cl sources under visible light irradiation. The process took place, in general, in good to excellent chemical yield during several hours in the presence of 1 mol% Ru(Phen)
3Cl as photocatalyst (
Figure 2) and K
2HPO
4 (0.3 equiv) as buffering reaction system. DCM and MeCN were the solvents of choice and other photocatalysts such as Ir(ppy)
3 and Ru(bpy)
3Cl
2·6H
2O also afforded good yields.
The scope of the method was studied showing, in general, high reactivity with terminal alkenes including functionalized olefins containing groups such as
N-Tosyl,
N-Boc, alcohols, esters, amides, and pyridines. 1,1-Disubstituted and internal alkenes also provided high yields in the chlorotrifluoromethylation process. The authors, moreover, indicated that this protocol was also appropriate for the late-stage difunctionalization of biologically active compounds such as the pesticide rotenone and the insecticide (+)-nootkatone. A mechanism was proposed, which starts with the first irradiation of the photocatalyst Ru(Phen)
32+ with visible light to generate Ru(Phen)
33+. The CF
3SO
2Cl radical anion collapses to ∙CF
3, SO
2 and Cl
−. Then, the CF
3 radical was added to the alkene to form, first, a radical intermediate
3, and then a carbocation
4, which was finally trapped by Cl
− to generate the target molecule (
Scheme 2).
More recently, Matsubara and coworkers studied the chlorotrifluoromethylation of substituted terminal olefins in the presence of highly coordinated square-planar cobalt(II) complexes, such as cobalt(II) porphyrin catalysts CoTPP (TPP= 5,10,15,20-tetraphenylporphyrinato) [
33]. After checking various cobalt and copper complexes, it was found that the combination of CoClTPP and CF
3SO
2Na was the most efficient catalyst when the reaction was carried out in MeCN as solvent. The authors also used trifluoromethanesulfonyl chloride (CF
3SO
2Cl) as a CF
3 radical source, generated after pyrolytic release of SO
2 gas, and a chlorine source (
Scheme 3). Modification of the reaction conditions showed that the combination of the Co
III porphyrin, easily prepared by air-oxidation of Co
II complexes, and Langlois’ salts (CF
3SO
2Na) provided good results in terms of yields, too. Under these optimized conditions, the scope of the method was studied using different terminal functionalized olefins and, in general, high chemical yields were obtained (
Scheme 3).
Interestingly, the use of vitamin B
12 (available as cyanocobalamin) was tested as Co
III catalyst, and it was found that the process worked quite well providing the target molecules in a reasonable yield. This procedure opens new opportunities for pharmaceutical applications and for the study of the regio- and diastereoselectivity in this kind of chlorotrifluoromethylation transformations. Thus, for example, cinchona alkaloid derivative
5 was converted into the target molecule
6 with improved chemical yield and selectivity (6.4:1) when vitamin B
12 (5 mol%) was used as catalyst instead of the CoClTPP (5 mol%) complex (1.4:1) (
Scheme 4).
A catalytic cycle was proposed to explain the obtained results (
Scheme 5).
Xiao, Lin et al. have recently focused on the development of simple and efficient protocols for the incorporation of various fluoroalkyl groupings, in particular a CF
3 substituent, using simple and inexpensive catalysts consisting of a mixture of copper(II) chloride (CuCl
2) and pyridine for the chlorotrifluoromethylation of simple aromatic alkenes, α,β-unsaturated alkenes, and aliphatic alkenes with CF
3SO
2Cl as CF
3 and Cl sources [
34]. Previously, Liu’s group had found that copper (I) complexes also reduced CF
3SO
2Cl and, under those conditions, they studied the successful aminotrifluoromethylation of alkenes. However, stoichiometric amounts of a silver salt were needed to eliminate side reactions, which limited the utility of the process [
35]. In this sense, Xiao’s group discovered that copper(II) chloride could also be an efficient catalyst in this kind of process, although their first attempts resulted in low efficiency (DMF, 7% yield). They observed that the reaction conditions such as temperature, concentration, presence of a base (e.g., pyridine) and, especially, the solvent played a crucial role in achieving high yields. After testing different solvents, they found that the use of 1,4-dioxane as a solvent, high temperature (100 °C) and pyridine (10 mol%) as base provided the best reaction conditions. Other solvents, bases, copper sources as well as different temperatures were also studied but lower yields were, in general, achieved. The scope of the process was studied, and they found a high level of functional group tolerance with styrene derivatives as well as with α,β-unsaturated amides, esters, and ketones. The process also worked with alkyl olefins but resulted in lower yields. The method can be applied to chloroperfluoroalkylation transformations (
Scheme 6).
A plausible reaction mechanism was proposed by the authors (
Scheme 7). Cu
II was initially reduced to Cu
I, which apparently is the real catalyst of the process. CF
3SO
2Cl was activated by forming a pyridinium salt, which through a redox process with Cu
I produces pyridine, CuCl
2 and a CF
3SO
2 radical, which easily underwent desulfonation to generate a CF
3 radical. Finally, this radical was trapped by the alkenes to afford a new radical
3, which after capture by CuCl
2, gave a Cu
III species
7. The final product
2 was obtained by reductive elimination and regeneration of the catalyst (
Scheme 7). Additionally, the chlorotrifluoromethylation was suppressed by the radical scavenger TEMPO (2,2,6,6-tetramethylpiperidine 1-oxyl), which supports a radical mechanism.
Atom transfer radical addition (ATRA) of R
FX (X = I, Br) to electron-rich alkenes is a well-established methodology. However, until recently, the efficiency of this strategy had not been demonstrated for additions to electron-deficient alkenes due to the low conversion, selectivity, and yield obtained. The formation of undesired by-products and, particularly, the electrophilic nature of the radical intermediate formed during the process represent additional obstacles (
Scheme 8). Thus, for example, due to their reducing ability, different Ru, Ir, and Cu species have not proved their efficiency in such reactions. In this context, Dolbier et al. recently studied photoinduced ATRA reactions of various fluoroalkyl-sulfonyl chlorides with electron-deficient alkenes to demonstrate the feasibility of this approach [
36].
Previously, the same group had encountered that different fluorinated radicals such as R
f = CF
3, CF
2H, and CH
2F, among others, could easily be generated from R
fSO
2Cl by photoredox catalysis under mild conditions as a way to obtain fluorinated 2-oxindoles by a tandem radical cyclization of
N-arylacrylamides [
37].
Dolbier first studied the reaction of CF
3SO
2Cl with electron-deficient alkenes using as a model system the α,β-unsaturated amide
N-methyl-
N-phenylacrylamide in the presence of different catalysts and reaction conditions. When Ru and Ir were used as catalysts either no reaction took place or low yields of products were obtained. The best results were achieved with Cu species, in particular, with the complex [Cu(dap)
2]Cl [dap = 2,9-bis(4-methoxyphenyl)-1,10-phenanthroline] for which good chemical yields were obtained when the reaction was carried out at room temperature in either DCM or DCE as solvents. Finally, the addition of K
2HPO
4 (20 mol%) provided an almost quantitative yield of the target chlorotrifluoromethylation product
9. Next, the scope of the process was studied with various electron-deficient alkenes such as α,β-unsaturated amides, esters, nitriles, and ketones under visible light using 0.5 mol% of [Cu(dap)
2]Cl. Moderate to excellent yields were obtained in all cases studied (
Scheme 9). In addition, CF
3Cl was, in general, detected as by-product, attributed to a competitive abstraction of the chlorine atom from CF
3SO
2Cl or Cu(II) species by the intermediate trifluoromethyl radical.
The scope was extended to other previously obtained fluoroalkylsulfonyl chlorides (R
f ≠ CF
3) with excellent results (
Scheme 9). These findings are particularly interesting results due to the biologically importance of the CF
2H group. In these cases, however, higher temperatures (100 °C) were required. Finally, the authors also explored synthetic applications of the target molecules. Thus, transformations such as the reduction, elimination, and substitution of the chlorine atom of the C-Cl bond by N
3, CN, amine, and sulfinate were studied with high yields (
Scheme 9).
Following this strategy, Reiser et al. also reported in 2015 an interesting study about the unprecedented trifluoromethylchlorosulfonylation of unactivated alkenes using the copper phenanthroline photoredox catalyst [Cu(dap)
2]Cl under LED irradiation [
38]. The authors provided evidence for an inner-sphere mechanism in contrast to other ruthenium, iridium and eosin Y catalyzed processes that give rise exclusively to trifluoromethylchlorination using the same alkenes. In the initial attempts, they observed, unexpectedly, that the green light irradiation (LED) of alkenes (using allylbenzene as a model system) with CF
3SO
2Cl in the presence of [Cu(dap)
2]Cl and acetonitrile as solvent resulted in an ATRA process without SO
2 extrusion providing the trifluoromethylchlorosulfonylation adduct
11 in moderate yield and only traces of the expected trifluoromethylchlorination product. Testing different reaction conditions such as catalysts, temperatures, and solvents showed that the presence of bases significantly improved the yield of
11. Thus, the optimized conditions are the use of 1 mol% of [Cu(dap)
2]Cl as catalyst, MeCN as solvent, addition of K
2HPO
4 (1 mmol), and LED irradiation (530 nm) (
Scheme 10). Next, the scope of the process was examined, and the authors observed that allylbenzene with moderate donor or acceptor substituents on the aromatic ring give rise to excellent regioselectivity and good yields. Internal alkenes, however, provided mixtures of regio- and diastereoisomers. On the contrary, styrenes and substrates bearing donor atom in close proximity to the alkene functionality afforded the trifluoromethylchlorination product
2 (
Scheme 10).
Despite the interest in the development of Cu-based photoredox catalysts, only a few Cu catalysts have been used in this kind of processes, with most of them containing one or two phenanthroline ligands. In this context, Xu and coworkers designed new copper complexes based on 4,6-disubstituted 2,2′-bipyridine ligands. In particular, the complex with Xantphos, [Cu
I L
n (Xantphos)][PF
6], turned out to be an efficient catalyst for the chlorotrifluoromethylation of terminal alkenes, especially styrenes, which had not been suitable in this processes [
39]. After testing several alternatives, it was concluded that complex
15 was the most efficient for this study. The synthesis of ligand
13 and complex
14 was prepared in three steps starting from 2,4,5-trimethylbenzaldehyde (
12) by mixing ligand
13 with [Cu (MeCN)
4][PF
6] and Xantphos in DCM as solvent (
Scheme 11).
The efficiency of complex
14 as photoredox catalyst was tested in the chlorotrifluoromethylation of styrenes and CF
3SO
2Cl as trifluoromethyl source. The result of this study was the formation of two main products, the expected chlorotrifluoromethylation derivatives
2 and another
15 resulting from the chlorotrifluorosulfonation of the starting alkenes
16 (
Scheme 12).
The process was very efficient with excellent conversion (100%) when the reaction was carried out at 40 °C using 1.2 equiv of CF3SO2Cl, 2 equiv of K2CO3 as a base, 0.5 mol% of catalyst in DCM and LED450 illumination. Under these conditions, a ratio of 15:1 was obtained in favor of chlorotrifluoromethylation product 2. Other Ru, Ir, Cu, and organic photocatalysts provided less efficient results. Interestingly, when the LED source was replaced by sunlight, a similar conversion and selectivity were obtained. During this study, the authors found that chlorotrifluorosulfonation product 15 was sensitive to water and eliminates HCl during the isolation to yield compound 16. The optimized conditions were successfully applied to a large number of substituted styrenes. Other aromatic groups such as naphthyl, pyridyl, and substrates derived from estrone, and α-tocopherol worked also well, proving the feasibility and tolerance of the method. The authors also studied the limitations of the process and found that internal alkenes were, in general, unreactive. In other cases, e.g., 4-vinylpyridine and 4-methoxy-1-vinylbenzene, polymerization was observed.
A mechanism was proposed based on previous studies with other terminal alkenes [
17]. Thus, the photoexcited state of the Cu* catalyst undergoes oxidative quenching by CF
3SO
2Cl to give the CF
3SO
2 radical, which is trapped by the olefin, extruding SO
2 to give a new CF
3 radical, which adds to the olefin. The addition product is an alkyl radical able to take different reaction pathways [
26].
Very recently, Qing and co-workers reported the first copper/zinc-copromoted chloro- and bromo-trifluoromethylation of alkenes and alkynes using inexpensive and simple materials such as trifluoromethanesulfonic anhydride (CF
3SO
2)
2O (Tf
2O) [
29,
40] as trifluoromethylating reagent [
41]. Thus, the reaction of alkenes and alkynes with Tf
2O in the presence of copper salts such as CuX
2 (X = Br, Cl), Zn powder and 2,2′-bipyridine provided the corresponding bromo- or chlorotrifluoromethylated products in good yields. The authors also showed the dual role that copper(II) salt plays in the process as catalyst and halogen source, whereas the 2,2′-bipyridine acts both as activating reagent of Tf
2O and the ligand to coordinate to CuX
2. With the optimized reaction conditions in hand, the scope of the copper/zinc copromoted process was studied. A variety of unactivated alkenes with a range of functional groups and styrenes with different substituents in the benzene ring were studied first, and the corresponding bromotrifluoromethylated derivatives
17 were obtained in good to excellent yields including biologically relevant compounds such as dicamba herbicide and estrone (
Scheme 13A). Internal alkenes, however, turned out to be unsuitable substrates in this process. Moreover, this protocol is also suitable for terminal and internal alkynes
18, which were converted into the substituted bromotrifluoromethylated alkenes
19 with good regio- and stereoselectivity and moderate yields (
Scheme 13B).
This Cu/Zn-process was successfully extended to chlorotrifluoromethylation reactions by simply replacing the CuBr
2 salt with CuCl
2 using similar conditions as before. In this case, however, addition of
n-Bu
4NCl was necessary (
Scheme 14). Unfortunately, this reaction could not be extended to fluoro- and iodotrifluoromethylation reactions.
The above results can be explained by the mechanism outlined in
Scheme 15. First, the reaction of Tf
2O and 2,2′-bipyridine (L) provides the
N-triflylpyridinium salt, which after SET reduction with Zn powder affords a radical species
20 that collapses to liberate SO
2, 2,2′-bipyridine and a CF
3 radical. At the same time, LCuX
2 (X= Cl, Br) is reduced by Zn powder to deliver LCuX, which acting as a reductant converts the
N-triflylpyridinium salt into the radical species
20. The addition of the CF
3 radical to the alkene/alkyne gives a new β-trifluoromethyl radical
22, which can progress through two possible pathways to the desired bromo/chloro final molecules (
Scheme 15).
Following similar strategies but, in this case, carrying out the reaction under visible light catalyzed process, Han and coworkers described in 2017 a one-step regio- and stereoselective photoredox protocol for synthesizing tetrasubstituted alkenes by chlorotrifluoromethylation of internal arylalkynes
18 using CF
3SO
2Cl as both CF
3 and chlorine source (
Scheme 16) [
42]. Tri- and, particularly, tetrasubstituted alkenes containing a CF
3 group are described as useful intermediates in the synthesis of pharmaceutical and agrochemical agents [
43]. After screening different ruthenium and iridium photocatalysts, bases and solvents, the optimal conditions were obtained with Li
2CO
3 as base, acetone as solvent and
fac-Ir(ppy)
3 (see,
Figure 2) as photocatalyst. The alkyne scope of the process was studied under the optimized conditions, and it was found that the process takes place, in general, with moderate to good yields. A variety of functional groups and substituents in the arene ring worked efficiently. Thus, for example, alkynes containing acetyl,
p-tosyl and Boc-protected amines in the arene ring, and 4-acetaminophenylalkynes containing various R groups at the nonaryl-substituted position provided moderate to good yields. The authors also highlighted that the reaction failed in the absence of visible light irradiation.
The above results were explained by the following mechanism: the excited state of the photoredox catalyst (*M
n), generated by visible light, reacts CF
3SO
2Cl to provide a CF
3 radical, SO
2 and a chloride anion. The addition of the CF
3 radical to the alkynes takes place regioselectively to produce an aryl ring stabilized the vinyl radical
22. Next, the vinyl radical
22 is oxidized to generate a new vinyl cation
23 and the regeneration of the photocatalyst. The last step is addition of chloride to the vinyl cation
23, finally yielding the target olefin
24. The authors explained that due to electrostatic repulsion, the addition of the chloride takes place preferentially anti to the CF
3 group (
Scheme 17).
The utility of this protocol for preparing more complex molecules was also studied in Suzuki coupling reactions with boronic acids. Thus, the chloro-atom was substituted, for example, by pyrazole, furan, and benzothiophene groups providing other interesting complex tetrasubstituted alkenes
25 (
Scheme 18).
Notably, an interesting contribution concerning the chlorodifluromethylation and chlorocarbomethoxydifluoromethylation reaction of unactivated alkenes under metal-free conditions was also developed in 2015 by Dolbier et al. [
44]. The incorporation of a CF
2H, as well as a CF
2CO
2R group, is considered of particular interest in the pharmaceutical and agrochemical industry [
45,
46].
A different and practical strategy for the iron-mediated chlorotrifluoromethylation of styrenes and aliphatic alkenes was presented by the Qing group in 2016 [
47]. The process uses inexpensive and solid materials such as ferric chloride (FeCl
3) as the Cl source and the Langlois reagent [
48] (sodium triflinate, CF
3SO
2Na) as the CF
3 source. After screening different metal salts, solvents, and temperatures, the authors found that FeCl
3 and MeCN as salt and solvent, respectively, were the best option. Other salts (Cu
+1and Cu
+2) and solvents (DMF, DMSO, etc.) were less efficient (
Scheme 19). The scope of the iron-mediated chlorotrifluoromethylation of alkenes showed that styrenes bearing electron-donating and electron-withdrawing groups reacted smoothly as well as unactivated aliphatic alkenes to give the target molecules
2 in good to excellent yields. This protocol was also efficient for biologically active compounds such as vinyl-
N-benzoyl-
L-tyrosine, showing that this method could be suitable for the “late-stage chlorotrifluoromethylation” of natural products and drugs.
The mechanism of the process was studied. Thus, a trifluoromethylradical is generated from Langlois reagent through a SET process under the treatment of FeCl
3 and K
2S
2O
8, which then reacts with the alkene to give the radical intermediate
3. Finally, the radical intermediate
3 was oxidized by FeCl
3 and K
2S
2O
8 to generate a cation
4, which undergoes a nucleophilic attack by the chloride anion to provide the final addition product (
Scheme 20). In the presence of TEMPO, the reaction was inhibited, suggesting a radical/cationic process.
The easy-handling Langlois reagent has also been used successfully in photoredox processes. Thus, quite recently, Feng Liu and coworkers reported a mild, one-pot, and transition-metal-free photoredox halo- and trifluoromethylthio-trifluoromethylation of simple alkenes with sodium triflinate [
49]. In this process,
N-chloro- and bromophthalimide (
26) and
N-trifluoromethylthiosaccharin (
27) were used as halogen and SCF
3 sources, respectively. An organic photoredox catalyst, 9-mesityl-10-methyl acridinium (Mes-Acr
+) perchlorate (
28), with an excited-state reduction potential of 1.8 V, was used under visible-light irradiation (white LED). The authors found that the optimized conditions are the use of 1,2-dichloroethane (DCE) as a solvent, TFA (trifluoroacetic acid) as acid additive, Langlois reagent as CF
3 source,
N-chloro- (NCP) and
N-bromoimides (NBP) as halo source, and Mes-Acr
+ as photocatalyst. It was also found that irradiation with blue LED gave satisfactory results. The scope of the process was studied, and it became apparent that many functional groups including amides, ketones, imides, esters, etc., were tolerated and smoothly converted into the desired final products
29 with high yields. The case of 2,2-diallylmalonate for which a radical cascade took place is particularly interesting. Simple control experiments with TEMPO supported the presence of radicals in the process. This protocol was extended to the trifluoromethylthiotrifluoromethylation of simple alkenes using
N-trifluoromethylthiosaccharin
27 as the SCF
3 transfer reagent with similar results. In this case, however,
p-toluensulfonic acid (PTSA) was the best acid additive (
Scheme 21).
A plausible mechanism was proposed to rationalize the formation of the desired product. According to the authors, the process was initiated by a single electron transfer of the Langlois reagent by the photoexcited Mes-Acr
+* generating a CF
3 radical and an acridine radical. The subsequent addition of the CF
3 radical to the olefin followed by halogenation with NXS (X = Cl, Br) lead to the final product
29 (
Scheme 22).
Following a similar strategy but using enynes instead of simple olefins, a cascade cyclization of enynes was recently reported by Hou, Chen, and Zhu’s group. Thus, a new visible-light-mediated photocatalytic chlorotrifluoromethylation and chlorotrichloromethylation of 1,6-enynes
31 in the presence of CX
3SO
2Cl (X= F, Cl) is described [
50,
51]. The process proceeds through an initial radical addition, followed by a cyclization/chlorination sequence. The use of terminal alkene-derived 1,6-enynes provided an array of carbo- and heterocycle derivatives
32 in a regio- and stereoselective manner (
Scheme 23). In this process, both Mes-Acr
+ ClO
4− and Ir(dtbbpy)(ppy)
2PF
6 were effective catalysts showing that the transition metal was not essential. Although different Ir and Ru complexes were tested, the best result was obtained when the organic dye Acr
+-Mes ClO
4 was used as catalyst and DCM as solvent under irradiation with a 23 W fluorescent bulb. Although the process worked quite well for nitrogen derivatives (X = NSO
2R
2) and all-carbon cyclic compounds, the method failed for oxygen derivatives (X = O).
The mechanism of the process for the chlorotrifluoromethylation is based on previous reports and it is outlined in
Scheme 24. Once the CF
3 radical is generated, its addition to the enyne double bond provides a tertiary carbon radical intermediate
33. The 5-
exo radical cyclization results in a new vinyl radical
34 formation, which finally reacts with CF
3SO
2Cl affording through a chlorine atom transfer reaction the final
Z-products
32 and regeneration of the sulfonyl radical. Interestingly, the 6-
endo radical cyclization was not observed.
Previously, Song Lin et al. [
52] had reported a convenient procedure for the preparation of chlorotrifluoromethylated pyrrolidine derivatives, which are interesting molecules in medicinal chemistry research, by electrocatalytic radical ene-yne cyclization. The process was carried out using anodically coupled electrolysis and the incorporation of a chelating ligand such as 2,2′-bipyridine (bpy) provided the final products
32 with high regio- and stereochemical control in favor of the (
Z)-isomer. The Langlois reagent (CF
3SO
2Na) and MgCl
2 were used as CF
3 radical and Cl
− sources via electrochemical oxidation as well as different manganese salts as catalysts. After checking different reaction conditions, it was found that the use of Mn(OTf)
2 as a catalyst, a mixture of HOAc/MeCN as solvent, MgCl
2 and CF
3SO
2Na salts as CF
3 radical and Cl
− sources, and the application of a constant current of 10 mA were the most efficient conditions in terms of chemical yield (up to 89%) and selectivity (
Z/E > 19:1). Interestingly, the addition of bpy as ligand increased the yield and selectivity notably. When the reaction was carried out in absence of the Mn catalyst, no product was obtained indicating the crucial role of the catalyst in the process (
Scheme 25). Concerning the scope, aryl, alkyl, silyl, and terminal alkynes were also compatible with the described electrocatalytic conditions.
The proposed mechanism of the process is outlined in
Scheme 26. The starting 1,6-substrates undergo a cascade reaction, which is a selective trifluoromethylation, radical cyclization (
5-exo-dig cyclization) and final chlorination to yield the target pyrrolidines
32. The mechanism entails the initial simultaneous anodic oxidation of the Langlois reagent (formation of the electrophilic radicals CF
3, anodic event A) and [Mn
III]-Cl (anodic event B, formed by mixing the Mn catalyst and MgCl
2). The final
Z/E-stereochemistry is sterically controlled by the conjugation between the aryl π-orbital and the single occupied molecular orbital in intermediate (
35). This orbital is shielded by the substituents at the quaternary carbon α, favoring the approach on the less congested side to the final
Z-isomer.
Using a similar strategy, the same group had simultaneously developed a new protocol based on the anodically coupled electrolysis for the regio- and chemoselective chlorotrifluoromethylation of simple alkenes [
53]. The optimized reaction conditions were studied, and it was found that the Langlois reagent and MgCl
2 were the starting materials of choice to carry out the process successfully. The most favorable reaction conditions are outlined in
Scheme 27. Thus, Mn(AcO)
2 + MgCl
2 generate the latent radical chlorination agent [Mn
III]-Cl, trifluoroacetic acid (TFA) as the sacrificial oxidant and proton source, LiClO
4 as the electrolyte in MeCN, and a constant current of 15 mA were the most efficient conditions found.
The scope of the process was also investigated, and it was found that cyclic and aliphatic alkenes and a variety of styrene derivatives with oxidative labile functional groups were compatible with those conditions. However, electron-rich styrenes were not effective in the process. Furthermore, small amounts of the corresponding dichloride derivatives as undesired compounds were also detected (
Scheme 27). This methodology was also applied to the difunctionalization of alkynes and the bromotrifluoromethylation of alkenes using, instead of MgCl
2, potassium bromide (KBr) as the bromine source (
Scheme 27). The mechanism of the process is similar to that described by the same group (see
Scheme 26), but using simple alkenes instead of 1,6-enynes as starting materials. Radical intermediates such as
3 (free radical addition to the alkene, fast) and
37 (metal-mediated radical addition to the alkene, slow) explained the obtained results (
Scheme 27).
In the chlorotrifluoromethylation of alkenes, several reagents (in particular, CF
3SO
2Cl and others) have been reported for that purpose. However, most of them suffer some problems in terms to find mild reaction conditions. An unprecedented and relatively new strategy makes use of the solid and bench-stable trifluoromethanesulfonyl hydrazides as trifluoromethylating reagent for the vicinal difunctionalization of terminal alkenes [
54]. Thus, Tian et al. reported in 2016 that TfNHNHBoc, prepared by reacting triflic anhydride with monosubstituted hydrazines, and NaCl are efficient substrates for the vicinal chlorotrifluoromethylation of styrene derivatives in the presence of CuCl as redox catalyst. The use of Me(CH
2)
15NMe
3Cl as a phase transfer catalyst, DMSO/water (4:1) as solvent at room temperature, and TBHP (
tert-butyl hydroperoxide) as oxidant provided the most effective conditions. Excellent regioselectivity and good chemical yield were, in general, obtained. This process was extended to the bromotrifluoromethylation of arylalkenes by simple substitution of NaCl and CuCl by NaBr and CuBr increasing the scope of the process (
Scheme 28). On the other hand, the process failed in the iodotrifluoromethylation of arylalkenes.
A plausible mechanism of the process is depicted in
Scheme 29. Thus, oxidation of TfNHNHBoc leads to the formation of a diazene
38, which decomposes into a CF
3 radical with extrusion of SO
2. Regioselective addition to the alkene followed by copper-catalyzed oxidation, formation of a carbocation and reaction with Cl
− gave the final product
2.
In 2019, Wang and coworkers reported the use of the CF
3-containing λ
3-iodane, PhICF
3Cl [
55], as a bifunctional reagent for the uncatalyzed chlorotrifluoromethylation of non-conjugated and conjugated alkenes [
56]. This process was also successfully extended to biologically important molecules such as ibuprofen, indomethacin, estrone, and oxaprozin derivatives among others. The best reaction conditions are the use of 1,4-dioxane as solvent, nitrogen atmosphere, and temperatures of 60 °C (
Scheme 30a). Moreover, the nature of the final product is highly dependent on the reaction conditions. Thus, when the reaction was carried out at higher temperatures (60–100 °C) and the solvent was DMF, the obtained products were identified as vinyl derivatives, being the result of the elimination of HCl of the initially formed chlorotrifluoromethylated product. This “side” reaction was particularly effective in the case of the styrene derivatives (
Scheme 30b).
A plausible mechanism was proposed for this process. Thus, the initial reaction of the alkene with PhICF
3Cl undergoes a SET process to generate a radical cation and a hypervalent iodine radical, followed by a coupling of the radical cation
40 with the generated CF
3 radical affording a new carbocation
4 and releasing of PhI. Final nucleophilic attack on the chloride anion to the carbocation provided the target molecule
39 (
Scheme 31). In this mechanism, an ionic pathway cannot be ruled out. Finally, vinyl C-H trifluoromethylation products were obtained by elimination of HCl in the case of conjugated alkenes.
Although the halotrifluoromethylation of alkenes has recently been studied in some detail, complex reaction conditions are required in most of the cases, including expensive photoredox catalysts (Ru, Ir and Cu complexes), high temperatures and the use of sophisticated bases. In this context, Jiang, Yang, and co-authors [
57] described a general, gram scale, three-component and efficient method for the intermolecular chloro- and bromotrifluoromethylation of electron-deficient and unactivated alkenes using simple CuBr
2 as catalyst and Togni reagent in combination of dihalo-sulfoxide (SOX
2, X = Cl, Br) as the trifluoromethylation and halogen sources, respectively. The optimized and mild reaction conditions are the use of CHCl
3 as solvent, room temperature, catalyst loading of 1 mol%, 1.5 equiv of Togni II reagent, and 1.0 equiv of SOX
2. The scope of the process is wide providing good to excellent results with cyclic pyrrolidinon derivatives, linear α,β-unsaturated amides as well as simple aliphatic alkenes. Interestingly, Umemoto reagent and Togni reagent I were either inactive or much less efficient in the process. In the case of the bromoderivatives, PBr
3 was used as bromine source in some cases instead of thionyl bromide (
Scheme 32). Control experiments showed that both the copper catalyst and the halo-source were essential for the success of the process. When the reaction was carried out in the presence of TEMPO, no final product was obtained indicating the radical nature of the process.
Considering the obtained results, a simple mechanism was proposed (
Scheme 33). Togni reagent is activated by the copper catalyst, generating a radical CF
3 (SET process) and release of a Cu
+3 species. The CF
3 radical was then added to an electron-deficient alkene generating a new radical
41, which underwent a second SET process, affording a carbocation
42 and regeneration of the active Cu
+2 catalyst. Finally, the formed carbocation is attacked by Cl
− anion of the thionyl chloride.
Although many reports have been published using different strategies and trifluoromethyl sources for the chlorotrifluoromethylation of olefins in the last decade, the same does not apply to the other halogens. In fact, very few protocols have been established for the general halotrifluoromethylation of alkenes, and a lot less in the case of styrene derivatives. In this sense, Masson et al. [
58] described in 2015 a quite general protocol for the three-component halotrifluoromethylation of olefins in a photoredox process catalyzed by the Ru(bpy)
3(PF
6)
2 complex as photosensitizer and Umemoto reagent as CF
3 source. Concerning the nature of the halogen, the process can be applied to chloro-, bromo-, and iodo-derivatives, and the described method is particularly efficient in the case of styrene derivatives for which moderate to good yields were obtained. Moreover, short reaction times and mild reaction conditions were required. In the case of the chloro-derivatives, TMSCl was used as chlorine source and HCCl
3 as solvent. These conditions failed for the other halogens for which a CsX (X = Br, I) salt as halogen source and MeCN as solvent were used (
Scheme 34).
A reasonable mechanism for this multi-component process is shown in
Scheme 35. The process is initiated by the formation of a strong reductant species *Ru(bpy)
32+, obtained from the photoredox initiator after excitation with visible light. Then, a single electron transfer generates an electrophilic CF
3 radical from Umemoto reagent, which next reacts with the olefin to give an α-aryl radical intermediate affording a new α-aryl carbocation. Final nucleophilic trapping of the carbocation by Cl
−, Br
−, or I
− leads to the trifluoromethylated product
29.
In 2014, Liu and co-authors described an effective and general methodology for the iodotrifluoromethylation of alkenes and alkynes using the Langlois reagent and iodine pentoxide (I
2O
5) as CF
3 and iodine sources [
59]. More recently, the same group extended this methodology to the bromotrifluoromethylation of unactivated olefins and styrenes using an appropriate bromine source. After having screened several bromides, they found that sodium bromate (NaBrO
3) along with the Langlois reagent were the most efficient reagents [
60]. The metal-free process worked particularly well when a mixture of DCM/H
2O (4:1) as solvent was applied. It is noteworthy that sodium bromate plays a dual role in the process acting both as a single-electron oxidant and a bromine source. Under these conditions, a wide range of unactivated alkenes with different functional groups, including nonterminal alkenes and styrene derivatives, were well tolerated and, in general, moderate to good yields were obtained (
Scheme 36A). The authors also demonstrated that a large-scale (gram level) was also possible, thus increasing the potential of this methodology in the chemical industry as well as the utility of the obtained molecules in synthetic organic chemistry (
Scheme 36B).
The formation of the target molecules was initiated by a single-electron oxidation of the Langlois reagent by NaBrO
3 to generate a CF
3 radical. Next, the CF
3 radical adds to the double bond to afford a new radical intermediate
3, which finally undergoes the bromination step to result in the target product
17 (
Scheme 37). Different mechanistic studies involving a radical-clock experiment and electron-spin resonance (ESR) detection supported the radical nature of the process.
In order to extend the utility of this methodology, the same group studied, one year later, the behavior of 1,6- and 1,7-enynes in this kind of processes. The result was, using the same reaction conditions (NaSO
2CF
3/ NaBrO
3), the facile access to a wide range of 5- and 6-membered heterocycles bearing a CF
3 group and Br atoms in their framework (
Scheme 38). This free-radical cascade process took place under mild metal-free reaction conditions with good chemical yields and high chemoselectivity [
61].
The mechanism of the process is quite similar to the one previously described for alkenes (see
Scheme 37). Initially, a CF
3 radical is generated by electron transfer from NaSO
2CF
3 to NaBrO
3 followed by release of SO
2 and Br
2. Next, radical addition and cyclization afford a new vinyl radical. Final Br-atom abstraction completes the process (
Scheme 39). Interestingly, the CF
3 radical was trapped by 2-methyl-2-nitropropane (MNP), which demonstrated the radical nature of the process.
A completely different and novel strategy was reported by Tan, Liu, et al. for the radical tandem trifluoromethylation-initiated remote cross-coupling of a variety of carbonyl compounds containing an alkene moiety
44 with simple nucleophiles such as halogens (Cl, Br, I) and a cyano group, using Togni reagent II as the CF
3 source (
Scheme 40) [
62]. The process provided the simultaneous formation of two new C-CF
3 and C-X (X= Cl, Br, I, CN) bonds, in which the key step is an intramolecular 1,5-H radical shift. After checking different reaction conditions, the best results were obtained in the case of the remote bromo-trifluoromethylation with CuBr as catalyst, KBr as inorganic salt, 2,2′-bipyridyl (
45) as ligand and EtOAc as a solvent. Similar reaction conditions were employed for chloro- and iodo-trifluoromethylation, but in this case using CuI as catalyst and inorganic salts such as LiCl or KI, respectively. In order to extend this methodology, the authors also studied the remote α-cyano-trifluoromethylation of carbonyl compounds containing an alkene in the ω-position. In this latter case, the optimal conditions are the use of the Cu(MeCN)
4PF
6 complex as catalyst, 1,10-phenanthroline (
46) as ligand, trimethylsilyl cyanide (TMSCN) as cyano source and MeCN as solvent. The process worked well with a variety of aliphatic and aromatics alkenyl carbonyl derivatives, including ketones, esters, amides, etc. In all cases, moderate to good yields were obtained and the final products
47 were easily transformed into valuable CF
3-containing aza-heterocycles.
After several control experiments including reactions with TEMPO or 2,6-di-
tert-butyl-4-methylphenol (BHT) and with deuterated compounds, the authors concluded that the process proceeds through an intramolecular 1,5-H radical shift. Thus, the CF
3 radical generated from the reaction of Togni reagent with Cu(I) attacks the alkene moiety providing intermediate
48, followed by a 1,5-H radical shift obtaining radical
49, from which a new Cu(III) species
50 originates. Finally, reductive elimination leads to the target product (path A). Another alternative is the formation of carbocation
51 by single-electron oxidation with Cu(II) species and nucleophilic addition of inorganic halogen salt or TMSCN (path B) (
Scheme 41). This strategy provides a novel and elegant procedure for synthesizing trifluoromethyl α-halo and α-cyano-carbonyl derivatives of great importance in the pharmaceutical industry.
3. Iodotrifluoroalkylation
Among trifluoromethylation-involved difunctionalization of unsaturated compounds, the iodotrifluoromethylation of alkenes and alkynes, has attracted much attention in the past decade [
63]. However, most all reported strategies for iodotrifluoromethylation of olefins and alkynes are based on using trifluoroiodomethane (CF
3I) as the trifluoromethylation source, which is a gas and hampers the handling and operation. Consequently, operationally safer, and more sustainable iodotrifluoromethylation of alkenes and alkynes have emerged as alternative methods for the same purpose.
In this regard, different iodotrifluoromethylation methods have been developed including visible-light-induced approaches based on the use of transition-metal complexes as photosensitizers [
64]. Most of these approaches are based on the use of iridium and ruthenium photocatalysts as they have efficiently catalyzed the formation of carbon-fluoroalkyl bonds under visible light irradiation [
65]. Photoredox catalysts based on ruthenium or iridium metal complexes from polybipyridyl ligands absorb blue light, and high yields were generally achieved with high chemoselectivities under mild reaction conditions.
Using this approach, in 2014, Cho and coworkers developed a controlled methodology based on the use of iridium and ruthenium photocatalysts for the iodotrifluoromethylation and hydrotrifluoromethylation of alkynes
18 (
Scheme 42) [
66]. The authors initially examined the iodotrifluoromethylation of terminal alkynes with CF
3I and
N,
N,
N’,
N’-tetramethylethylenediamine (TMEDA) in MeCN under visible light irradiation (blue LEDs) in the presence of a wide range of iridium and ruthenium photocatalysts, including
fac-[Ir(ppy)3], [Ir(ppy)
2(dtb-bpy)]PF
6, [Ru(bpy)
3]Cl
2, and [Ru(phen)
3]Cl
2. Despite all catalysts favoring the generation of the iodotrifluoromethylation product in high yields and high
E/Z ratios (from 17:1 to 20:1), [Ru(phen)
3]Cl
2 was chosen as the optimal catalyst for evaluating the scope of iodotrifluoromethylation because of its low cost and simplicity of reaction. In this study, the iodotrifluoromethylation of a variety of aromatic and aliphatic alkynes bearing several functional groups (13 examples) was accomplished with excellent
E/Z stereoselectivities.
The authors proposed a plausible mechanism for the hydrotrifluoromethylation of alkynes in which visible light photoexcitation of [Ir(ppy)
3] affords the excited species*[Ir(ppy)
3], which is then reductively quenched by 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) to produce the anionic [Ir(ppy)
3]
−• radical and the DBU radical cation. Next, the radical iridium anion performs a single-electron reduction of the F
3C−I bond, regenerating the photocatalytic cycle and delivering a CF
3 radical. Subsequently, addition of this radical to the alkyne produces the vinyl radical
54, which through a direct hydrogen abstraction by the vinyl radical generates the desired alkenyl-CF
3. Alternatively, the alkenyl–CF
3 product could also be followed by competitive iodide abstraction from CF
3I by the vinyl radical to give the alkenyl iodide as an intermediate, which supports the iodotrifluoromethylation of alkynes. The iodotrifluoromethylation is followed by de-iodination of the trifluoromethylated alkenyl iodide intermediate which is coupled to the same photocatalytic system as confirmed by an experimental transformation of the alkenyl iodide into the desired alkenyl–CF
3 under the conditions for the hydrotrifluoromethylation (
Scheme 43).
On the other hand, for the hydrotrifluoromethylation reaction, iridium catalysts displayed more effective activity than ruthenium catalysts with superior activity for
fac-[Ir(ppy)
3]. The choice of base was found to be critical, as the base also acts as a hydrogen donor. Wide scope of terminal alkynes (10 examples) underwent the hydrotrifluoromethylation under visible-light irradiation (blue LEDs) with DBU in MeCN (1:1) yielding a mixture of the
E and
Z hydrotrifluoromethylatedcompounds in good to excellent yields. Interestingly,
fac-[Ir(ppy)
3] also catalyzed the trifluoromethylation reaction of aromatic alkynes but showed to be inefficient in the trifluoromethylation of aliphatic alkynes. Aromatic alkynes were converted into trifluoromethylated alkynes with CH
3I and
fac-[Ir(ppy)
3] under visible light irradiation (blue LEDs) using such bases as
tBuOK and Cs
2CO
3, but with lower yields (60–68%) than those for the iodo- and hydrotrifluoromethylation reactions. The reactions also afforded the bis(trifluoromethylated) products in 10–20% yield (
Scheme 42).
As an alternative to visible-light-induced reactions which require the use of expensive ruthenium, iridium, and copper photocatalysts, organic dyes as photocatalysts have gained significant interest due to the less toxic and more sustainable properties [
67,
68,
69]. In the last decade, several families of organic dyes, such as flavins, acridiniums, cyanoarenes, xanthenes, diaryl ketones, thiazines, among others have been exploited in the field of organic photocatalysis [
70].
In contrast to the widely used transition-metal complexes as photosensitizers in the iodotrifluoromethylation reactions, only a few examples have been described using organic sensitizers as catalyst in this kind of organic transformations. One of the rare examples in this field was described in 2017 by Cho’s group, who reported the trifluoromethylation of alkenes and alkynes with trifluoroiodomethane (CF
3I) as fluorine source in the presence of the inexpensive organic dye Nile Red (
56, 9-diethylamino-5H-benzo[α]phenoxazine-5-one) [
71]. In this light-assisted process, the selective transformation of alkenes into either alkenyl-CF
3 compounds or trifluoromethylated alkyl iodides
55 was achieved depending on the reaction conditions, in particular, the appropriate selection of the base and the solvent, which notably affected the selectivity of the process (
Scheme 44). After assaying different organic dyes, bases, and solvents, the authors found that the environmentally benign organic dye Nile Red was very efficient as a photosensitizer and it was used for the first time in organic transformations. Furthermore,
N,
N-diisopropylethylamine (DIPEA) as base in acetonitrile was shown to be the most appropriate solvent in the iodotrifluoromethylation of alkenes, while DBU and DMF were the optimal base and solvent in the synthesis of alkenyl-CF
3 derivatives, respectively. The scope was also studied and under the optimized conditions, aliphatic alkenes with a wide range of functional groups were easily converted into the target molecules with good chemical yields. Interestingly, the process was not appropriate for aromatic alkenes. This study was also extended to alkynes with similar results but in this case, 2 equiv of
N,
N,
N’,
N’-tetramethylethylenediamine (TMEDA) as base in MeCN were required under either yellow LED or with a compact fluorescent lamp (CFL) irradiation (
Scheme 44). Additionally, this protocol was also efficient in the trifluoromethylation of some
N-heterocycles, such as
N-methyl pyrrole and 3-methylindole.
A quite simple mechanism was proposed for the above transformations. Thus, a CF
3 radical was generated by photocatalysis with Nile Red, which rapidly reacts with the alkene moiety to form a trifluoromethylated radical species
3, followed by an atom transfer radical addition between this new radical and CF
3I, which allows obtaining the target molecule
55. This, after elimination of HI by the action of the base produces the alkenyl-CF
3 derivative. Alternatively, the final iodotrifluoromethylated derivative can be obtained from a carbocation intermediate
4 generated by photocatalytic oxidation of the initial radical (
Scheme 45).
Vincent and co-workers studied, for the first time, the use of Togni reagent as the source of CF
3 and iodine and benzophenone as a photosensitizer for the light-promoted iodotrifluoromethylation of alkenes (
Scheme 46) [
72,
73]. They observed a fast reaction of the benzophenone triplet-state (
3BP) with isopropanol to generate an α-hydroxyisopropyl radical, which could be used to reduce the Togni reagent to afford a reactive trifluoromethyl radical, 2-iodo benzoic acid, and acetone. Several control experiments were carried out to show that both
i-PrOH and light were crucial to obtaining the best yields and that benzophenone notably increased the reaction rate. Concerning the scope (18 examples), the process worked well with a wide range of olefins although the chemical yields were only moderate. In some cases, however, the process failed particularly with styrene, and electron-deficient alkenes (i.e., phenyl vinyl sulfone and others), for which unexpected hydrotrifluoromethylated products were almost exclusively obtained.
The authors proposed a quite sophisticated mechanism, in which an α-hydroxyisopropyl radical was initially generated by the reaction of isopropanol with BP triplet state (
3BP). Next, α-hydroxyisopropyl radical reacted with the Togni reagent to produce CF
3 radicals and 2-iodobenzoic acid through the anion-radical intermediate
57. The CF
3 radicals react very quickly with
i-PrOH to yield CF
3H and regenerate α-hydroxyisopropyl radical, thus maintaining the radical chain cycle. Additionally, through the decomposition of intermediate
66, trifluoroiodomethane (CF
3I) was formed along with benzoic acid radical
58, which later reacts with
i-PrOH to produce benzoic acid regenerating α-hydroxyisopropyl radical. The CF
3 radical adds, next, to the olefin achieving, through of the α-CF
3 alkyl radical
3, the target molecule after final abstraction of an iodine atom from 2-iodobenzoic acid (pathway a). Alternatively, the CF
3I generated also in situ from intermediate
57 can also react with radical
3 to obtain the final iodotrifluoromethylation molecule
55 and regenerate the CF
3 radical (pathway b) (
Scheme 47).
One year later, the same group reported [
74] that catalytic amounts of chloride ions (i.e., NaCl or Bu
4NCl) could promote the iodotrifluoromethylation and iodoperfluoroalkylation of alkenes and alkynes under low intensity UVA irradiation in deoxygenated methanol solutions (
Scheme 48). Additionally, in another paper by Matile and coworkers [
75], the authors demonstrated that chloride ions can be transported through membranes using perfluoroalky iodides (R
FI) as transporters and, therefore, the chloride ions might be used to weaken the R
f-I bond through destabilization, thus facilitating the homolytic cleavage of the R
f-I bond by the halogen bonding interaction between R
fI and the chloride ions under direct photolysis. It is noteworthy that the direct photolysis of R
fI using a conventional TLC lamp was shown to be inefficient (<5% yield), but the addition of a stoichiometric amount of Bu
4NCl allowed the isolation of the desired compound in high yield (>95%). The optimized reaction conditions require the use of less than 10 mol% of the catalyst in a short irradiation time (2–4 h) and the use of methanol or ethanol as solvents. Interestingly, among all the halogens tested in the study, chloride ions were shown to be the most efficient (chloride salts such as NaCl and Bu
4NCl exhibited similar catalytic activity) but in the case of the bromide and iodide ions, no reaction occurred. Fluorine ions, on the other hand, provided only a modest efficiency (32% yield). The scope of the process was also studied, and found that linear alkenes with different functional groups and internal alkenes were efficient and the final products were obtained in good (up to 89%) chemical yields (
Scheme 48). This protocol was also successfully extended to terminal alkynes and good chemical yields were obtained in a short reaction time but with low to moderate stereoselectivity. However, CF
3I was found to be unreactive towards alkynes.
The authors proposed a plausible mechanism based on experimental observations: the catalytic amount of the chloride ions has a strong impact on the production of the perfluoroalkyl radicals (R
F.) when the experiment is carried out in absence of alkenes (
Scheme 49). Thus, the initially perfluoroalkyl radical generated from R
FI and Cl
− ions and light irradiation would initiate a radical-chain reaction, which reacts rapidly with methanol to generate R
FH and an α-hydroxymethyl radical. Then, this newly formed radical reacts with R
FI to produce either iodomethanol or formaldehyde to regenerate the initial perfluoroalkyl radical and/or HI reinitiating the catalytic cycle. Both iodomethanol and/or formaldehyde are finally converted into formaldehyde methyl acetal by reaction with methanol. In the presence of an alkene, the formation of R
FH was not observed, indicating a faster rate of addition of R
F. on the double bond than the hydrogen atom abstraction of methanol.
Interestingly, iodoperfluoroalkylation of unactivated olefins and alkynes can also be accomplished by photo-induced-ATRA of perfluoroalkyl iodide using simple organic compounds, such as enamines and amines as catalysts. In a recent publication, Yajima and coworkers explored the photo-assisted-ATRA reaction of alkenes and alkynes with perfluoroalkyl iodides catalyzed by an in situ generated enamine species and free amine (
Scheme 50) [
76]. In this regard, the authors found that the combination of diphenylacetaldehyde with pyrrolidine (10 mol%) efficiently catalyzed the iodoperfluoroalkylation reaction under visible light irradiation using a 23 W fluorescent lamp. Different light sources were also investigated, and the use of a green LED decreased the yield to 89%; whereas blue or white LEDs led to excellent yields and no reaction took place in the dark. Under the optimized conditions, the scope of a variety of perfluoroalkyl iodides and olefins/alkynes was explored and a high functional group tolerance was achieved. Interestingly, reaction with preformed enamine in the absence of a free amine did not proceed efficiently but the addition of a free amine improved the yield suggesting the role of the free amine in catalyzing the reaction.
The authors proposed a mechanism via the formation of an Electron Donor-Acceptor (EDA) complex between the perfluoroalkyl iodide and the in situ formed enamine from pyrrolidine and the aldehyde, as confirmed by optical absorption spectra. This EDA complex
59 generates a fluoroalkyl radical upon irradiation with light, which is added onto the alkene/alkyne to deliver a radical intermediate and an amine radical cation
60. The iodoperfluoroalkylated product is formed by partial iodine transfer from another perfluoroalkyl iodide. Simultaneously, the iodide ion reacts with the enamine radical cation to produce the iodine radical, and the enamine is regenerated. The iodine radical forms a complex with the free amine and this iodide–amine complex can effectively deliver the iodine radical to radical intermediate (
Scheme 51).
Hu and coworkers reported an Ag-mediated vicinal trifluoromethylation−iodination protocol of arynes to afford
o-trifluoromethyl iodoarenes
65 (
Scheme 52) [
77]. In this approach, trifluoromethylsilver (AgCF
3) was prepared in situ by mixing TMSCF
3 and AgF in 1:1 ratio and 1-iodophenylacetylene (
64) was added as electrophile of benzyne to promote the iodination of benzyne. The optimized reaction conditions also need an external additive/ligand such as the hindered secondary amine 2,2,6,6-tetramethylpiperidine (TMP), to accelerate the iodination process and favor the formation of the iodinated product over the protonated one. The scope of the reaction was evaluated through a variety of structurally diverse aryne precursors bearing functional groups such as acetal, bromo, and allyl. Furthermore, high regioselectivity was observed in the reaction with 3-substituted benzynes as a single isomer was formed; however, 4-substituted benzynes gave almost 1:1 ratio of regioisomers.
The in situ-generated aryne
67 reacts with the formed AgCF
3·TMP via a carboargentation reaction to give intermediate
68, which can afford intermediate
69 via a hydrogen bond formation with another TMP, which after deprotonation generates anionic intermediate
70. Then, iodophenylacetylene can coordinate to intermediate
80 via halogen bonding forming intermediate
81, which undergoes nucleophilic attack to form intermediate
72, which readily affords the desired substituted
o-iodotrifluoromethylbenzene and silver phenylacetylide as a byproduct (
Scheme 53).
A copper-mediated halotrifluoromethylation of alkenes was reported by Jung, Han, and coworkers in 2015 using Umemoto reagent and a copper(I) halide salt, which would form a transition metal halide that generates the active CF
3 species and at the same time act as the halogen source (
Scheme 54) [
78]. The main advantage of this approach is the high stability, commercial availability, and relatively low cost of copper halides. With this approach, bromotrifluoromethylation, chlorotrifluoromethylation, and iodotrifluoromethylation of terminal alkenes was achieved with moderate to high yields (60–99%) simply by selecting the copper(I) halide, CuBr, CuCl, and CuI, respectively. The use of bis(pinacolato)diboron (B
2pin
2) as an additive was found to be very efficient in terms of increasing the yield of the β-halotrifluoromethylated product. It is noteworthy that iodotrifluoromethylation using CuI and Umemoto reagent stands as an alternative to the problematic use of gaseous CF
3I. Finally, 1,1-disubstituted and internal alkenes were also converted into the corresponding halotrifluoromethylated products in high yields, tolerating the presence of other functional groups such as α,β-unsaturated aldehydes and ketones.
The authors proposed the mechanistic pathway displayed in
Scheme 55. Initially, the copper halide activates the CF
3 source upon reaction with Umemoto reagent by oxidation of Cu(I) to the Cu(II) species (
73) and generating the CF
3 radical. This CF
3 radical reacts with the alkene to give an alkyl radical, which is recombined with the previously generated Cu(II) species to afford the Cu(III) species (
74). Finally, reductive elimination delivers the desired halotrifluoromethylated product (
Scheme 55).
Hu and coworkers reported an iron-catalyzed 1,2-addition of perfluoroalkyl iodides (R
FI) to alkynes with a wide substrate scope (up to 34 examples) and high functional-group tolerance (
Scheme 56) [
79]. This iron-based methodology is based on the use of FeBr
2 at low loadings (5 mol%) and Cs
2CO
3 (0.8 equiv) as base, despite other transition metal catalysts such as CuI, CoBr
2, Cu(OTf)
2, or NiCl
2·dme, were also effective but in lower yields. The role of the base and running the reaction under a nitrogen atmosphere were found to be critical, the reaction did not occur either without a base or under air. The use of Et
3N as an alternative base gave modest yields but no conversion was observed when using other bases such as
tBuOLi,
tBuOK, and pyridine. Interestingly, this iodoperfluorination reaction also occurred with only Cs
2CO
3 as the base and without the iron catalyst, but reaction conditions required a high concentration of reagents and lowering the base loading to 0.25 equivalents. Under the optimized conditions, alkyne (0.5 mmol), R
FI (1.5 equiv), FeBr
2 (10 mol%), Cs
2CO
3 (0.8 mmol), in 1,4-dioxane at 60 °C, a great number of substrates with diverse functional groups were efficiently iodoperfluorinated using C
4F
9I, C
6F
13I, but also C
6F
12ClI and EtOOCCF
2I. The addition was found to be
E selective, with an
E/Z ratio higher than 5:1, and completely stereoselective with aryl alkynes, which yielded only
E isomers. The authors demonstrated that the reaction mechanism is consistent with the formation of a perfluoroalkyl radical and an iodide radical by reaction of the perfluororoalkyl iodide with the base and its subsequent addition to the unsaturated C-C, which is activated by the iron catalyst to form an alkenyl radical. This radical reacts with the previously generated iodide radical to deliver the iodoperfluorinated product. The iron-catalyzed iodoperfluorination was also expanded to both terminal and internal alkenes with a wide tolerance of functionalities (ester, keto, aryl-halide, and ether groups), which delivered the iodoperfluorinated product in moderate to high yields (64–95%) and in the case of internal olefins, stereoisomers were obtained in a 1:1 diastereomeric ratio.
In addition, Cu-catalyzed halotrifluoromethylation reactions of substrates with allylic C-H bonds using Togni reagent were reported by Szabó and co-workers [
80]. In this work, iodotrifluoromethylation with a stoichiometric amount of CuI was accomplished, affording allylic trifluoromethylated
76 and addition products
77 in moderate yields (
Scheme 57). Szabó’s group also reported a copper-mediated cyanotrifluoromethylation of styrenes using the Togni reagent as the CF
3 source in the presence of catalytic amounts of PCy
3 [
81]. However, iodotrifluoromethylation was afforded when using CuI instead of the CuCN employed for the cyanotrifluoromethylation reaction.
Interestingly, a product control in alkene trifluoromethylation using Togni reagent was reported by Sodeoka and coworkers [
82]. In this work, the hydrotrifluoromethylation, vinylic trifluoromethylation, or iodotrifluoromethylation product could be selected by tuning the reaction conditions (
Scheme 58). On one hand, the desired hydrotrifluoromethylation product
78 was achieved by using Togni reagent in the absence of any transition metal catalyst using K
2CO
3 as a base and DMF as solvent. Under these conditions, hydrotrifluoromethylation of several alkenes was accomplished in the presence of various functional groups, such as carbonyl groups, ester, and aryl halides, affording the corresponding products in good to moderate yields and high selectivity. Studies with deuterated solvent allowed to establish that the hydrogen source of the hydrotrifluoromethylation was the solvent, i.e., DMF. On the other hand, the vinylic trifluoromethylation product was formed with high selectivity using Togni reagent and tetra-
n-butylammonium iodide (TBAI) in 1,4-dioxane with the
E/Z ratio of the product up to 8:1. As also observed in the hydrotrifluoromethylation, various functional groups were tolerated in the vinylic trifluoromethylation. However,
E/Z ratio was found to be low (2:1 ratio) in substituted alkenes. Finally, the iodotrifluoromethylation product
55 was obtained in good to high yields under the same reaction conditions for the vinylic trifluoromethylation but simply replacing of TBAI by KI. Under the optimized conditions, iodotrifluoromethylation also tolerated several functional groups; however, internal alkenes were unsuitable for the iodotrifluoromethylation due to generate of regio- and stereoisomers.
Iodotrifluoromethylation of alkenes and alkynes can also be achieved via single electron oxidation of a CF
3-derived anion by an inorganic hypervalent iodine oxidant. In a work by Liu and coworkers, oxidation of sodium trifluoromethanesulfinate (NaSO
2CF
3) generated the CF
3 radical, which added to the unsaturated bonds, followed by the capture of the reducing substance iodine to form β-CF
3 iodides (
Scheme 59) [
59]. Among several inorganic iodine sources, iodine pentoxide (I
2O
5) was found to be more efficient than other organic hypervalent iodine compounds such iodic acid (HIO
3). Regarding the solvent, a 1:1 mixture of CH
2Cl
2/H
2O was shown to be the best choice. Under the optimized conditions, iodotrifluoromethylation of a wide variety of alkenes and alkynes was carried out by using stable and inexpensive solid reagents CF
3SO
2Na and I
2O
5 in aqueous medium, instead of using the typical and hard to handle CF
3I gas. A wide tolerance of functional groups (halogens, amide, ether, carbonyl, carboxylate, hydroxyl, nitro, sulfonate, and sulfamide) was also observed. In this regard, aryl- and alkyl-substituted alkenes afforded β-CF
3 alkyl iodides in moderate to high yields (40 examples, up to 90% yield). Terminal and nonterminal alkenes show also good compatibility, but when both double bonds are present in one substrate, the iodotrifluoromethylation selectively occurred at the terminal position. Electron spin resonance (ESR) studies allowed confirming that the reaction occurred via a free-radical process through characterization of two key intermediates such as CF
3 and the β-CF
3 alkyl radicals, which were trapped using 2-methyl-2-nitrosopropane (MNP) as a radical spin trap. Finally, the iodotrifluoromethylation of alkynes was also accomplished by this methodology affording €-β-CF
3 alkenyl iodides
52 with
E/
Z ratios up to 4:1 but with the assistance of NaHCO
3 and a mixture of 4:1 of DCE/H
2O as solvent.
The groups of Yang, Kim, and Cho reported in 2014 an interesting and environmentally benign methodology for the hydrotrifluoromethylation and iodotrifluoromethylation of unactivated alkenes and alkynes with CF
3I in the presence of inorganic electrides (ionic salts with electrons as the anions) [
83], such as polycrystalline [Ca
2N]
+∙e
−, which were used as an electron source through a SET pathway [
84]. The authors indicated that the solvent plays a crucial role in the method. After checking different solvents, they found that alcoholic solvents such as ethanol and acetonitrile (1:1) were the mixture of choice to carry out the reaction successfully. Initially, they studied the behavior of alkenes under the optimized reaction conditions and found that the process worked well but only with aromatic derivatives providing exclusively the hydrotrifluoromethylation products (
Scheme 60A). With aliphatic alkenes, an inseparable mixture of iodotrifluoromethylated
52 and hydrotrifluoromethylated compounds
78 was obtained. However, the situation for the alkynes was different. In this case, the authors observed that under the optimized conditions, the process worked also well, for both aromatic and aliphatic alkynes, providing only the hydrotrifluoromethylation product when using 3.0 equiv of the electride [Ca
2N]
+∙e
−. On the other hand, by controlling the electride amount, i.e., using only 1.5 equiv of [Ca
2N]
+∙e
−, the reaction led to the selective formation of the iodotrifluoromethylation product with good yields and excellent E/Z selectivity (
Scheme 60B).
The authors proposed a tentative mechanism starting with a single electron transfer from [Ca
2N]
+∙e
− to CF
3I to generate first a CF
3 radical and iodide anion, which after addition to the alkene or alkyne provided a new alkyl or vinyl radical
62. Final iodine abstraction from CF
3I generated the target iodotrifluoromethylated product
52 and a CF
3 radical. The hydrotrifluoromethylated products
39 were formed after a second SET from [Ca
2N]
+∙e
− to the radical intermediate
62, followed by protonation with ethanol, which was used as an electride activator and hydrogen donor (
Scheme 61).
It should be mentioned that there were also some related reports on the halo-perfluoroalkylation of unsaturated bonds which just contained one example about trifluoromethylation [
84,
85,
86,
87,
88,
89,
90,
91,
92,
93,
94,
95].
5. Cyano-trifluoromethylation
The cyano group belongs to an important carbon-containing unit for synthesis because this group can be easily further converted into other functional moieties. Due to its synthetic versatility, cyanotrifluoromethylation of alkenes also represents high-valued reaction and has attracted much attention over the past years.
In 2013, Szabó and coworkers reported a Cu/phosphine-promoted cyanotrifluoromethylation of aryl alkenes with Togni II reagent as trifluoromethyl source (
Scheme 67) [
81]. They successfully employed CuCN as the cyano source, which smoothly reacted with a diverse set of aryl alkenes to give the corresponding difunctionalized product
92. The reaction was carried out at room temperature and moderate to good chemical yields (50–87%) were obtained after 18 h. It should be mentioned that deuterated chloroform was used as the solvent for this reaction, as the authors investigated the crude reaction mixture with
1H and
19F NMR spectroscopy.
As shown in
Scheme 68, a radical mechanism was proposed for this Cu-catalyzed cyanotrifluoromethylation reaction. Initially, CuCN is oxidized by Togni II in the presence of phosphine ligand to give the Cu(III) complex
93, which undergoes the homolytic cleavage to generate the CF
3 radical and followed by addition to C=C double bond. Thus, the radical intermediate
3 is obtained as well as the formation of Cu(II) complex
94. Subsequently, the radical intermediate
3 oxidizes the Cu(II) complex
94 to form the Cu(III) complex
95. Finally, reductive elimination of the Cu(III) complex
95 affords the desired cyanotrifluoromethylated product
92 as well as the Cu(I) complex for the next catalytic cycle.
In addition to the use of CuCN as the cyano source, TMSCN has also been developed as the cyano source for the cyanotrifluoromethylation of alkenes. In 2014, Liang and co-workers reported an efficient cyanotrifluoromethylation reaction of alkenes using Togni II and TMSCN as trifluoromethyl and cyano source respectively under ligand-free and additive-free conditions (
Scheme 69) [
102]. Comparing with the work from the Szabó group, this difunctionalization reaction was carried out with a catalytic amount of Cu(OTf)
2 and no phosphine ligand was needed for the conversion. Furthermore, this reaction features a good functional group tolerance and mild conditions, affording a variety of useful CF
3-containing nitriles in 38–90% yield. Styrene worked well in this reaction and generated the desired product
92 in 83% yield. In addition, an alkyl alkene, such as cyclohexene, was a suitable substrate for this system with moderate yield and good diastereoselectivity (10:1 dr). Additionally, the authors also performed further synthetic transformation of the obtained CF
3-containing nitriles, and the corresponding CF
3-containing amine and carboxylic acid were obtained in good yields.
A possible mechanism for this Cu-catalyzed reaction with TMSCN as cyano source was postulated by the authors as shown in
Scheme 70, which is similar with the previous report. First, Togni reagent oxides Cu(II) catalyst to generate Cu(III) complex
96, which releases a trifluoromethyl radical. Then, addition of trifluoromethyl radical to C-C double bond gives the benzylic radical intermediate
3. Subsequently, the radical
3 is oxidized by Cu(III) to give cation
4, which then directly couple with TMSCN to form the desired product
92.
Afterward, the same group extended this Cu-catalyzed intermolecular cyanotrifluoromethylation reaction to alkynes (
Scheme 71) [
103]. This Cu(OAc)
2-catalyzed reaction was carried out under similar reaction conditions, and employed TMSCN and Togni reagent II as cyano and trifluoromethyl precursor, respectively. However, addition of 2,2’:6’,2’’-terpyridine ligand was essential for the transformation. Aryl alkynes were suitable substrates and good yields were obtained. On the sharp contrary, this reaction outcome was sensitive to the substitution on the C-C triple bond. A dramatically lower yield (30%) was found when the C-C triple bond features an alkyl group, instead of an aryl group. This reaction also showed excellent stereoselectivity, and the ratio of E/
Z was from 16:1 to >20:1.
In a related process, Liu and co-workers reported an alternative mild cyanotrifluoromethylation reaction of alkenes by use of 10 mol% CuBr as a catalyst with up to 98% yields in 2014 [
104].
The following research interests in the area of cyanotrifluoromethylation of alkenes mainly focused on the development of new trifluoromethyl sources. Xu, Wang, and co-workers developed a cyanofluoroalkylation reaction of alkenes with fluoroalkyl iodides as fluoroalkylation sources (
Scheme 72) [
105]. This strategy used trifluoromethyl iodide as substrate, which could be efficiently converted into trifluoromethyl radical under photoredox conditions. Interestingly, the alkyl substituted alkenes were suitable substrates and the corresponding vicinal trifluoromethyl nitriles were obtained in good yields.
The authors also conducted some control experiments to get insight into the mechanism. They found that no corresponding cyanotrifluoromethylated product was formed when a radical inhibitor TEMPO was added, which indicates the putative radical mechanism. The proposed mechanism was provided in
Scheme 73, which starts from ligand exchange of Cu(II) catalyst to generate Cu(II) complex
98. Then, reduction of Cu(II) complex
98 by DIPEA happens to form the Cu(I) complex
99, which is excited
99* under photo irradiation. Subsequently, reaction of CF
3I with complex
99* affords trifluoromethyl radical
3 as well as Cu(II) complex
98. Addition of trifluoromethyl radical to alkene, followed by reduction by Cu(II) complex
98 generates intermediate
100, which undergoes reductive elimination to give the corresponding difunctionalized product
92 as well as Cu(I) complex
99 for the next catalytic cycle.
Then, sodium trifluoromethanesulfinate was developed as trifluoromethyl radical for the related Cu-catalyzed intermolecular cyanotrifluoromethylation of unactivated alkenes (
Scheme 74) [
106]. The reaction was conducted in a co-solvent (DMSO/water) with Na
2S
2O
8 as an oxidant under microwave conditions. This reaction proceeded smoothly at 90 °C and afforded the corresponding product
92 in good chemical yields. In particular, unactivated alkenes were employed as substrates, which represents an alternative and efficient method for the preparation of β-trifluoromethyl nitriles.
The authors also performed control experiments with addition of TEMPO and BHT, which dramatically suppressed the reaction. Thus, the reaction proceeded through a similar process with trifluoromethyl radical as a key intermediate. The first step is the generation of trifluoromethyl radical via oxidation of sodium trifluoromethanesulfinate by Na2S2O8. Then, the reaction proceeds through the same oxidative addition and reductive elimination steps to form the desired product.
Trifluoromethylsulfonyl−pyridinium salt (
101, TFSP) is an air stable reagent, which can be easily prepared from the reaction of Tf
2O with 4-(dimethylamino)-pyridine. Very recently, Xiao and co-workers employed this TFSP reagent as a trifluoromethyl radical precursor and successfully applied it in the cyanotrifluoromethylation difuctionalization reaction of alkenes (
Scheme 75) [
107]. This reaction was conducted in the presence of blue LEDs with Cu
2O and Ir(ppy)
3 as co-catalyst, affording the corresponding trifluoromethylated nitriles
92 in moderate to good chemical yields. It should be mentioned that only styrene and its derivatives have been used as alkenes substrates, in particular, an estrone derivative also was a suitable substrate for this reaction.
The proposed mechanism for this photo-promoted difunctionalization reaction with TSFP as a radical source is presented in
Scheme 76, which is similar to the previously reported transformations after generation of trifluoromethyl radical. TFSP was reduced by the excited photocatalyst, Ir(ppy)
3* via a single-electron transfer affording the radical intermediate
102. Then, homolysis of intermediate
102 gives CF
3SO
2 radical, which further gives trifluoromethyl radical after release of SO
2. After generation of trifluoromethyl radical, the reaction proceeds through similar steps to form the final product
92.
Asymmetric radical difunctionalization of alkenes remains a great challenge due to the short life of radicals and the extreme difficulty of stereochemical control of reactive radical intermediate. On the other hand, asymmetric radical reactions catalyzed by the combination of Cu salt/chiral ligand have been developed as an efficient strategy for the synthesis of varieties of chiral trifluoromethyl-containing compounds, as those reactions usually provide high stereoselectivity due to the formation of chiral Cu(III) complex intermediate.
The pioneering work on enantioselective radical cyanotrifluoromethylation of alkenes was developed by Liu and coworkers in 2016 (
Scheme 77) [
108]. This asymmetric radical difunctionalization reaction used Cu(CH
3CN)
4PF
6 as catalyst and 3-bis-2-oxazoline derivate ((3a
S,3a’
S,8a
R,8a’
R)-2,2’-(cyclopropane-1,1-diyl)bis(8,8a-dihydro-3aH-indeno[1 ,2-d]oxazole))
103 as chiral ligand, affording trifluoromethylated alkylnitriles
92 in excellent chemical yields and high enantioselectivities. Interestingly, this reaction features a quite broad substrate scope which was generally not sensitive to the substitution pattern. It should be mentioned that the reaction only tolerated aryl substituted alkenes with good reaction outcomes; however, the terminal alkenes with an alkyl substituent could still provide the corresponding product but with poor enantioselectivities.
As indicated by the authors, this Cu-catalyzed reaction was also triggered by a SET to generate CF3 radical, followed by an addition to styrene to generate a benzylic radical intermediate. Then, the CF3 radical was trapped by Cu(II) to form a key chiral Cu(III) intermediate 104, which proceeds through a reductive elimination to give the desired product.
Notably, the Han group also reported independently a similar catalytic enantioselective cyano-trifluoromethylation of styrenes with CuF
2/chiral bidentate bis(oxazoline) ligand as catalyst to give trifluoromethyl nitriles in good yields and excellent enantioselectivities [
109].
Afterward, the same group demonstrated a related asymmetric catalytic system with Cu(OAc) and chiral bis-oxazoline ligand
106 as catalyst in 2020 to access a series of chiral α-cyano amides, α-cyano esters by use of enamides, and vinyl esters
105 as starting alkene substrates (
Scheme 78) [
110]. The key success of this reaction is the use of alkenes adjacent to a nitrogen atom or an oxygen atom, and the reaction proceeds via a key asymmetric coupling step of carbon-centered radical adjacent to heteroatom, affording the corresponding products
107 in good to excellent yields (70–98%) and enantioselectivities up to 98%.
Considering the advantages of the Cu/chiral ligand-catalyzed radical rely on strategy, then, the same group extended this asymmetric system from enamides to alkyl-substituted terminal alkenes featuring carbonyl unit, including ketone, amide, and ester in 2020 [
111]. The reaction showed a wide substrate scope and a variety of substituents was tolerated to afford the corresponding product in excellent yields, however, with slightly lower stereoselectivities.
Due to the advantage of introducing two groups at the same time (namely, cyano and trifluoromethyl), as well as the good functional group tolerance, and mild reaction conditions, the cyanotrifluoromethylation difunctionalization strategy also gained wide recognition in other chemical platforms, such as remote cyanotrifluoromethyl difunctionalization [
112,
113,
114] and construction of functionalized cyclic compounds [
115,
116,
117].



 ,
, 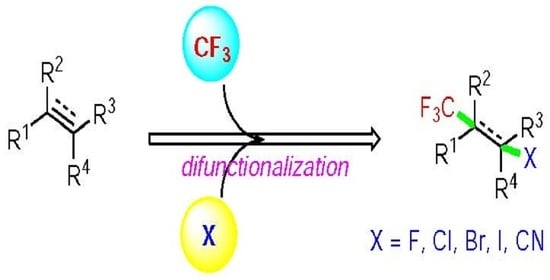
{kind=link}
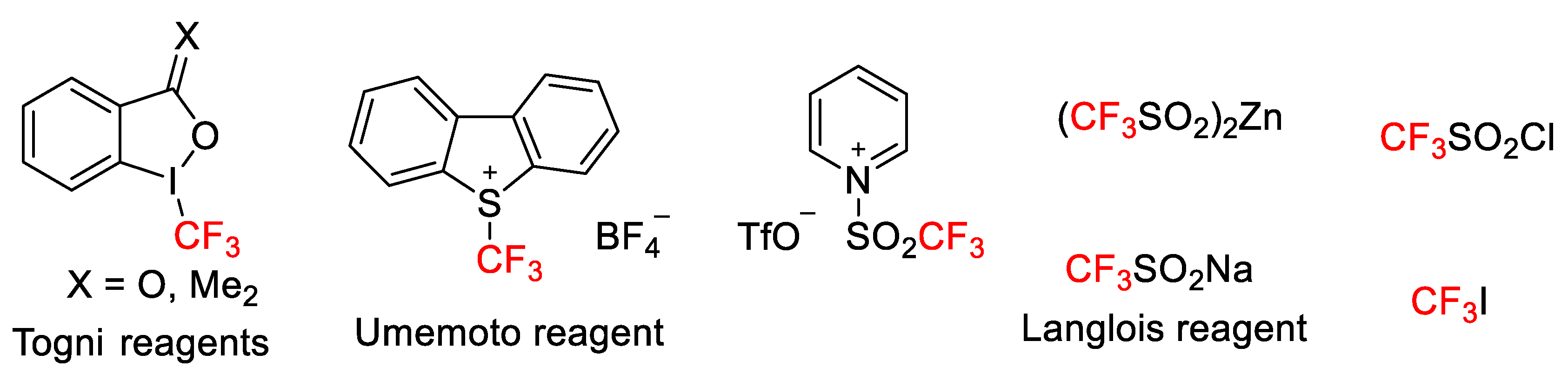{kind=link}
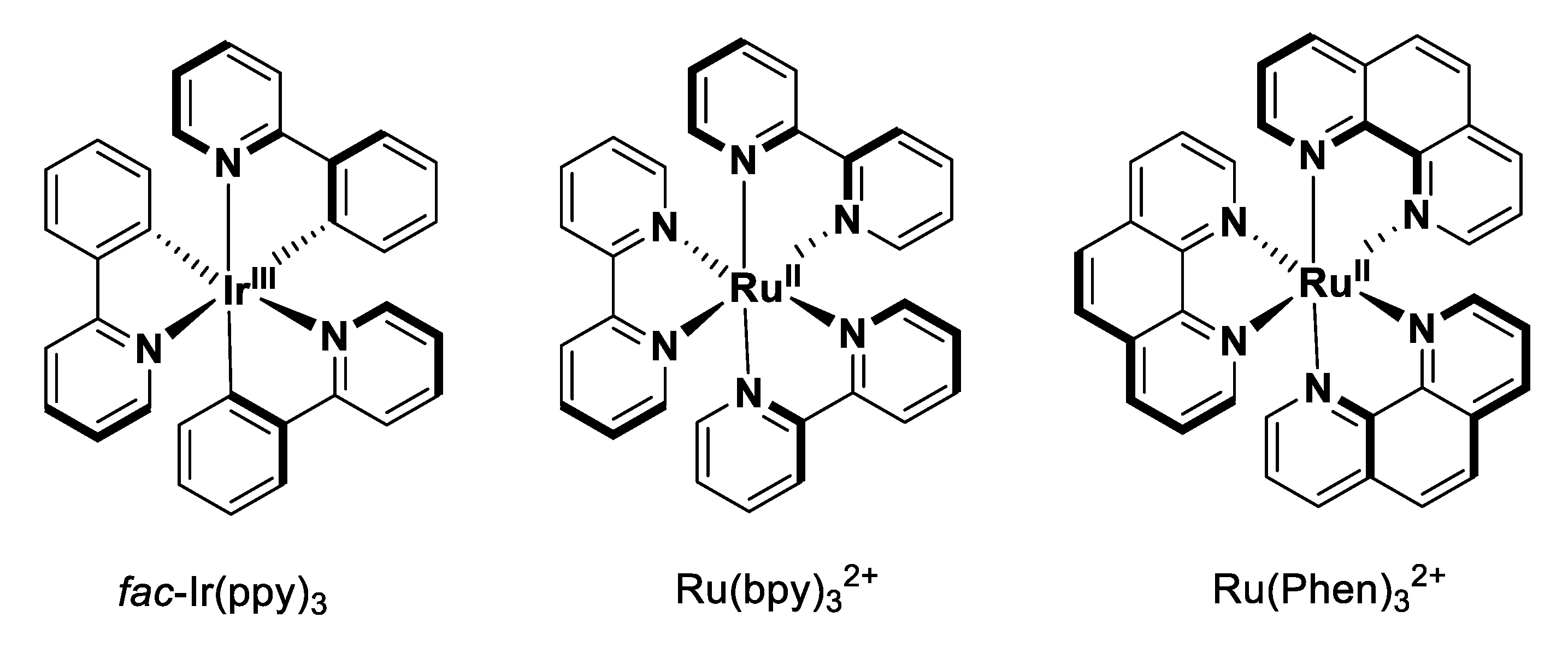{kind=link}
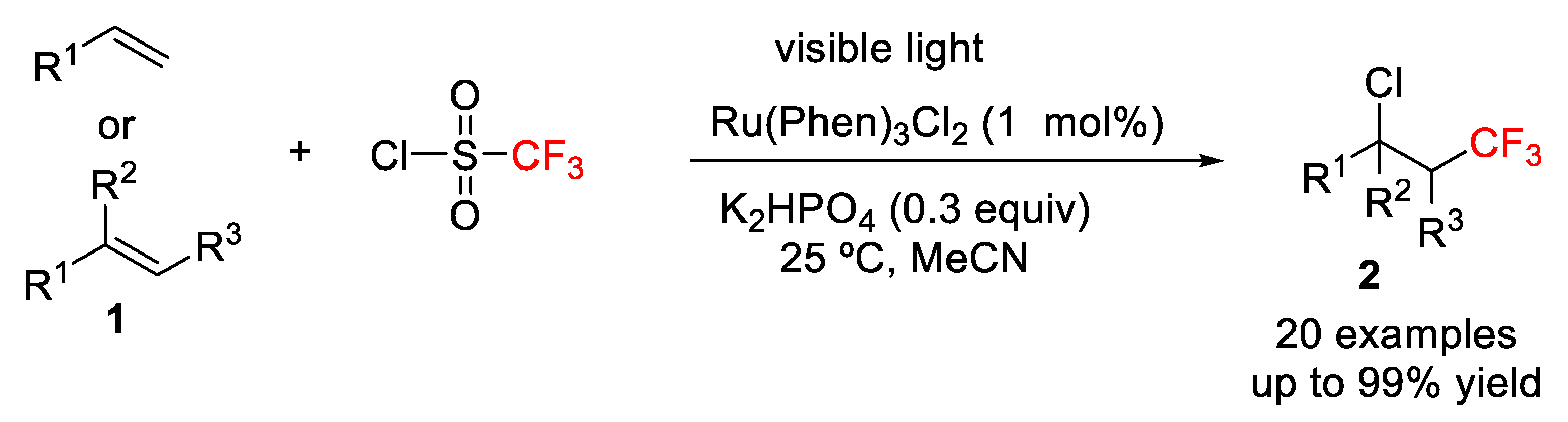{kind=link}
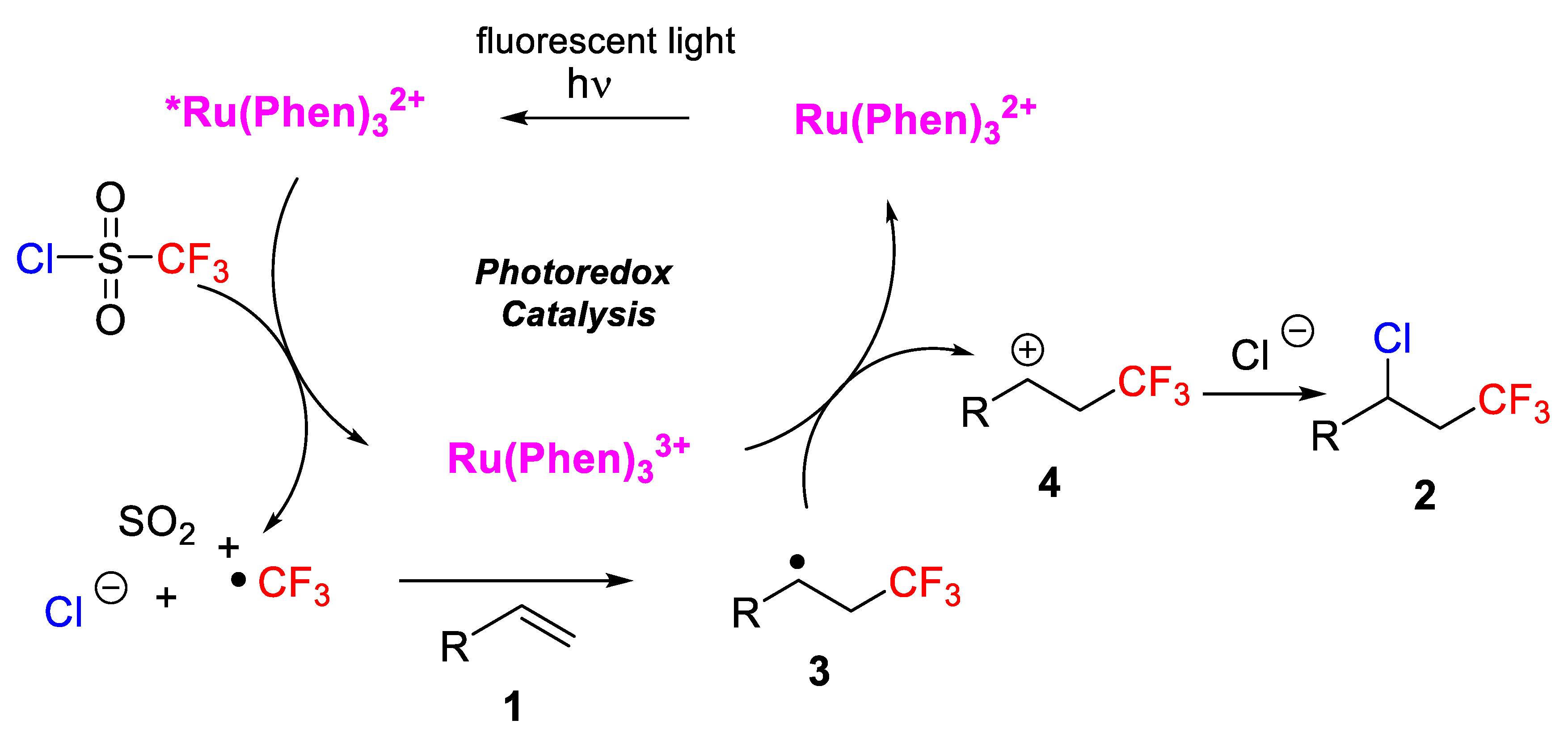{kind=link}
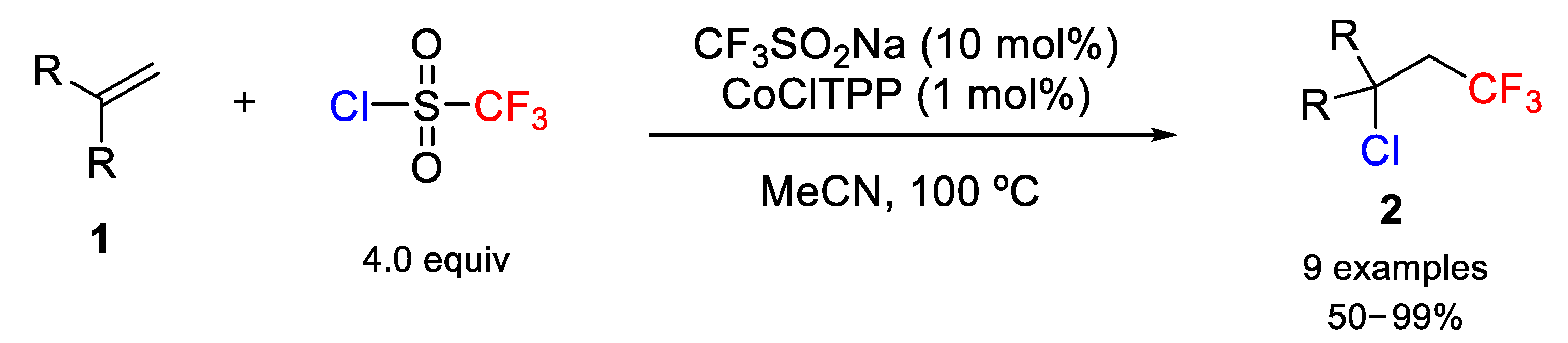{kind=link}
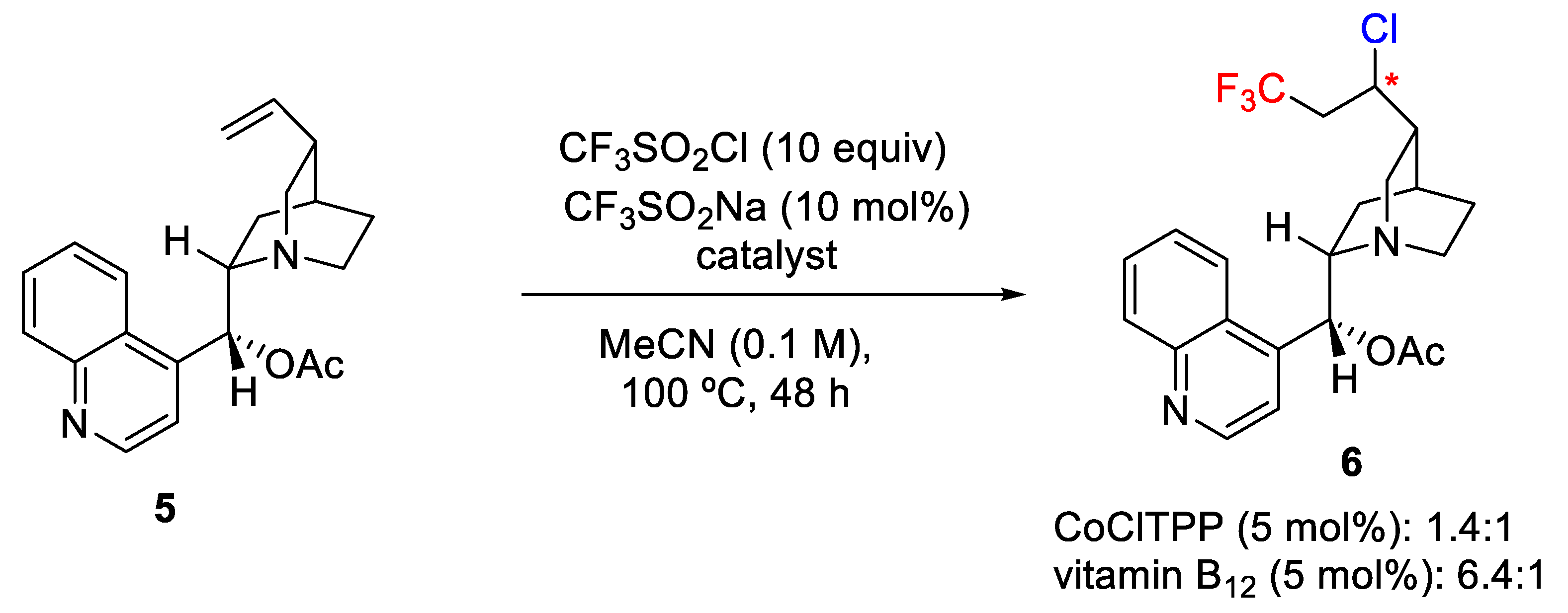{kind=link}
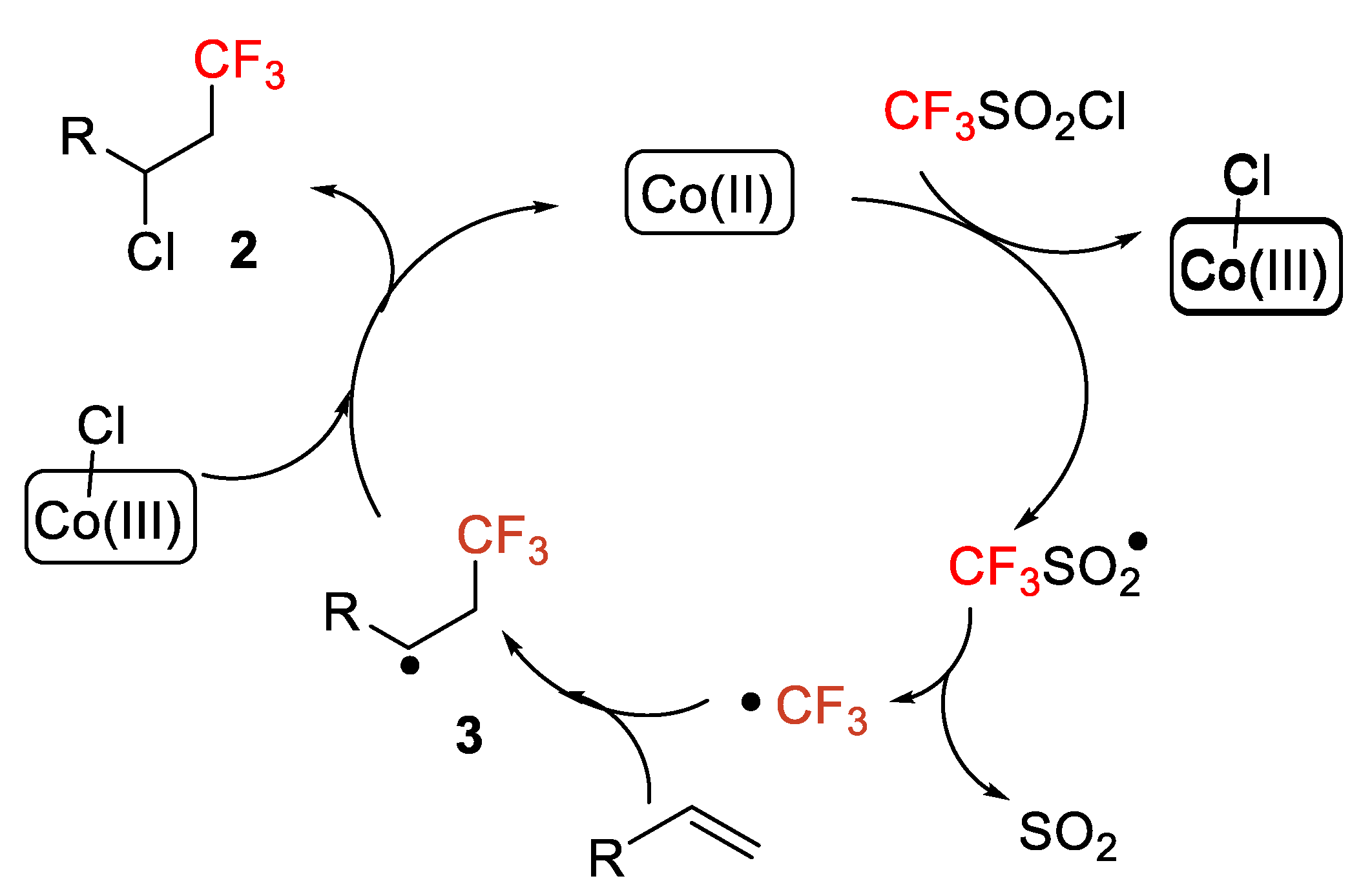{kind=link}
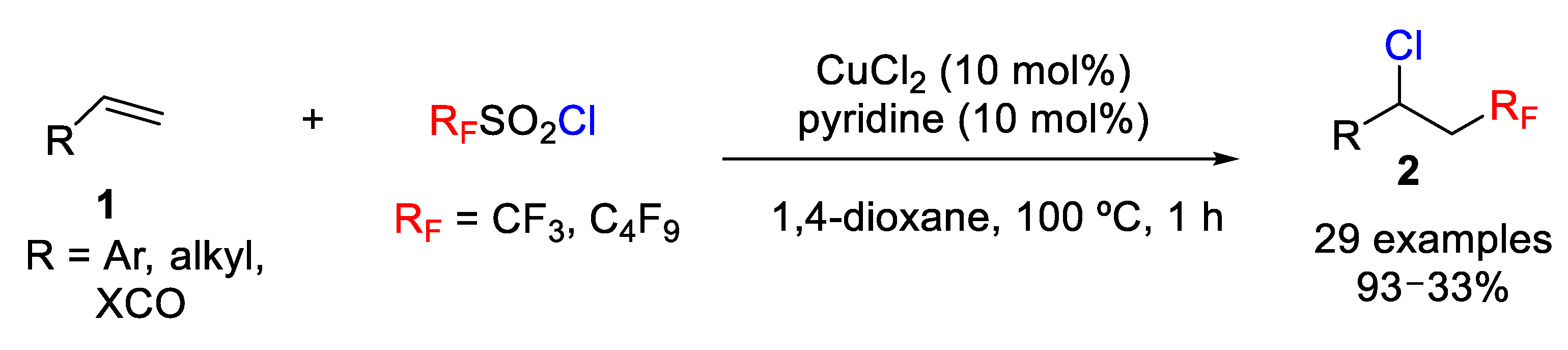{kind=link}
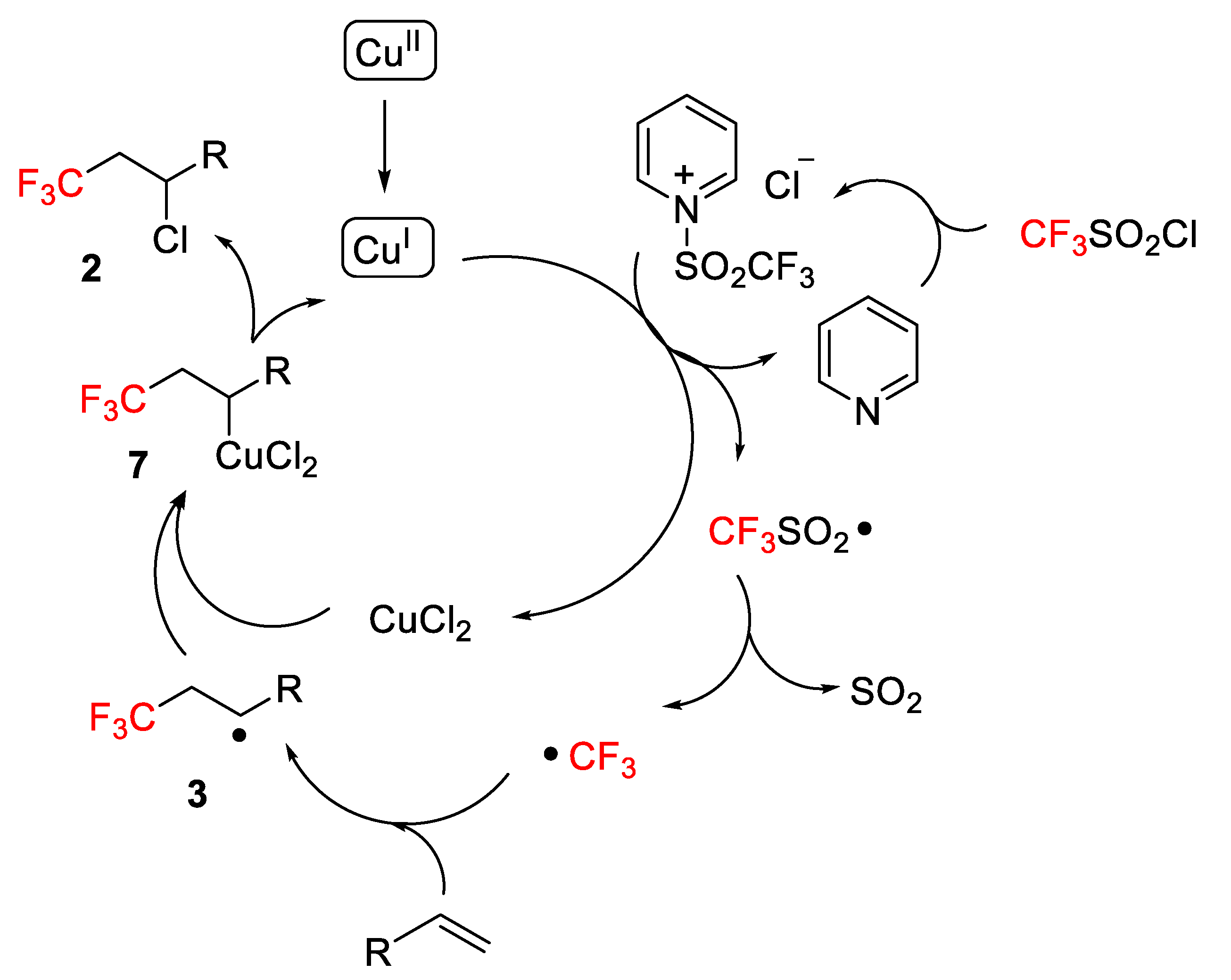{kind=link}
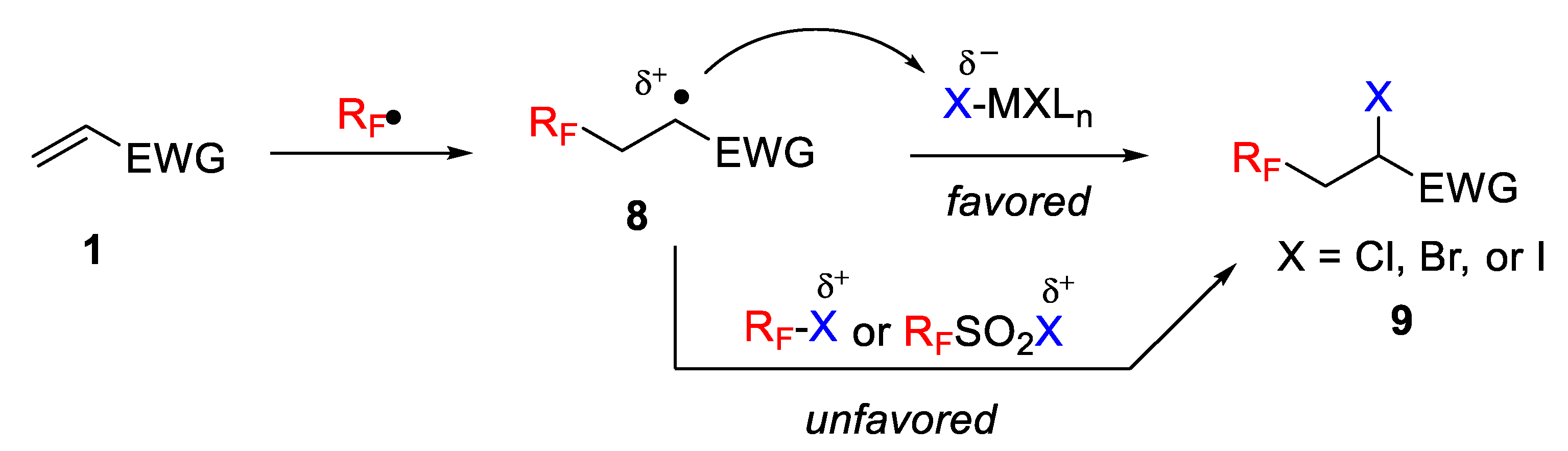{kind=link}
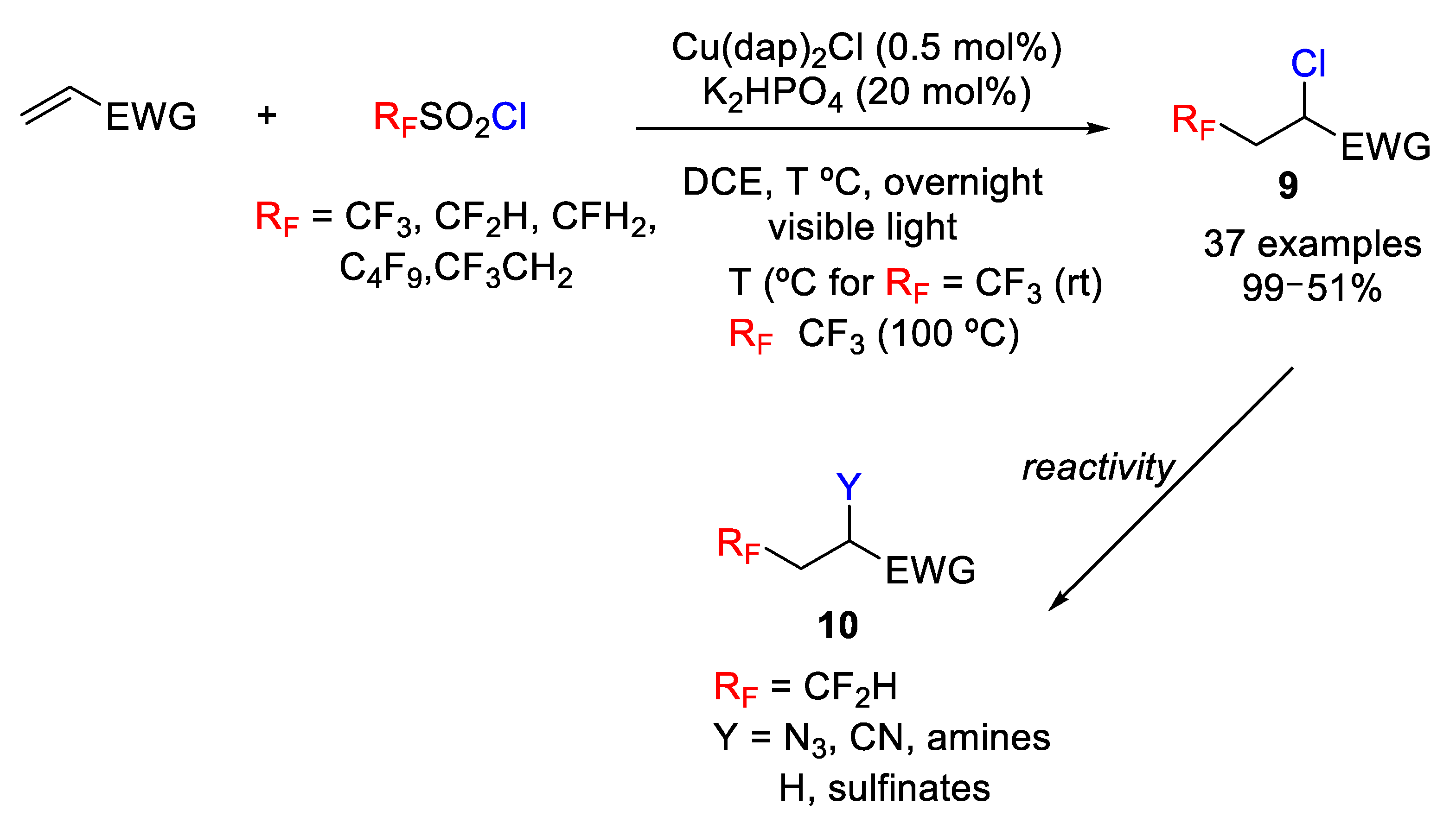{kind=link}
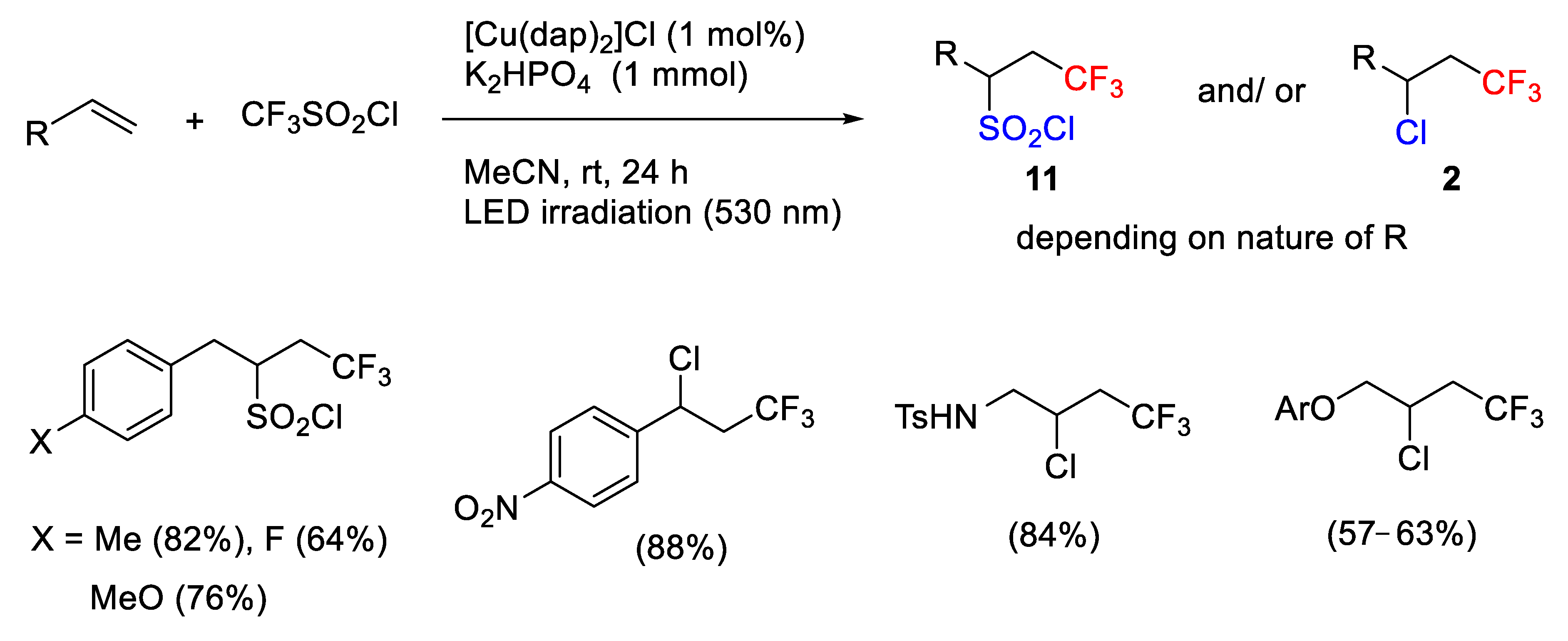{kind=link}
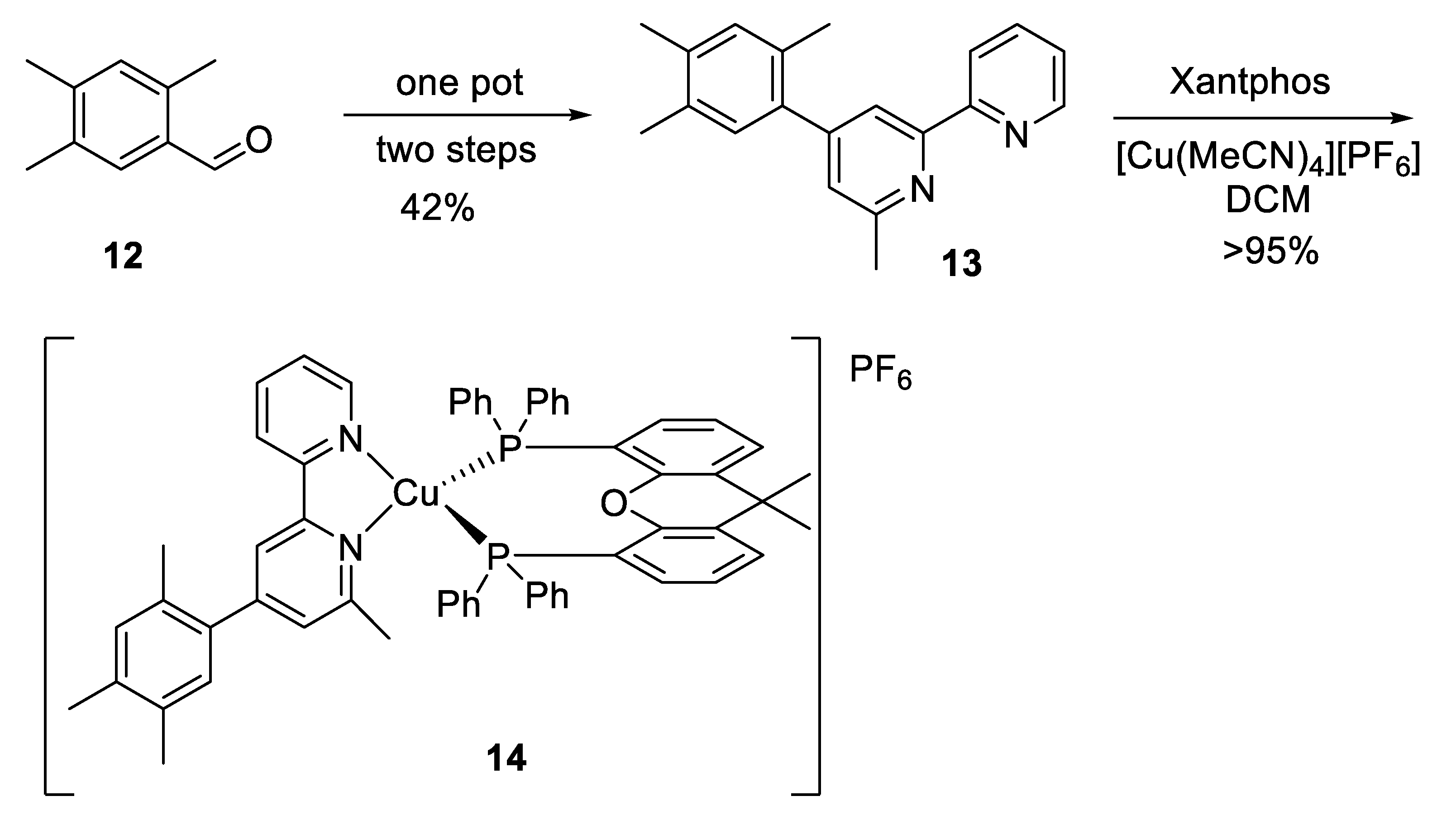{kind=link}
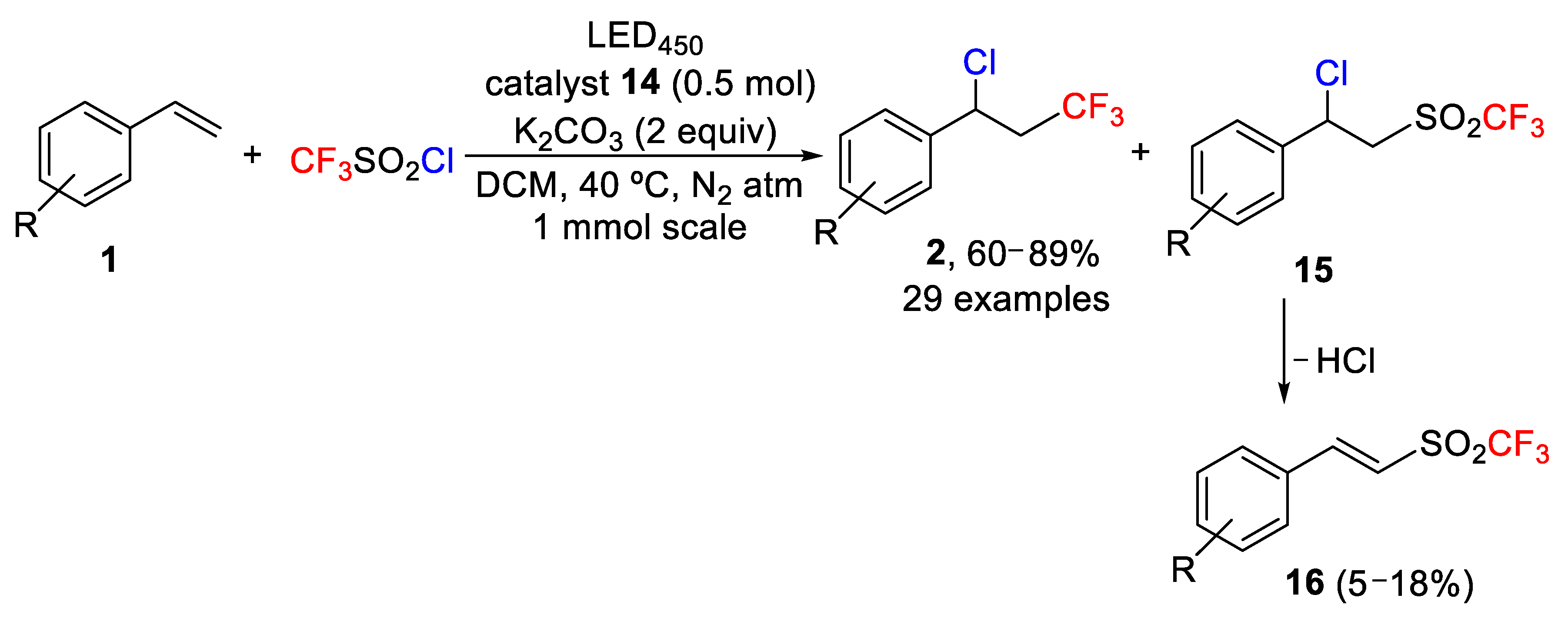{kind=link}
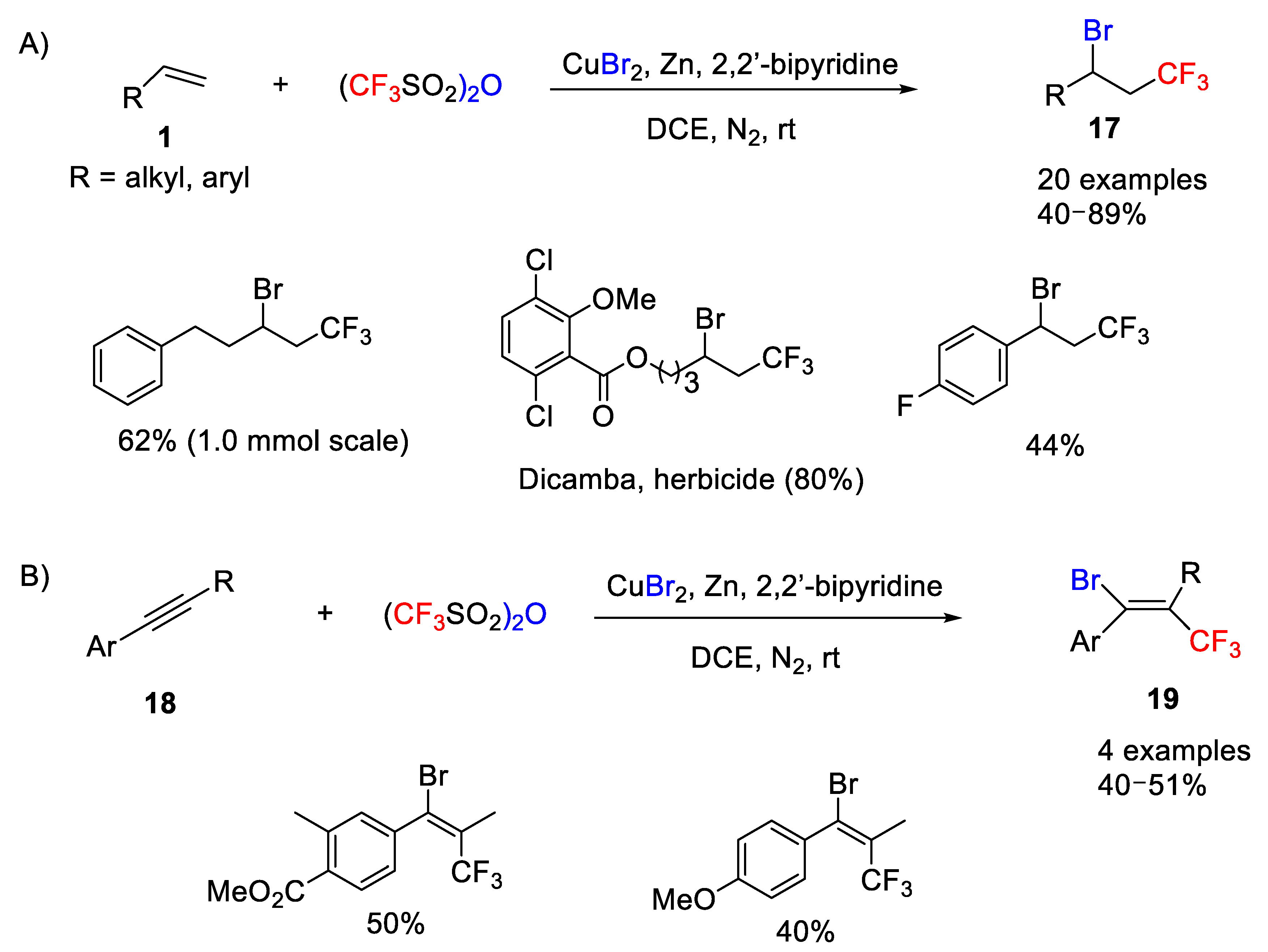{kind=link}
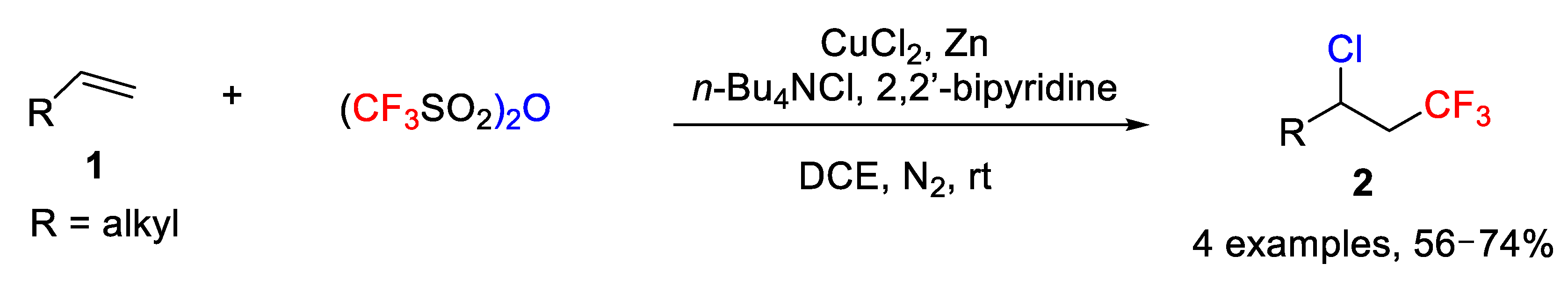{kind=link}
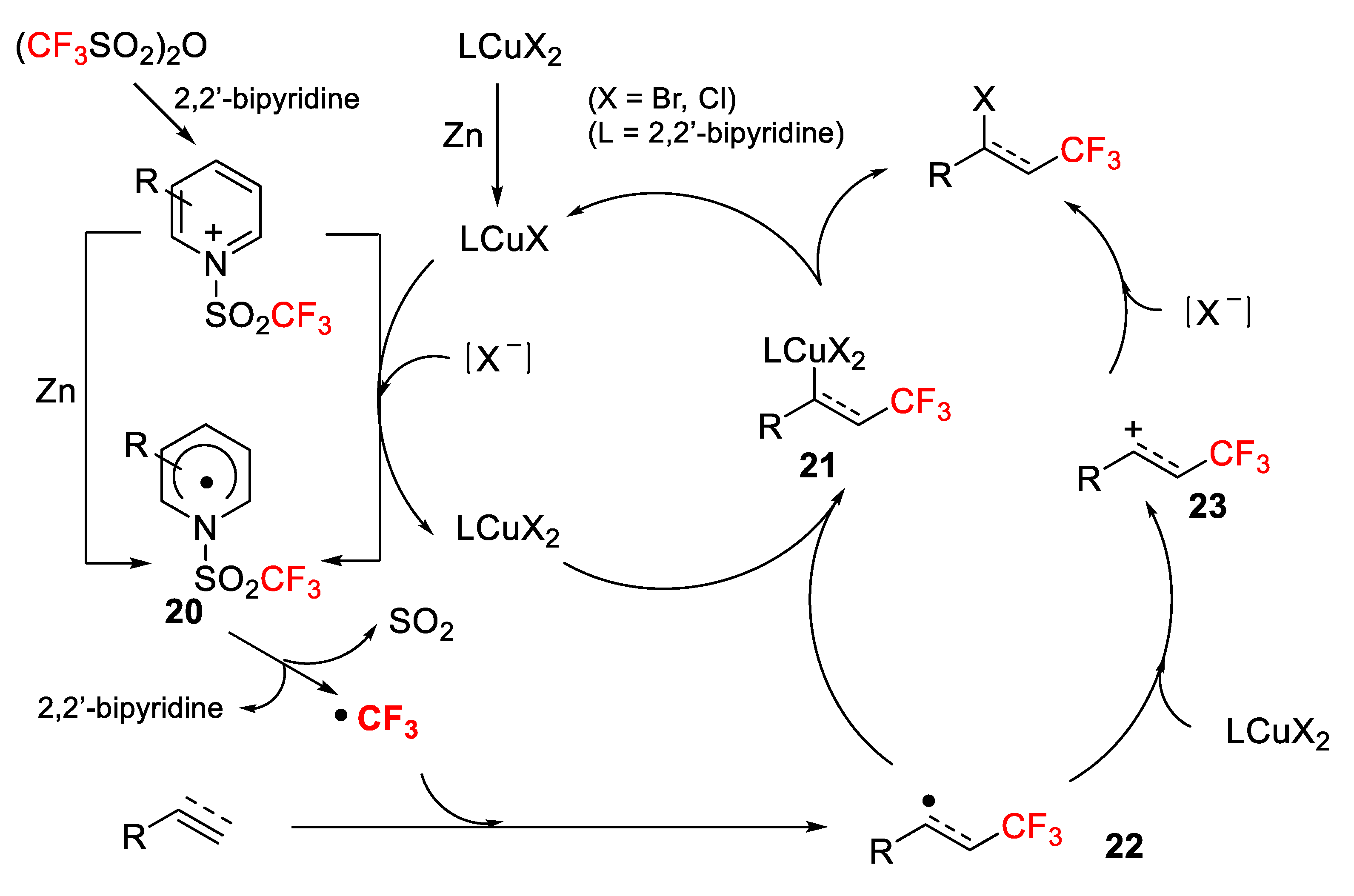{kind=link}
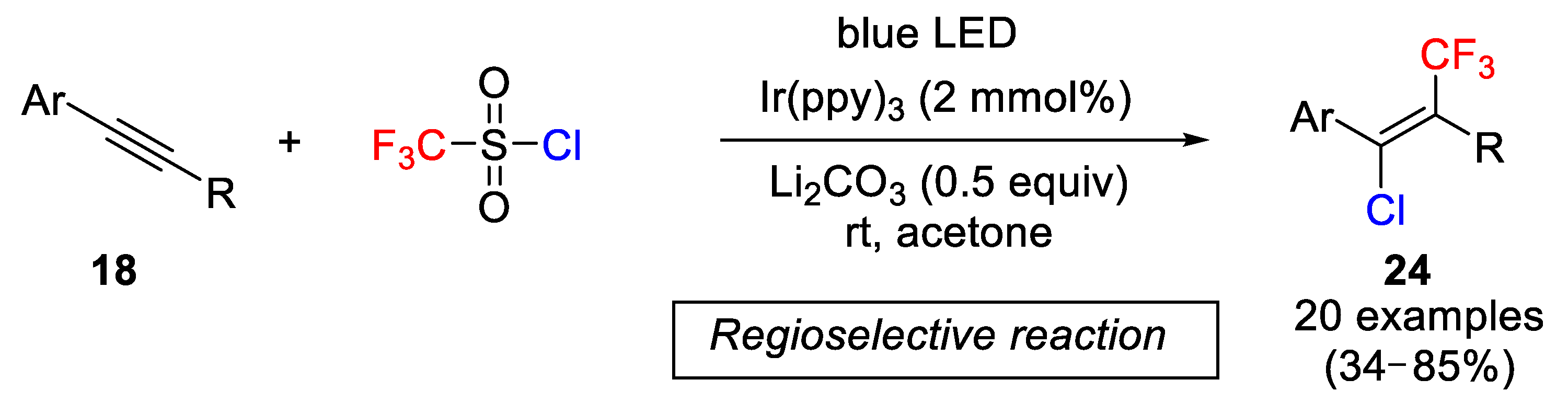{kind=link}
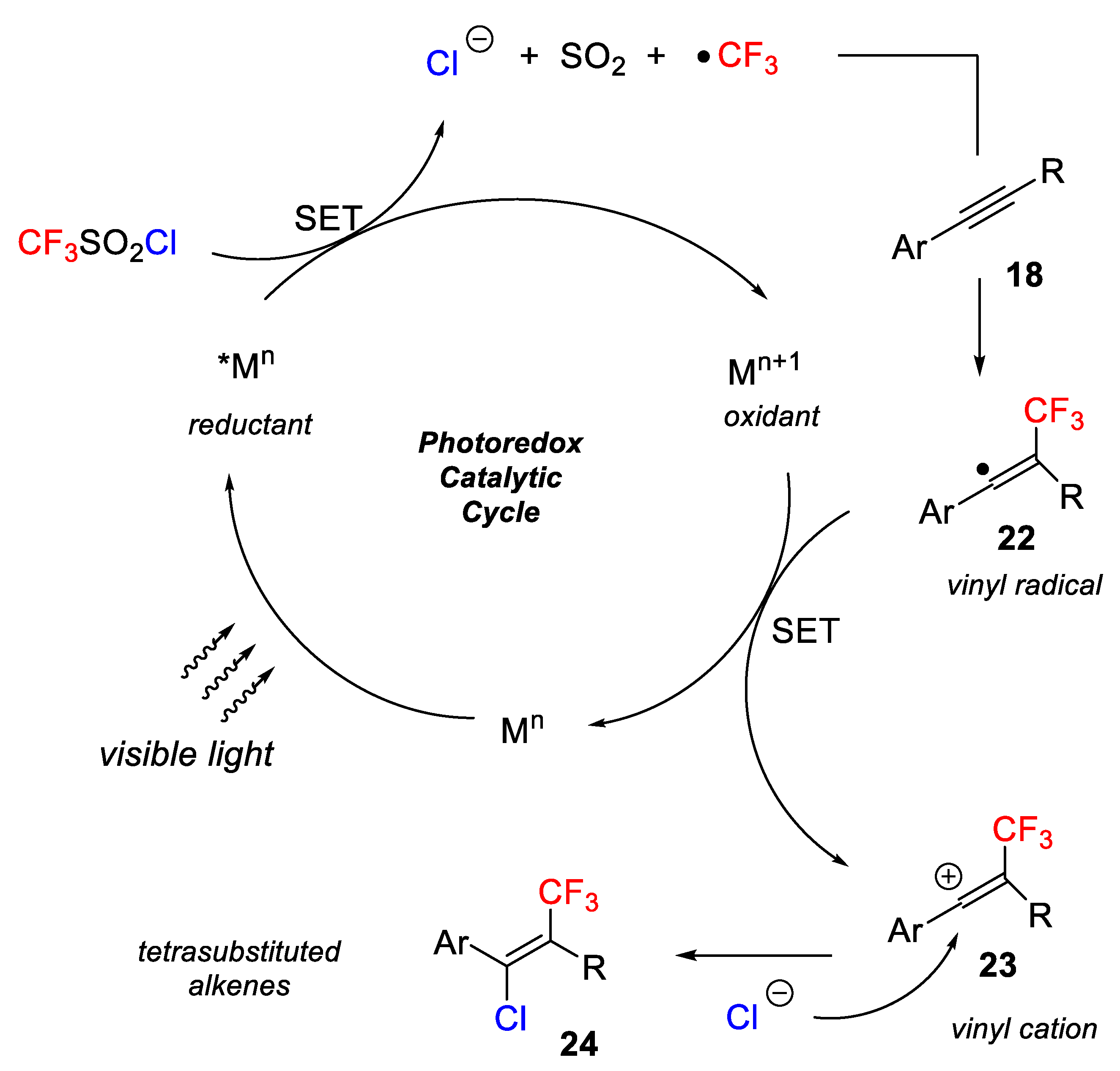{kind=link}
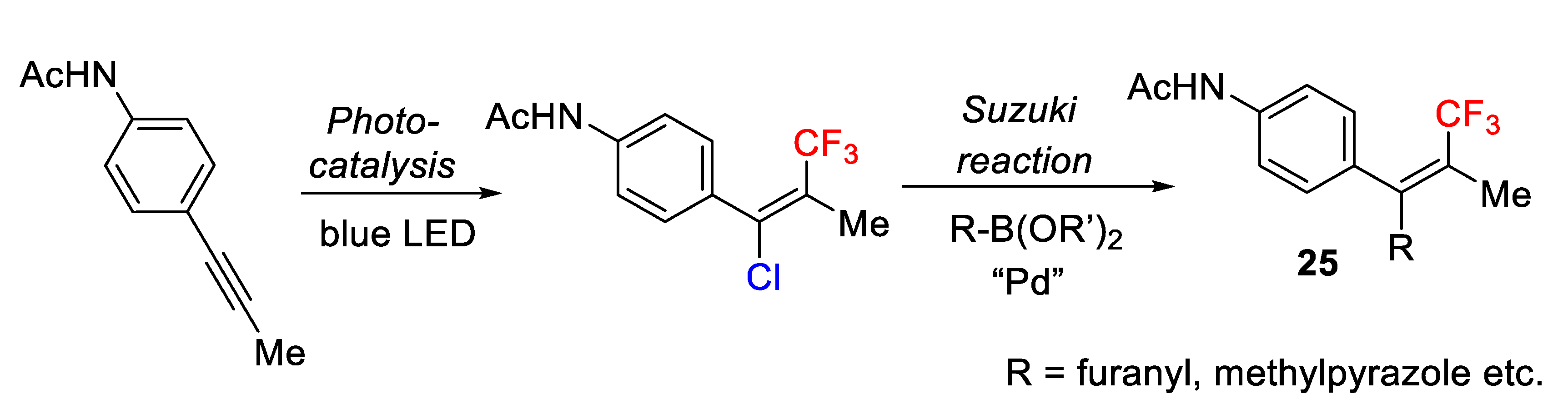{kind=link}
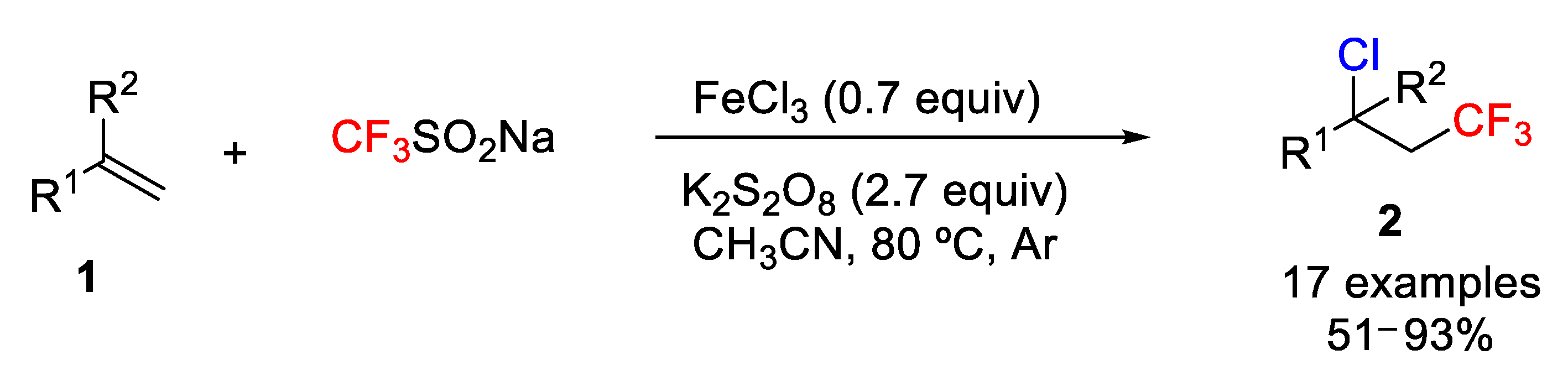{kind=link}
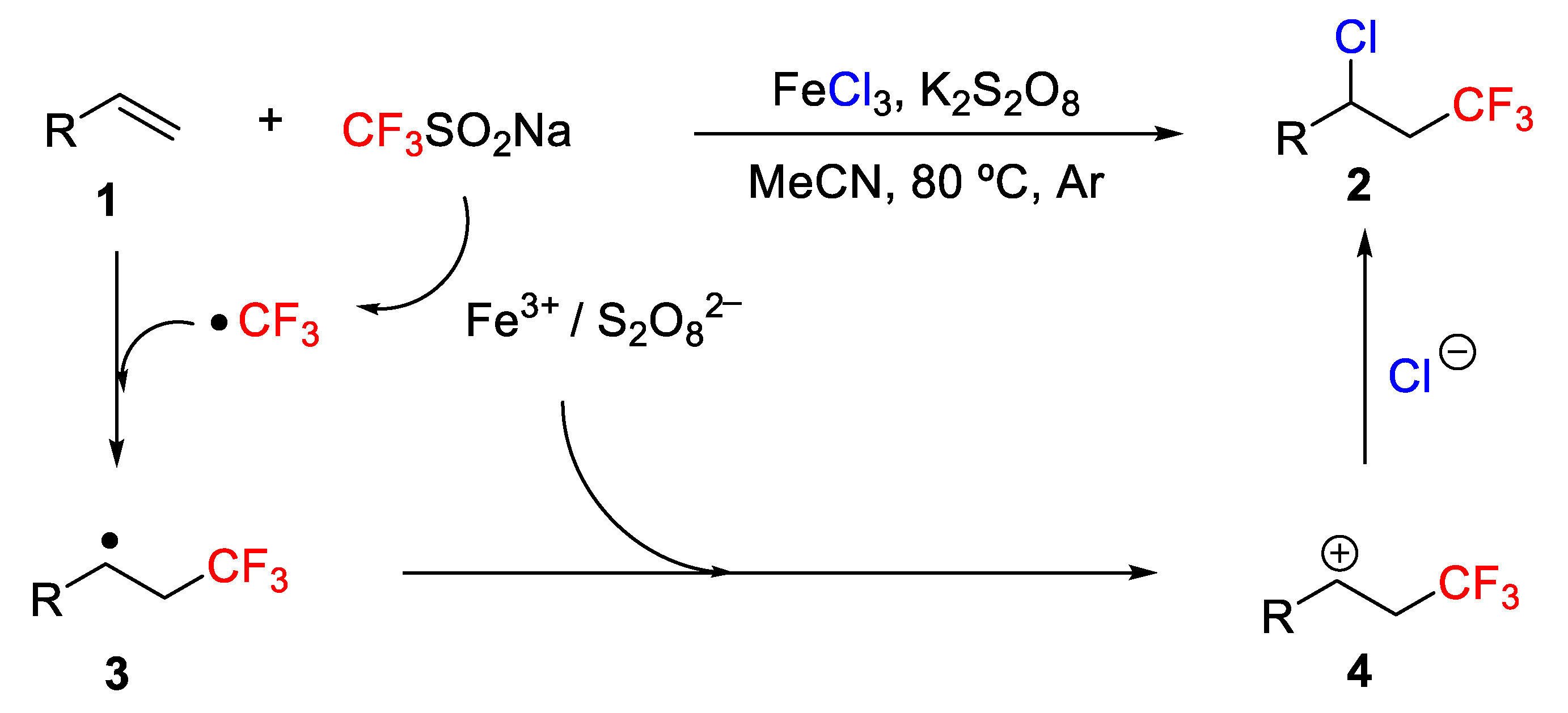{kind=link}
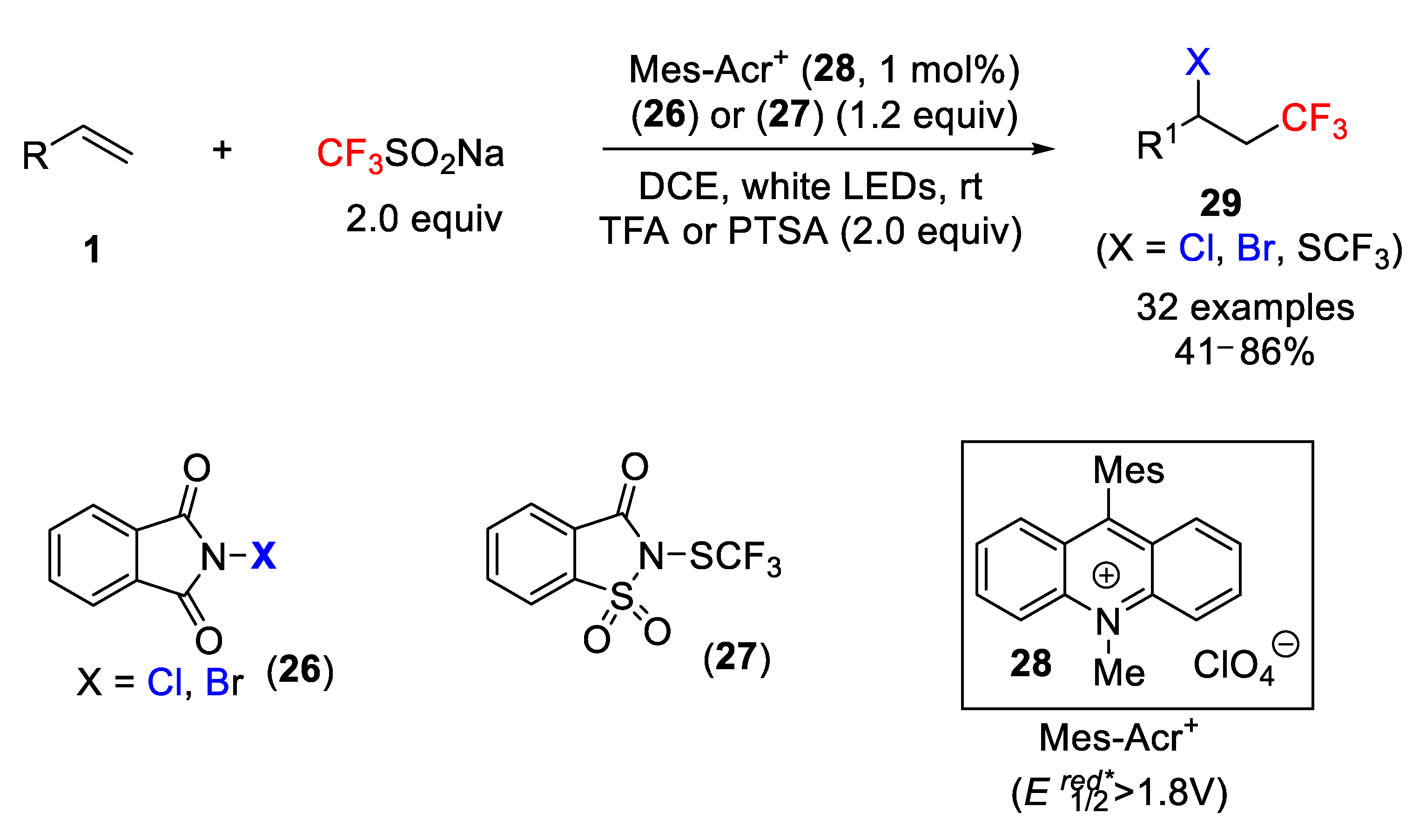{kind=link}
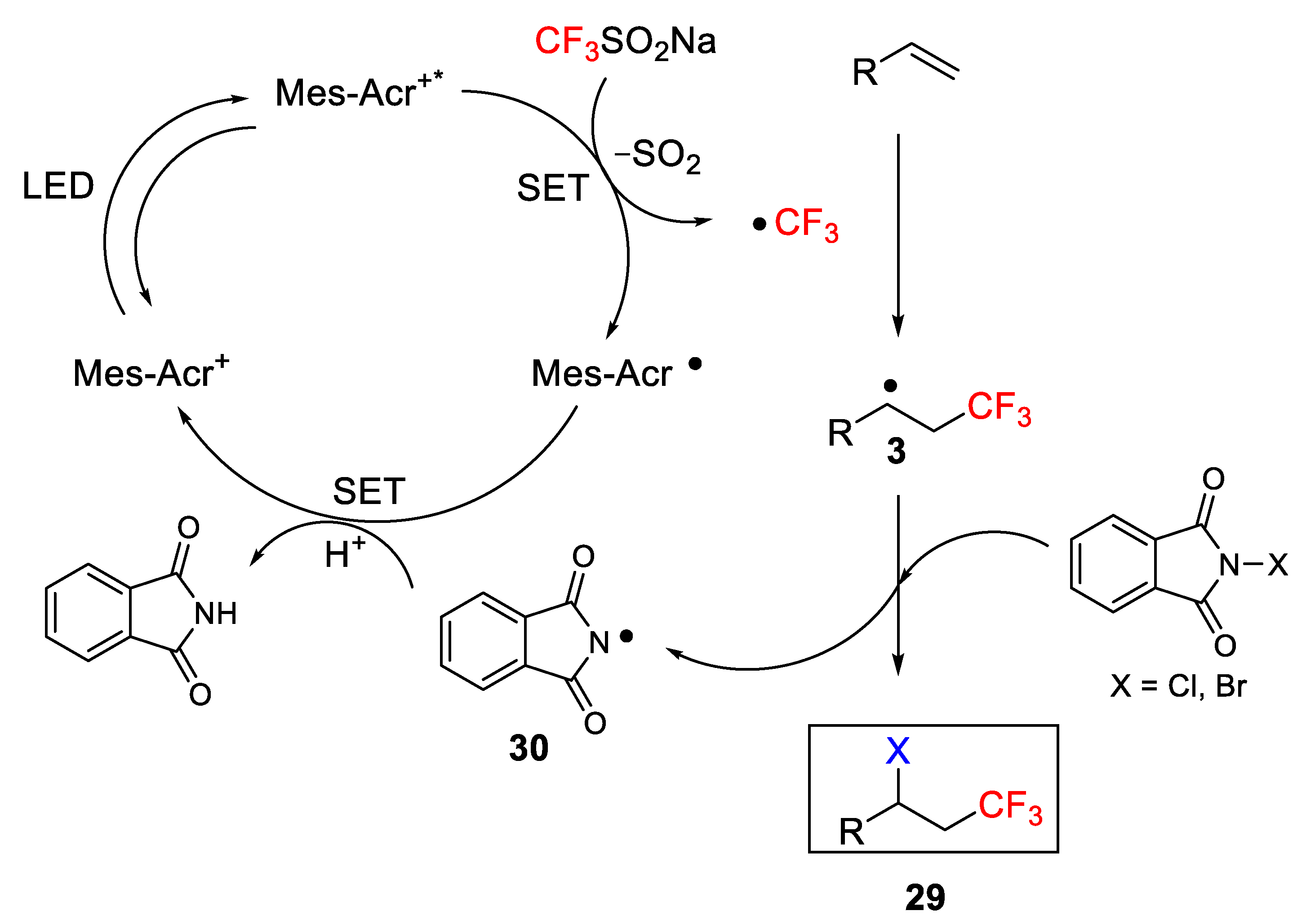{kind=link}
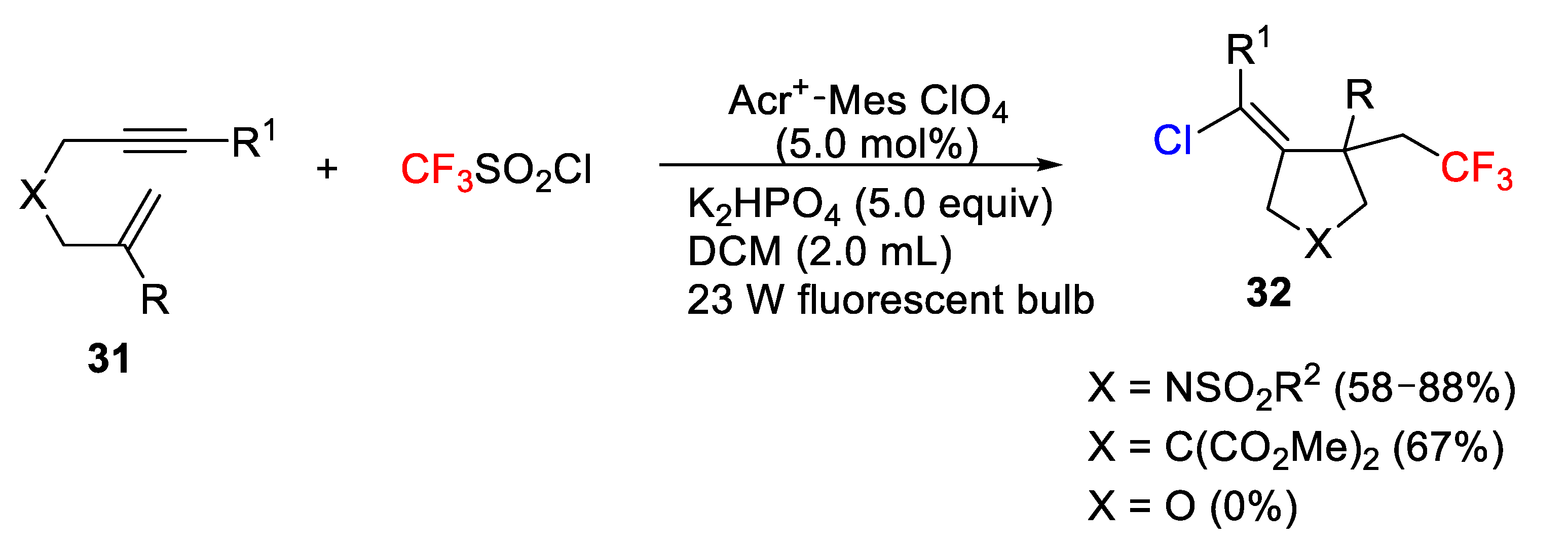{kind=link}
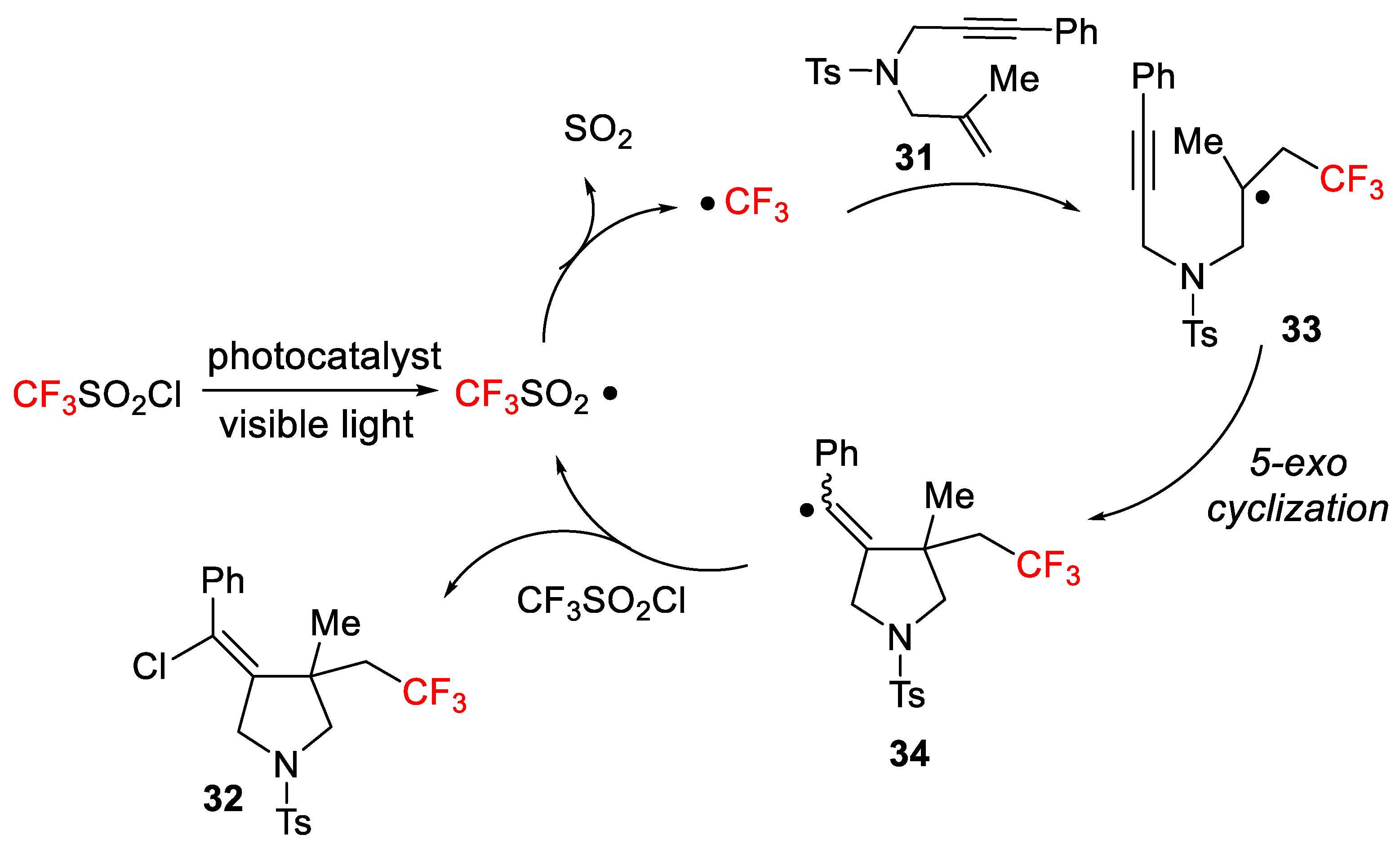{kind=link}
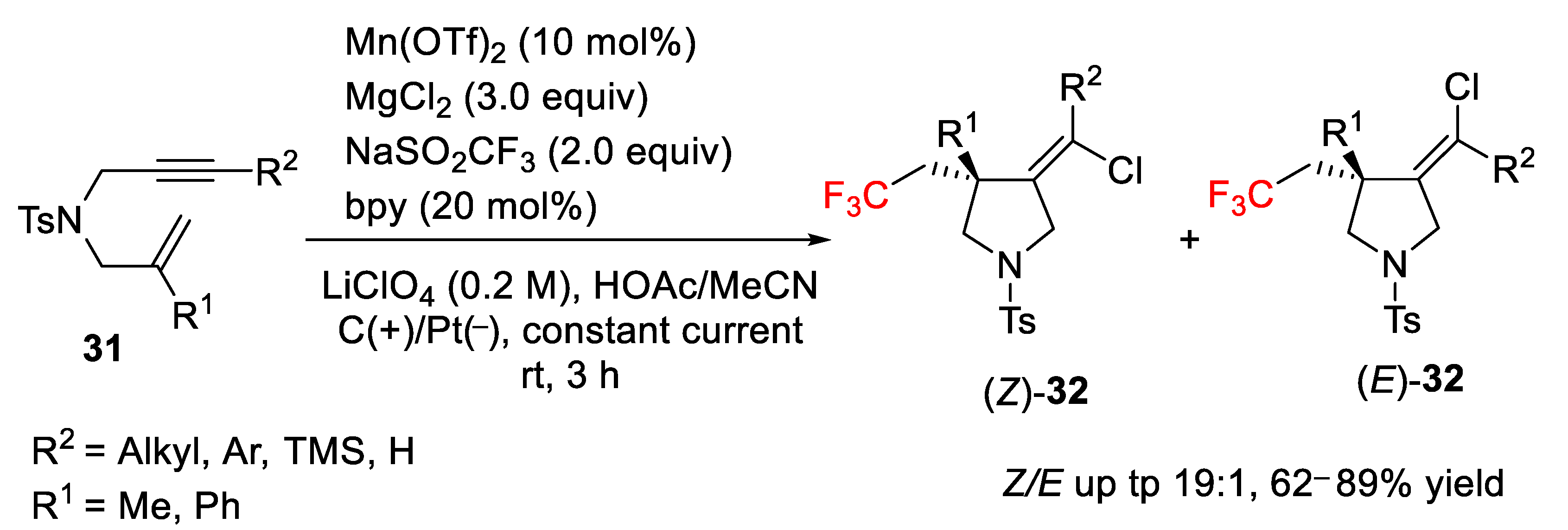{kind=link}
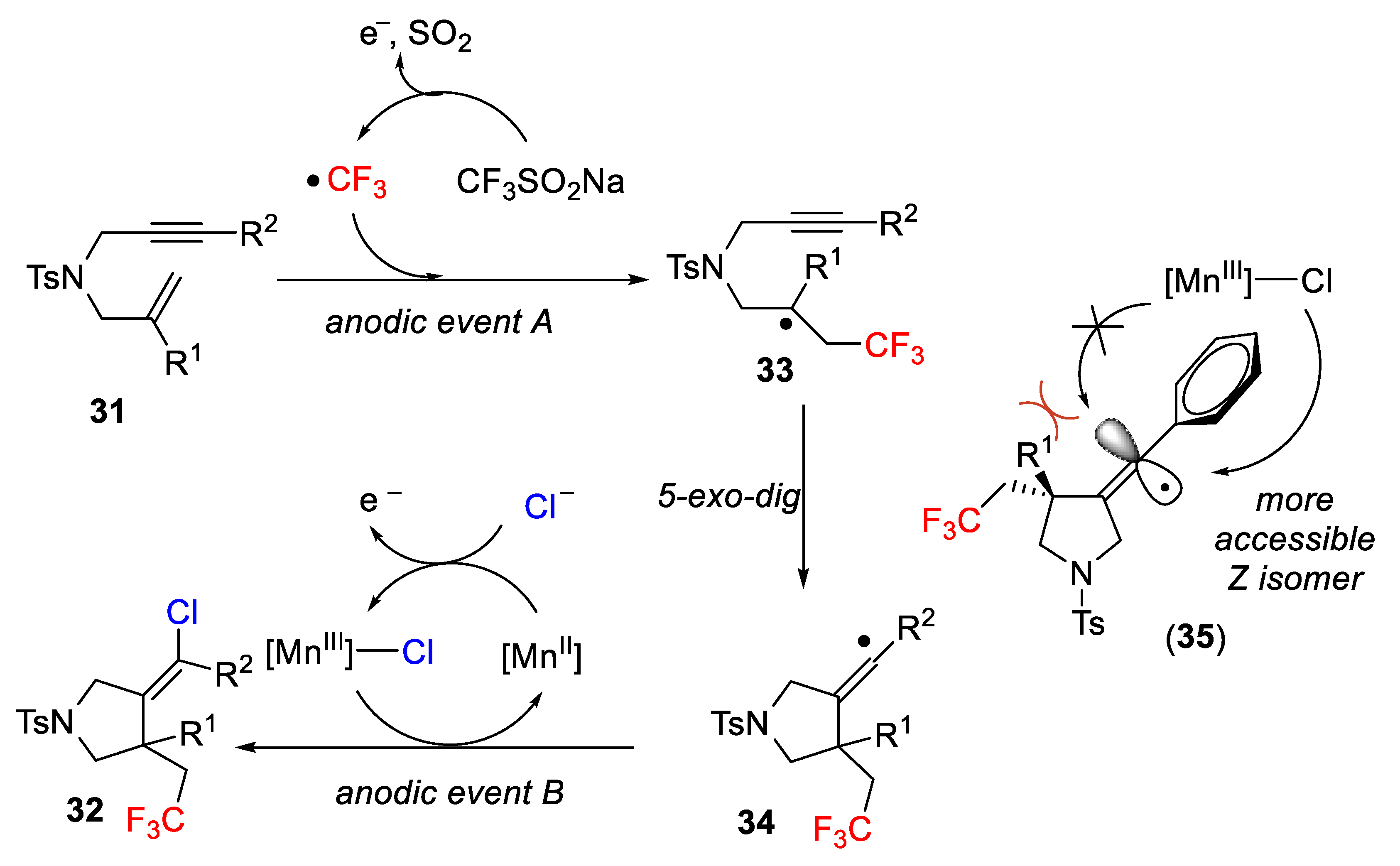{kind=link}
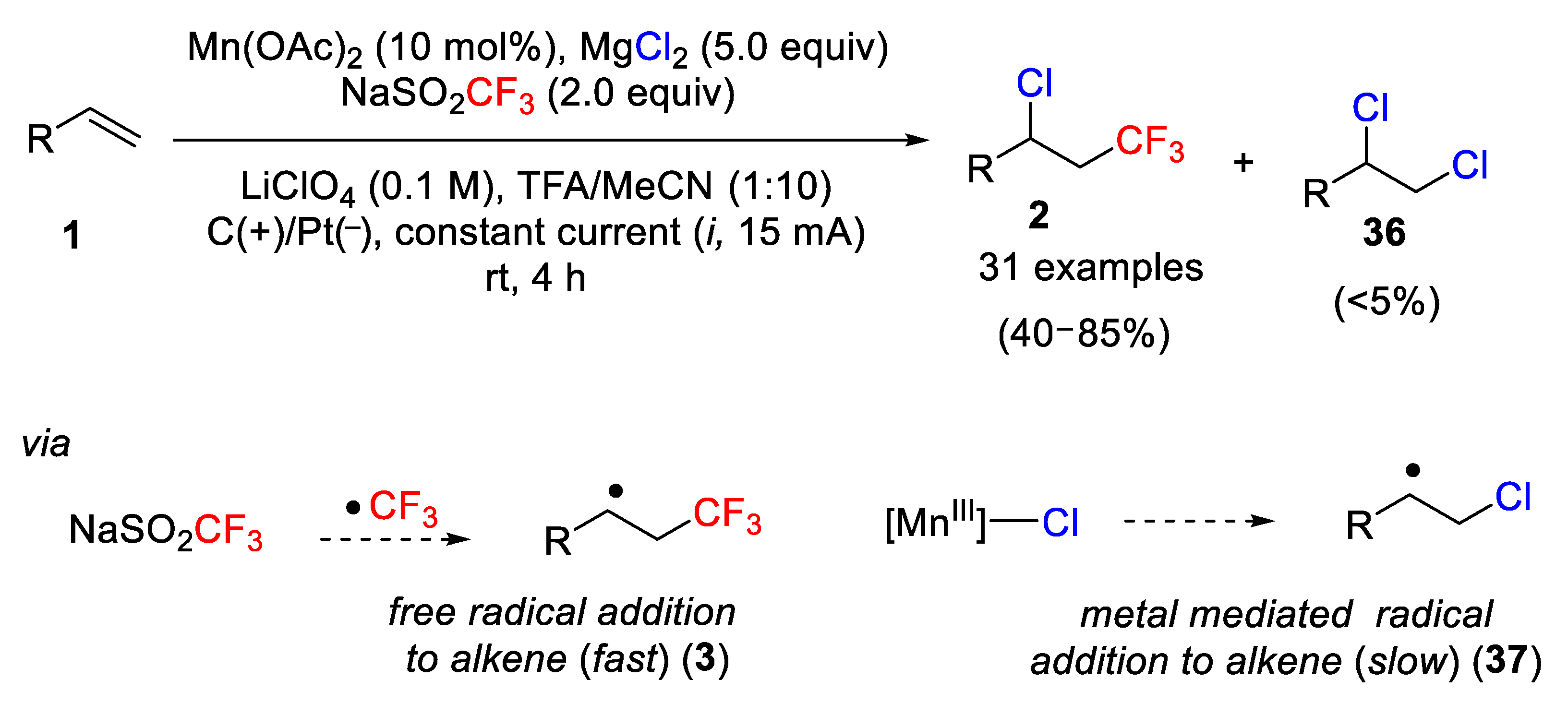{kind=link}
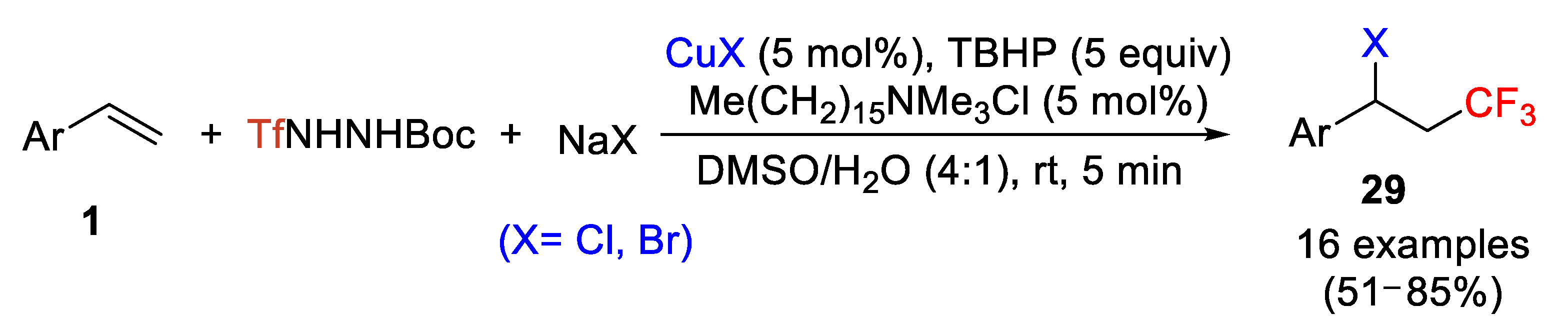{kind=link}
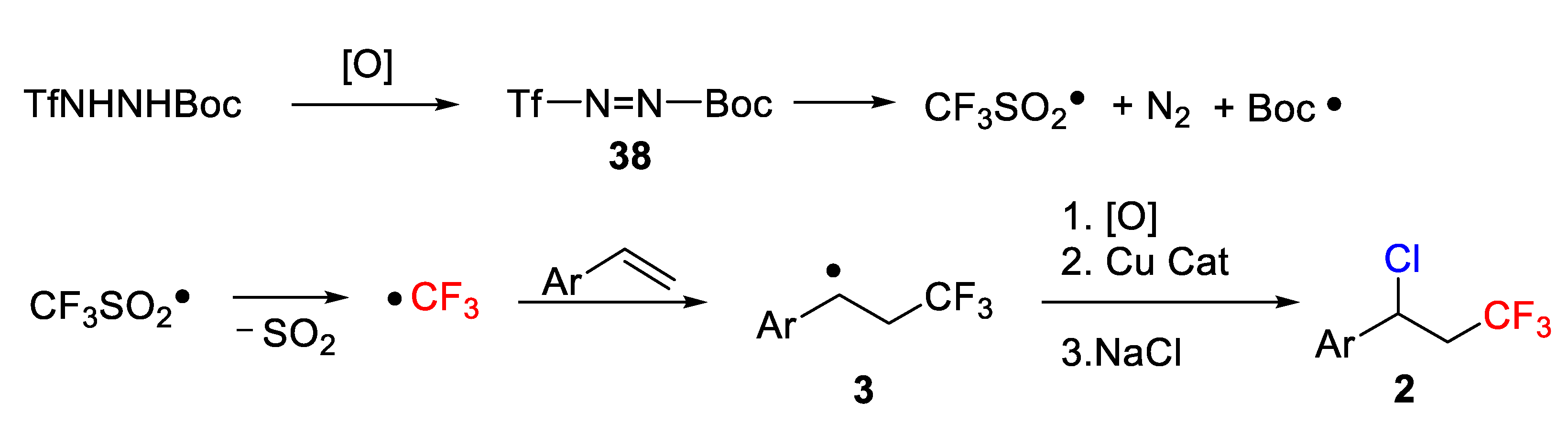{kind=link}
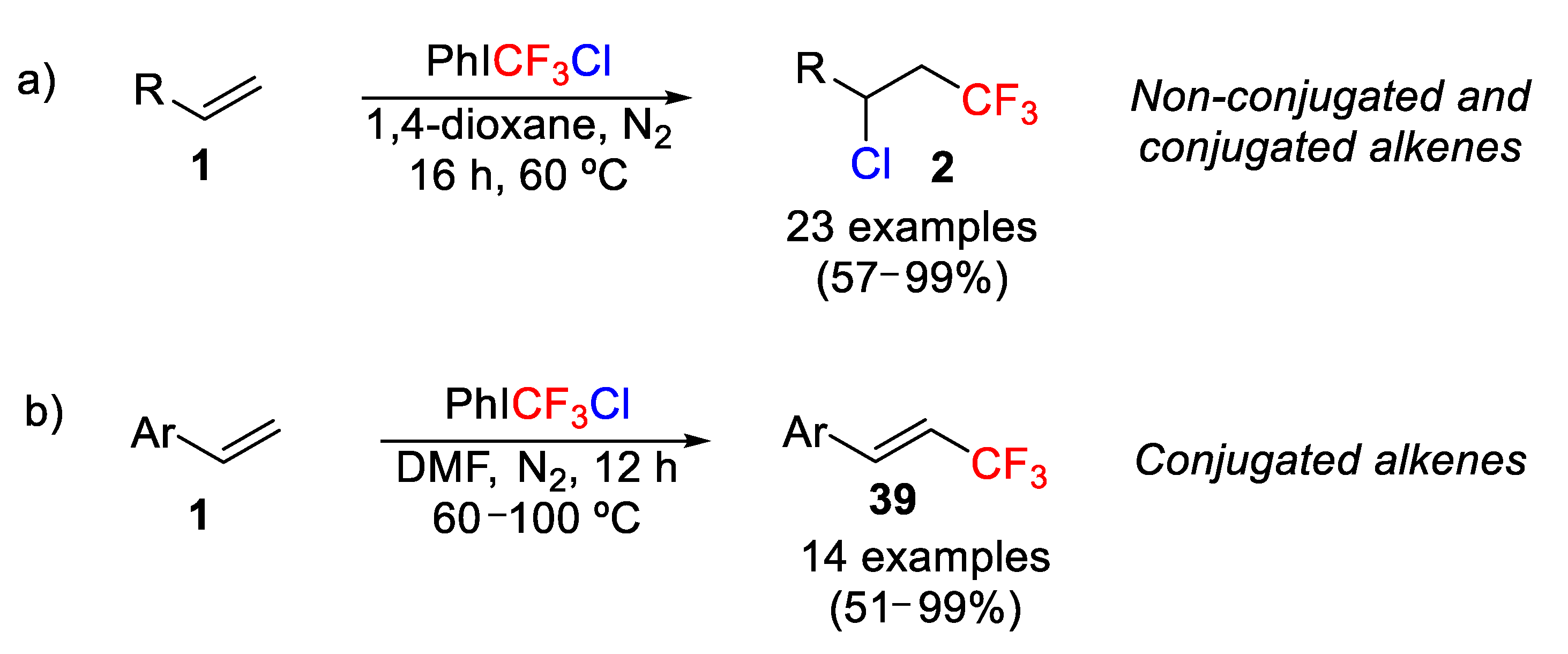{kind=link}
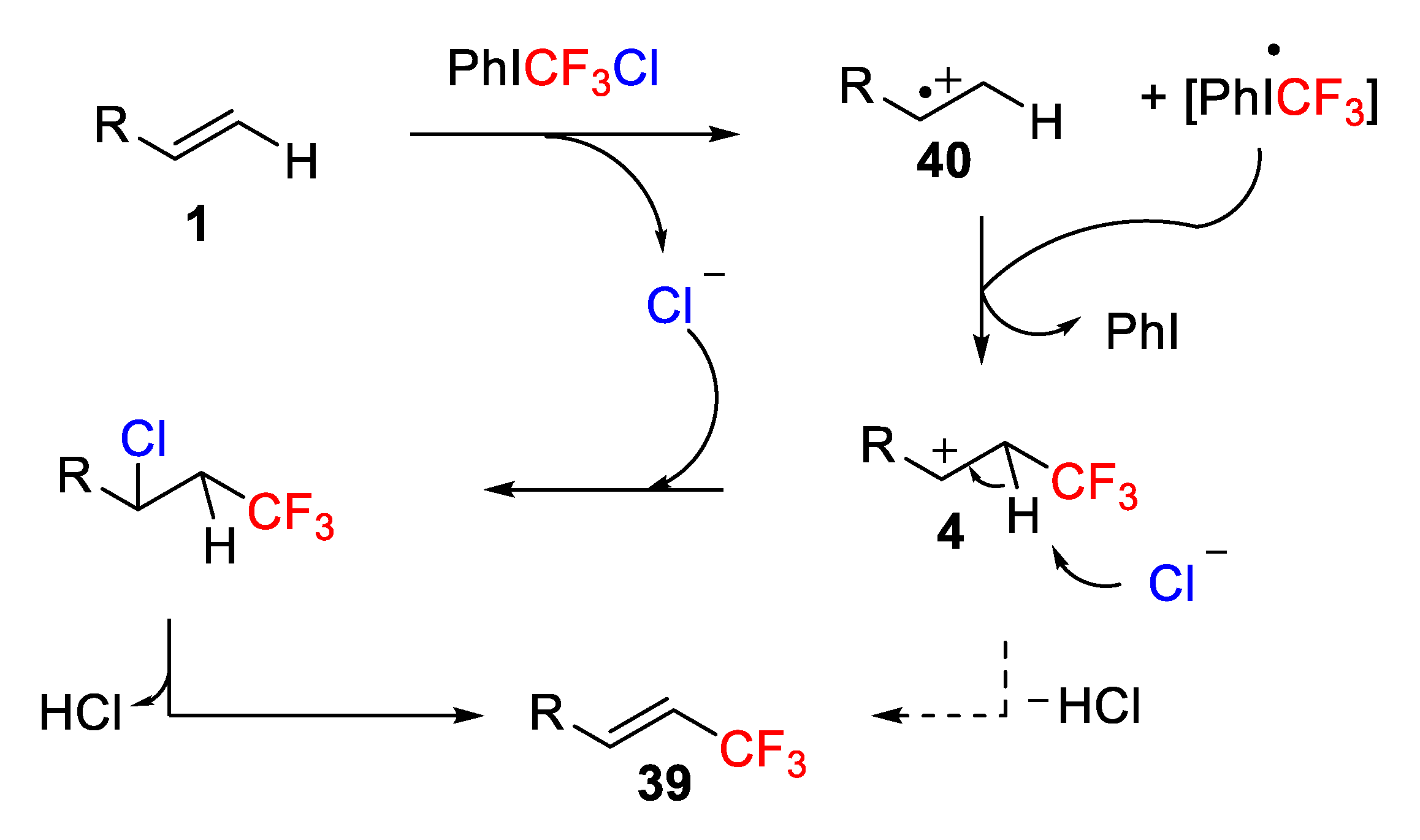{kind=link}
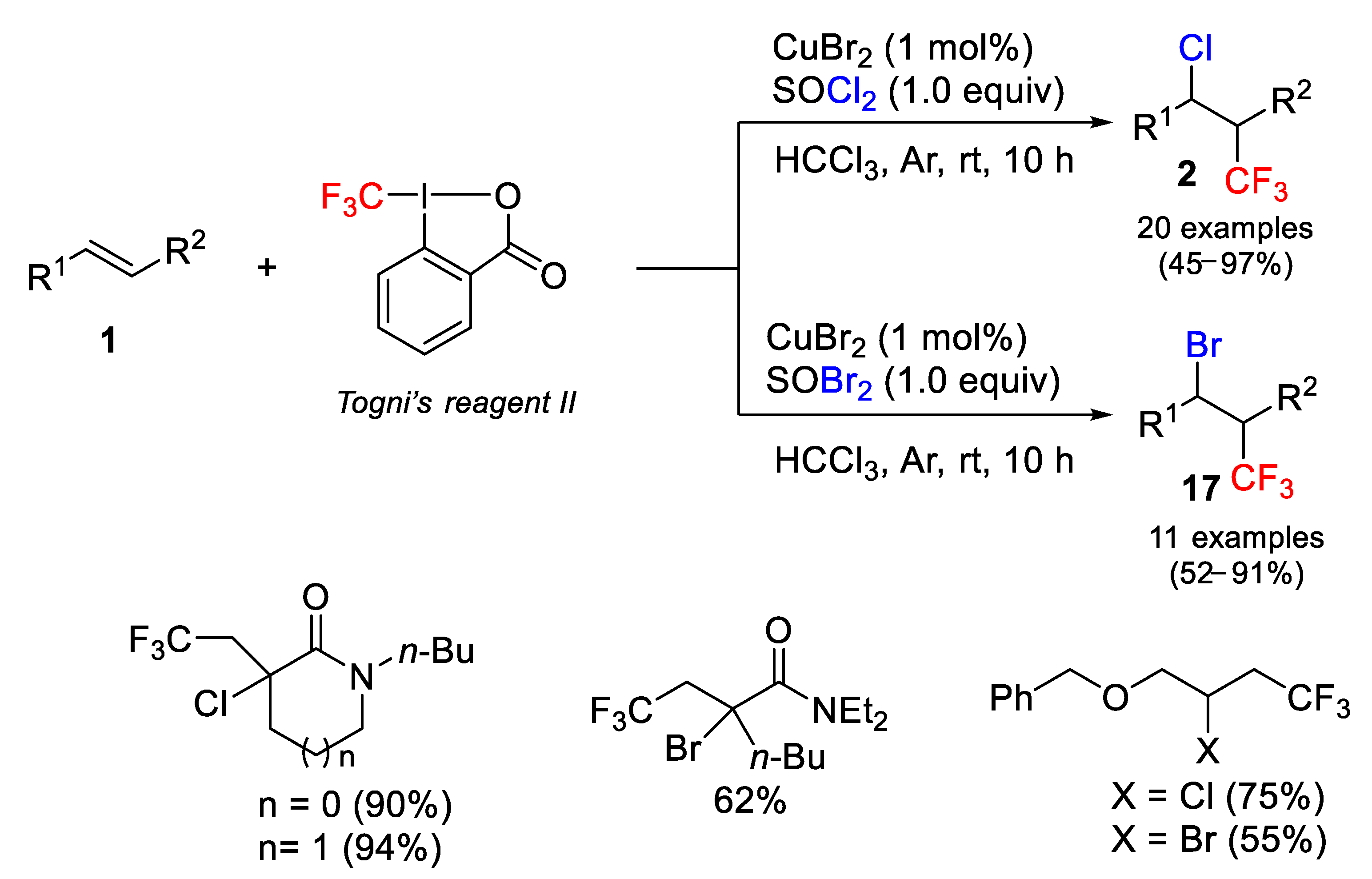{kind=link}
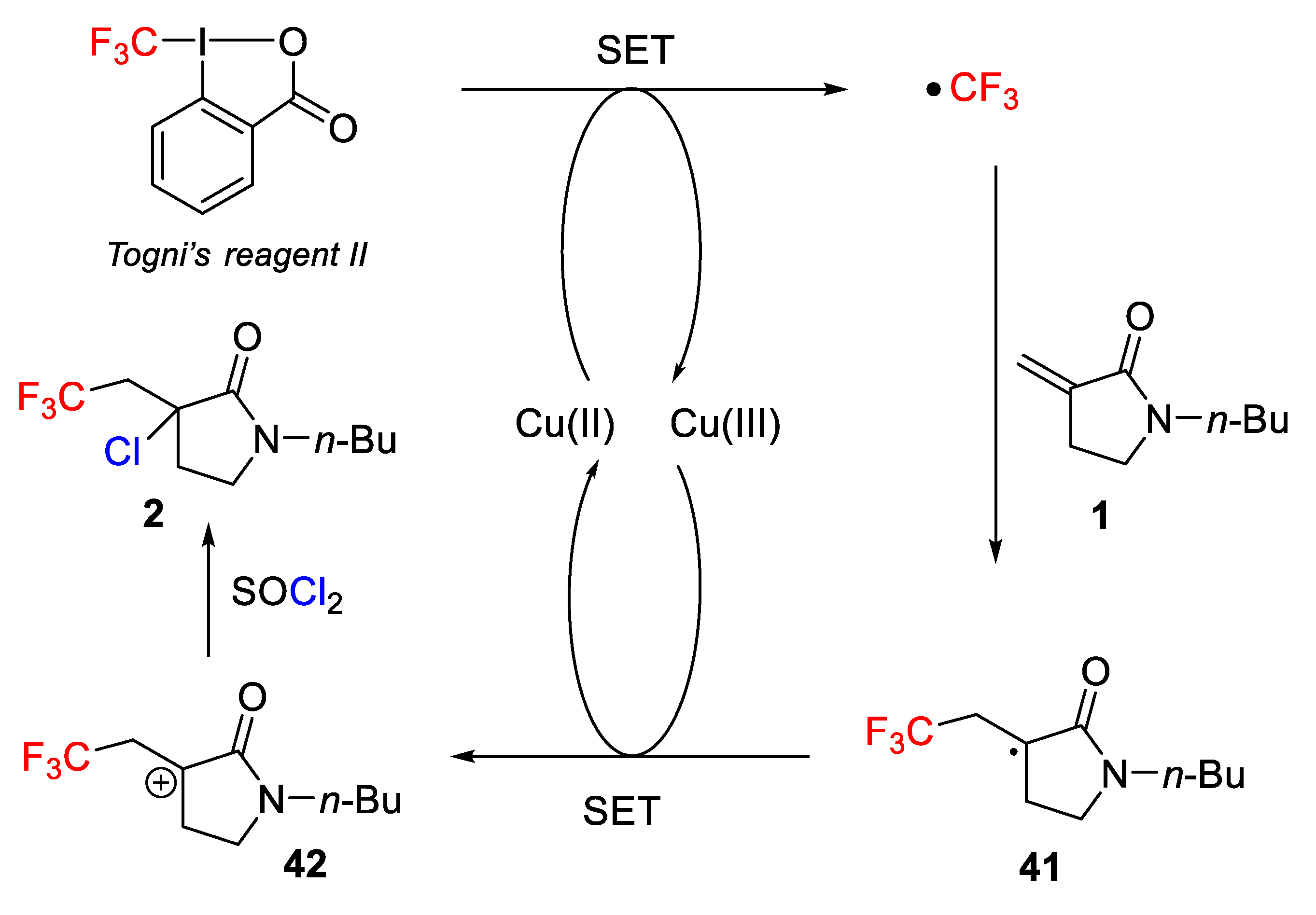{kind=link}
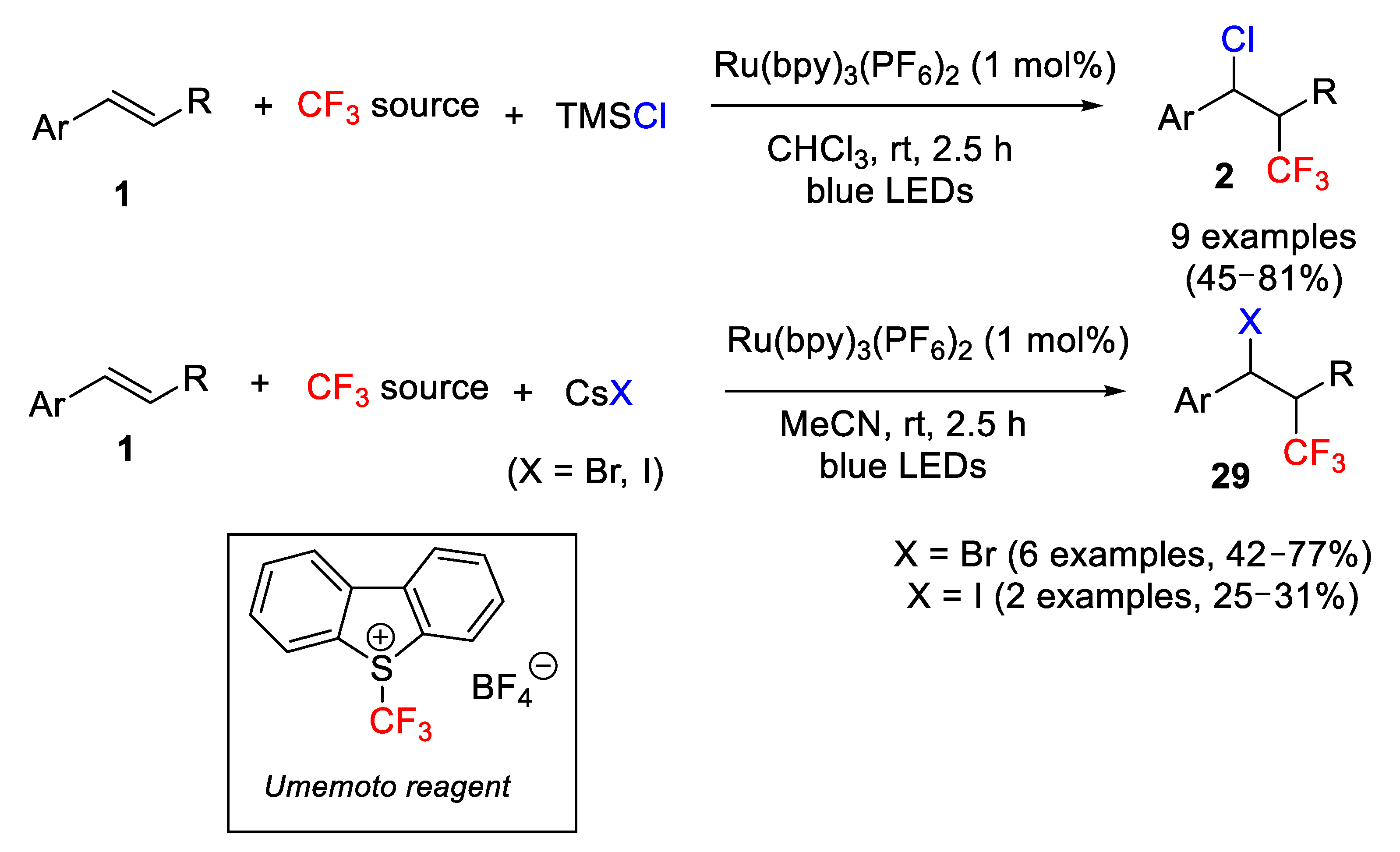{kind=link}
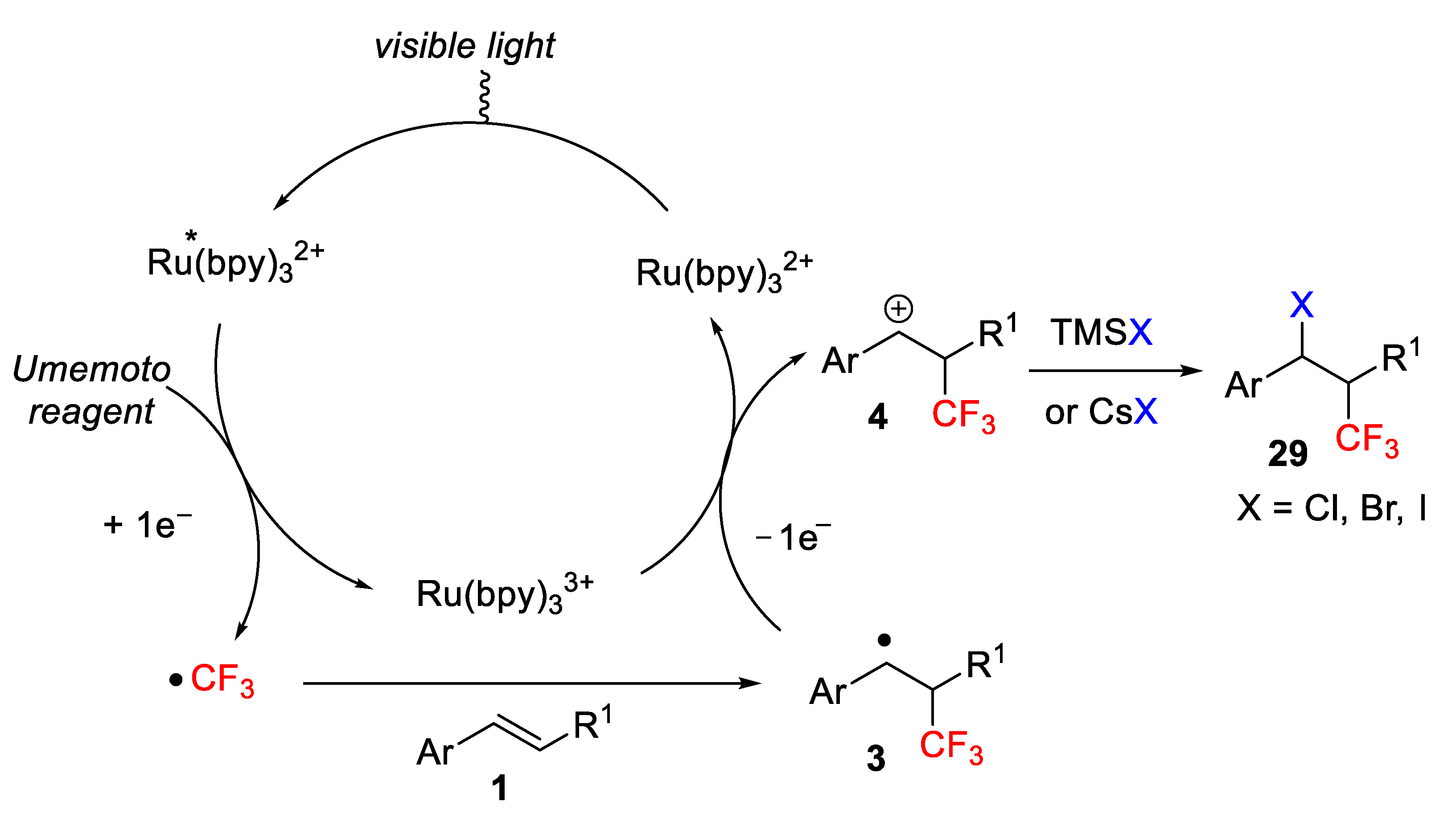{kind=link}
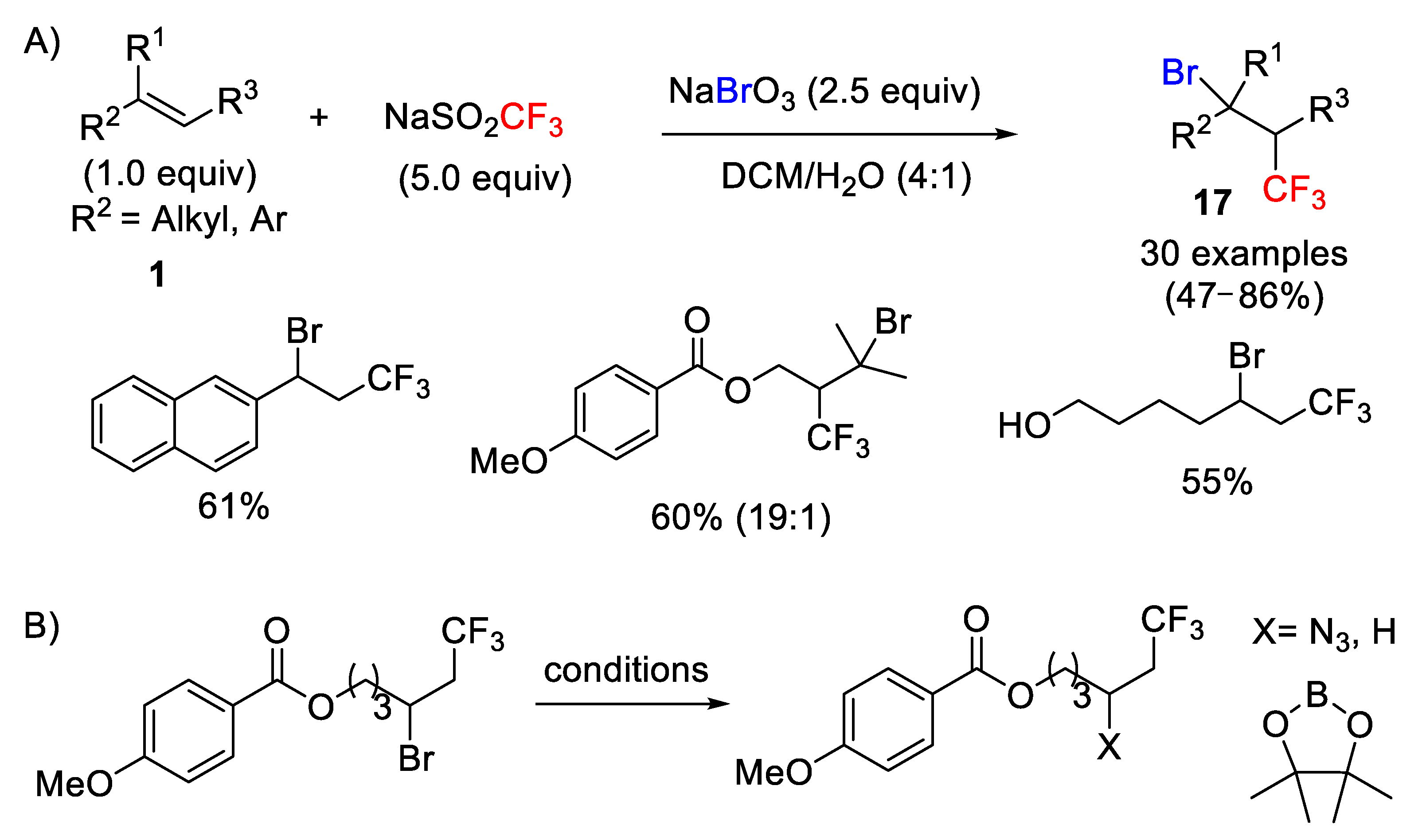{kind=link}
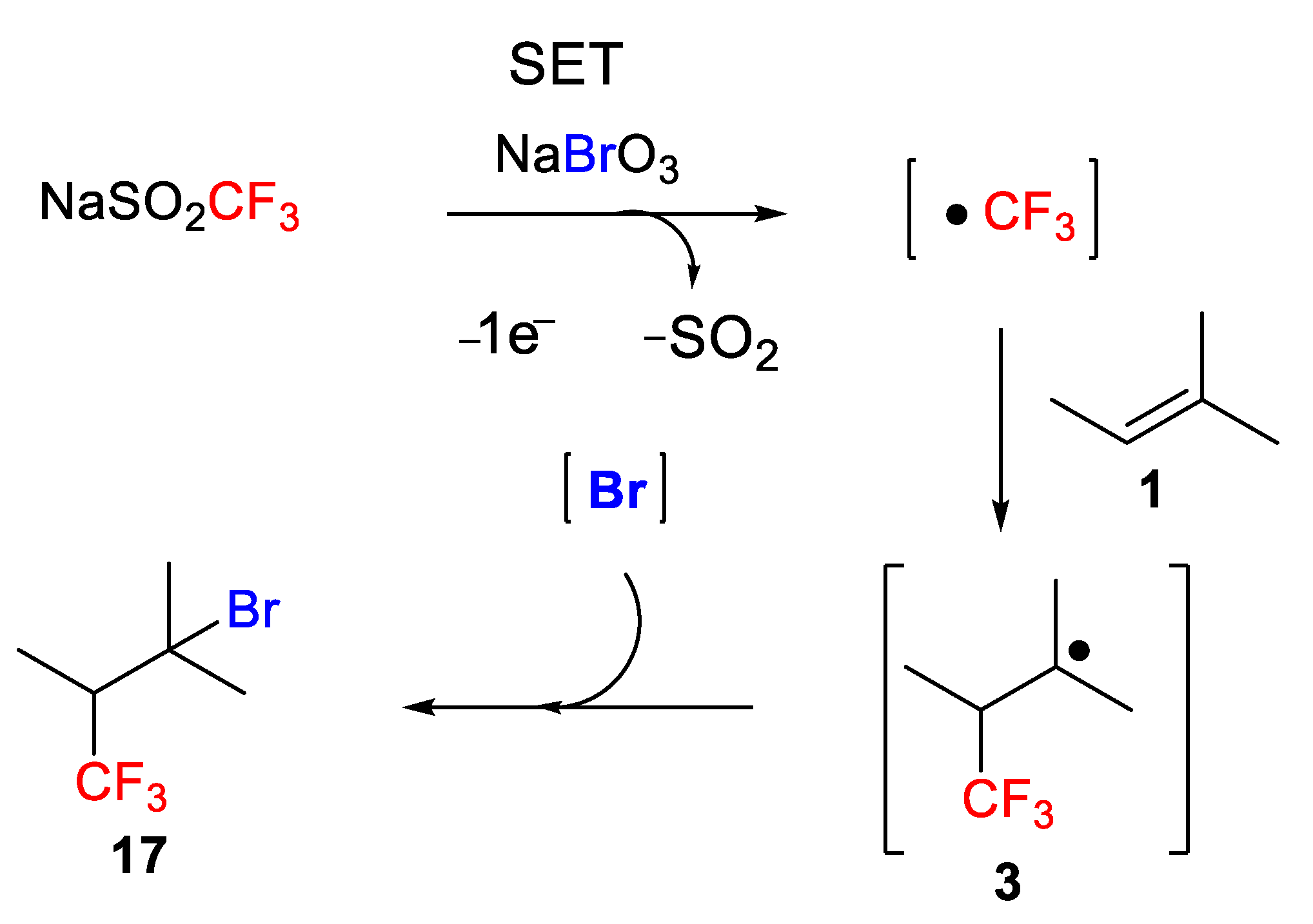{kind=link}
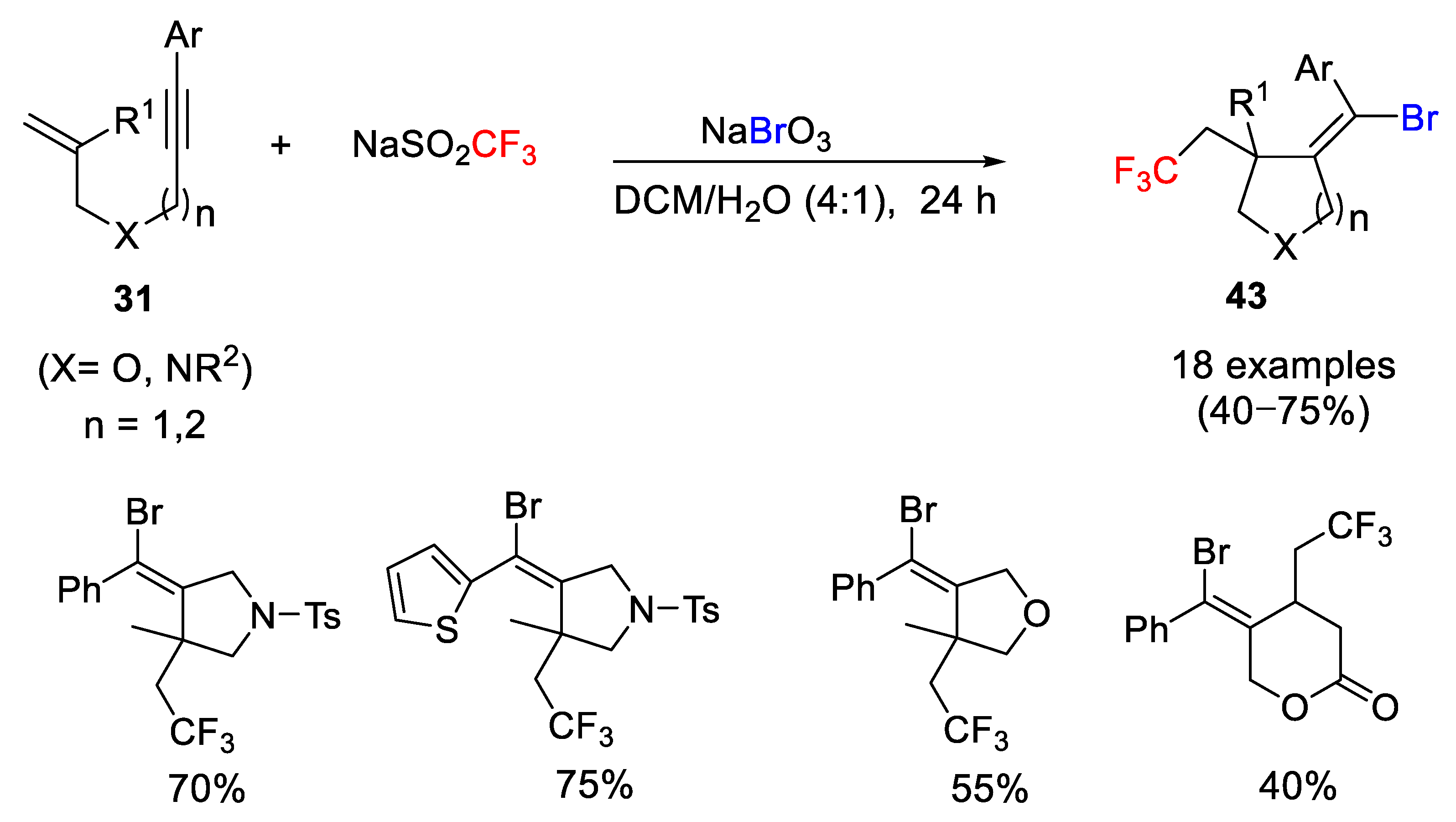{kind=link}
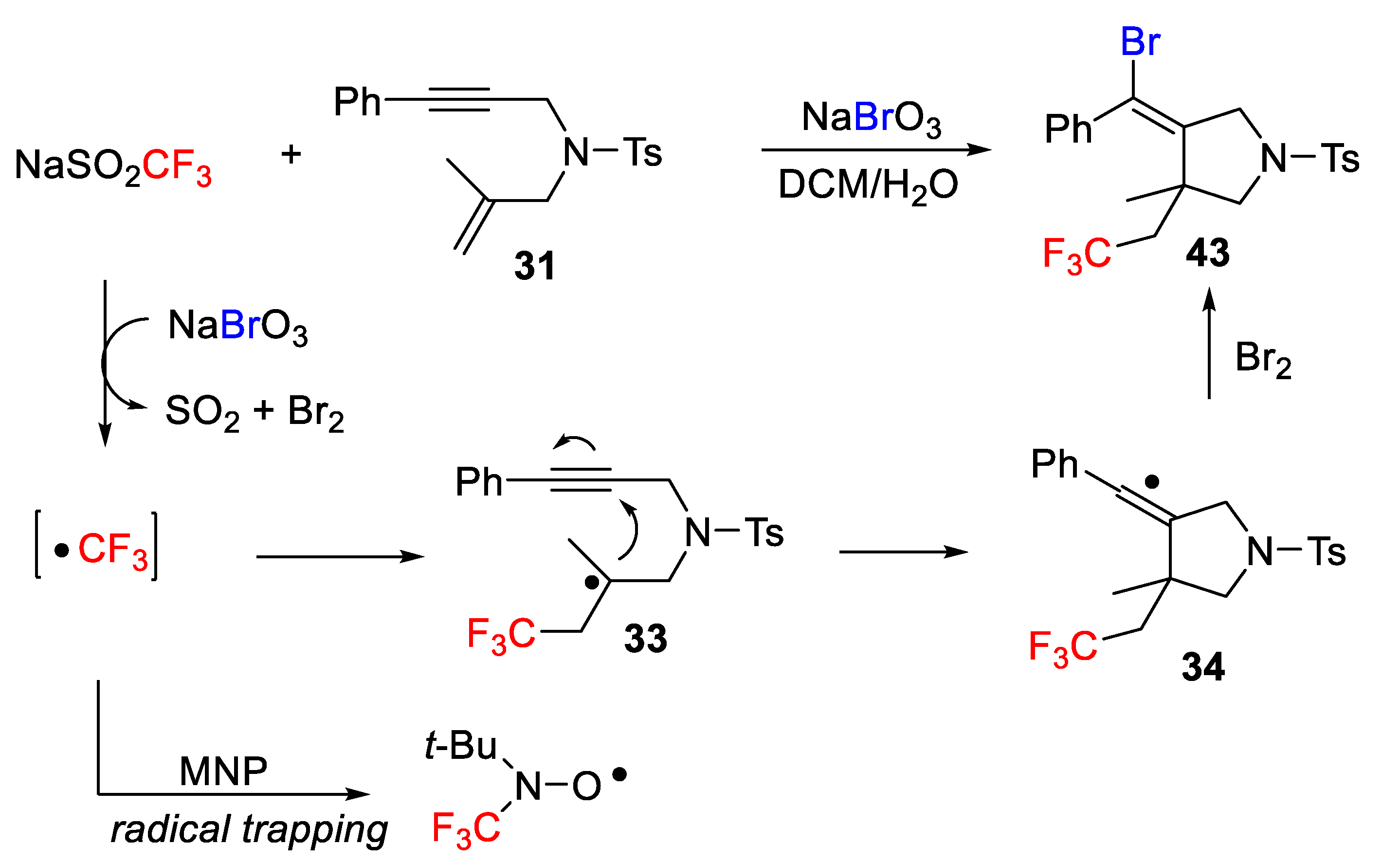{kind=link}
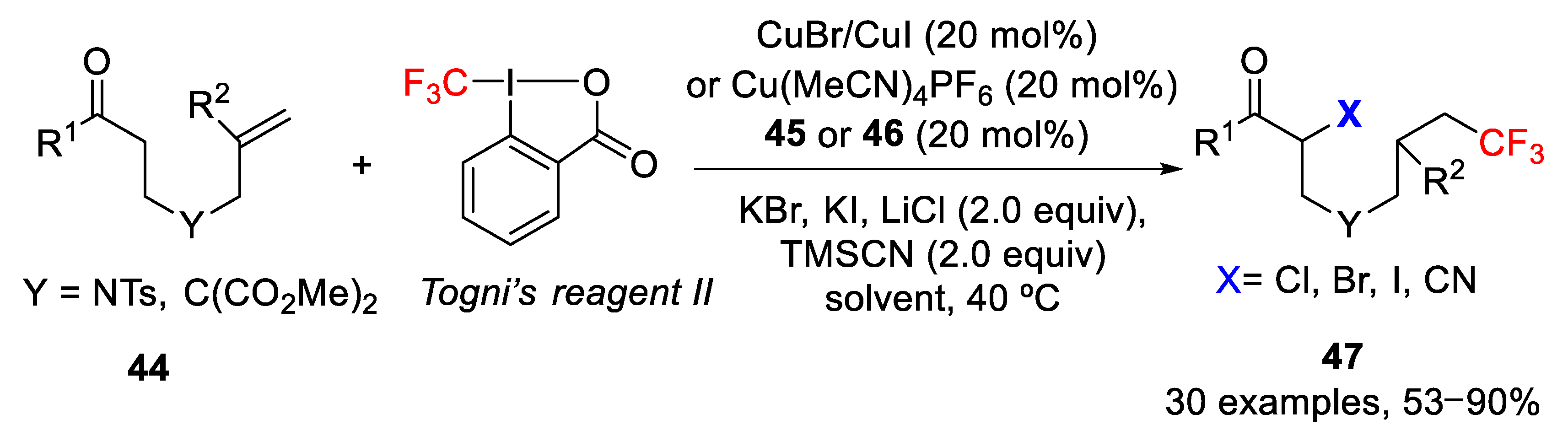{kind=link}
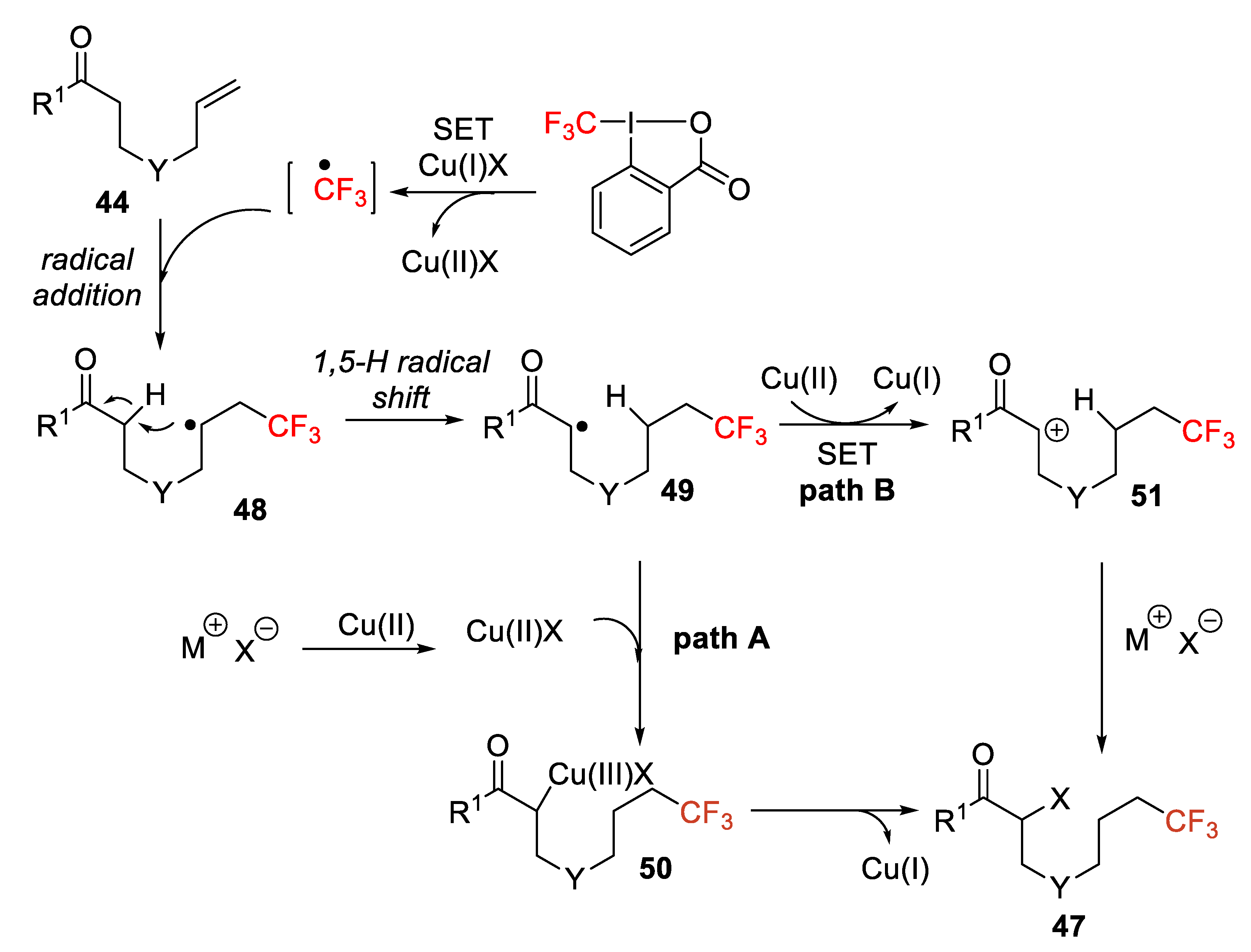{kind=link}
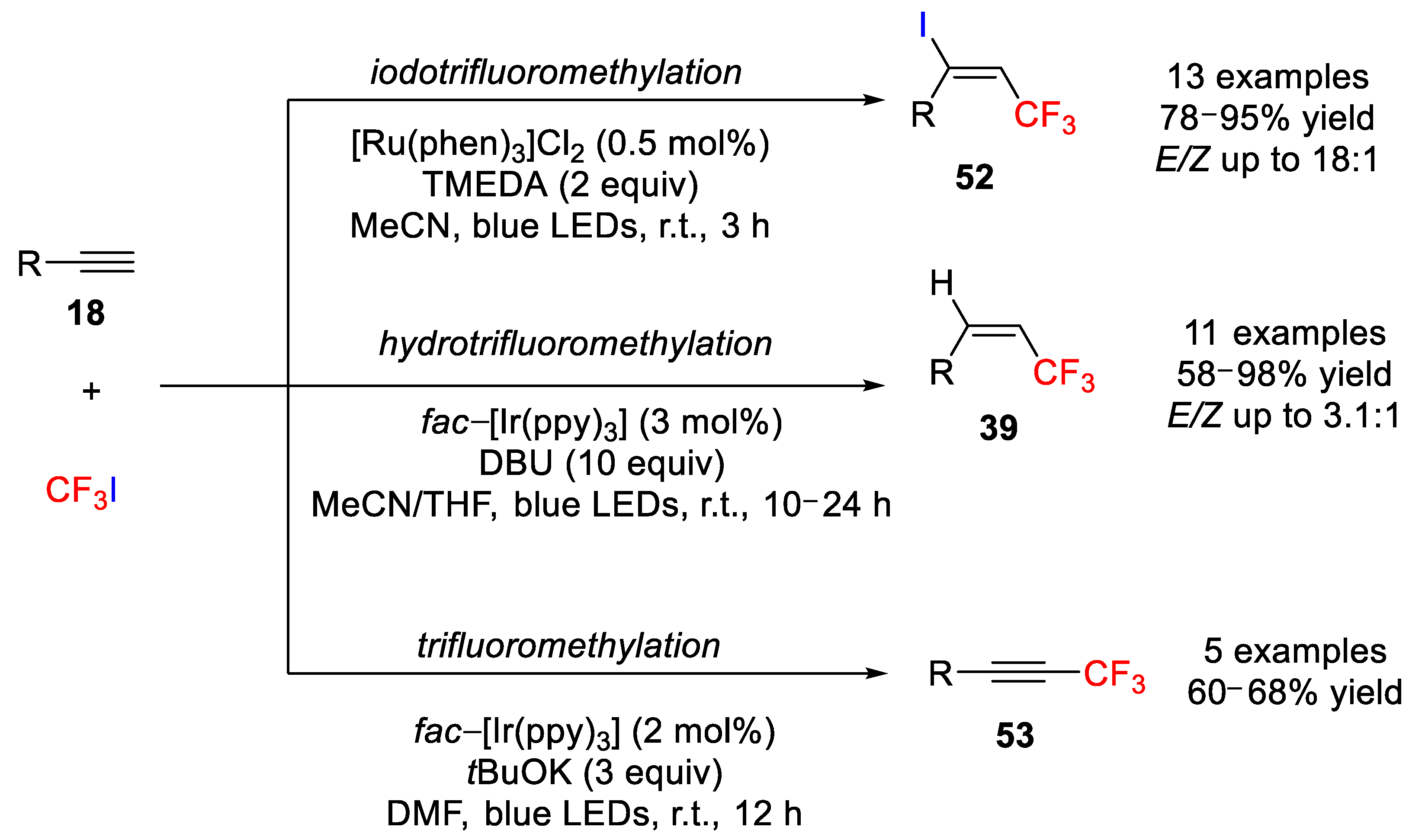{kind=link}
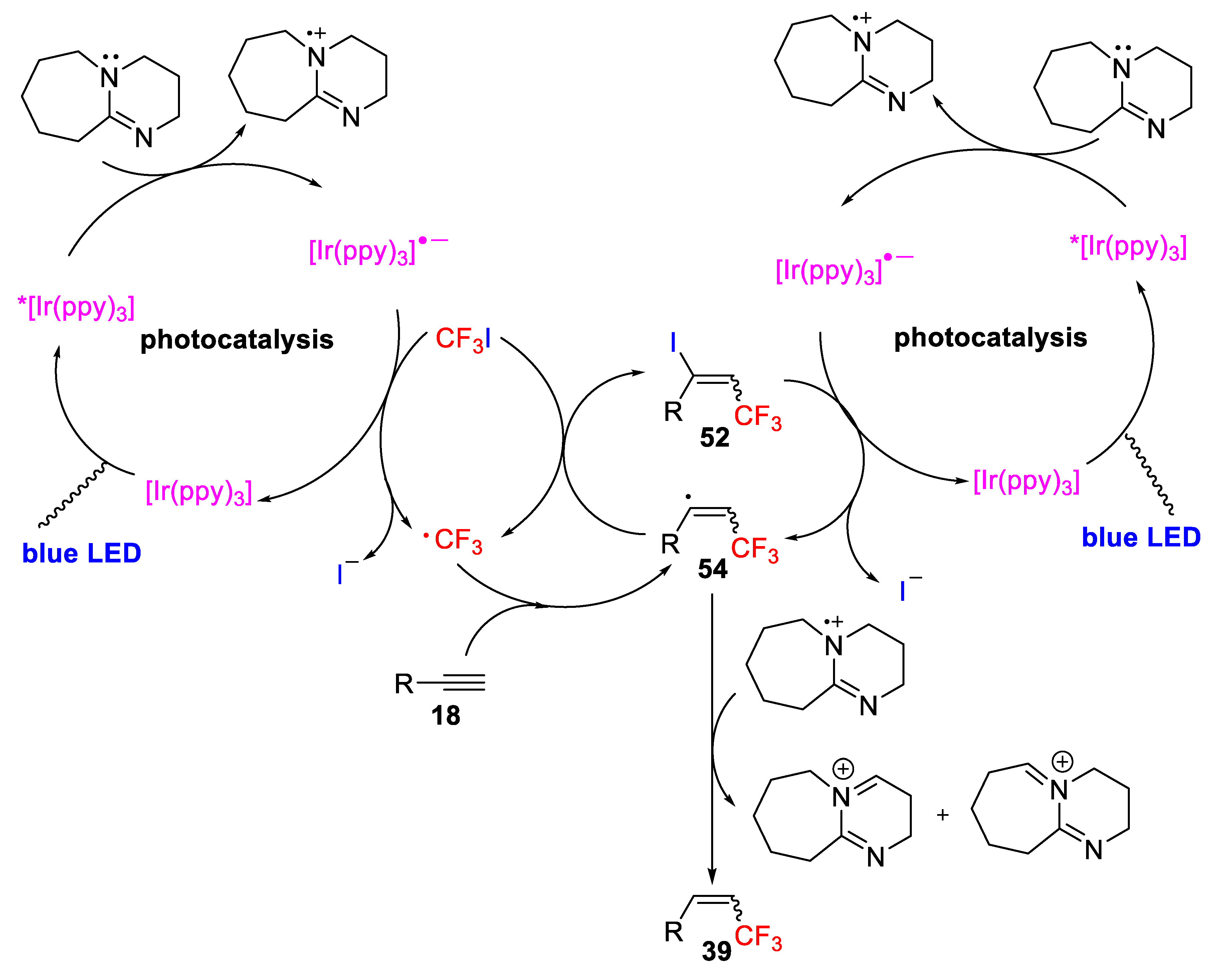{kind=link}
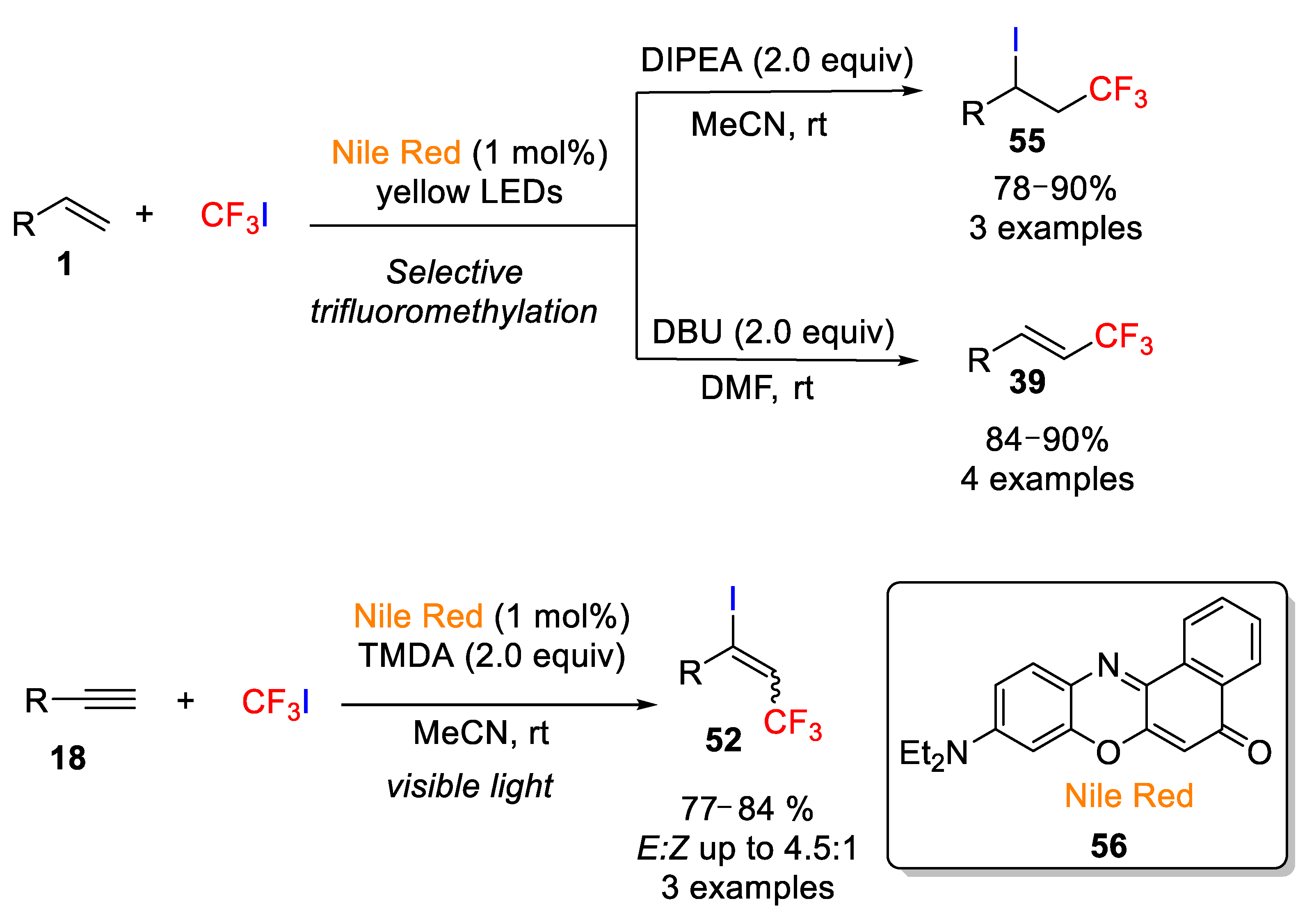{kind=link}
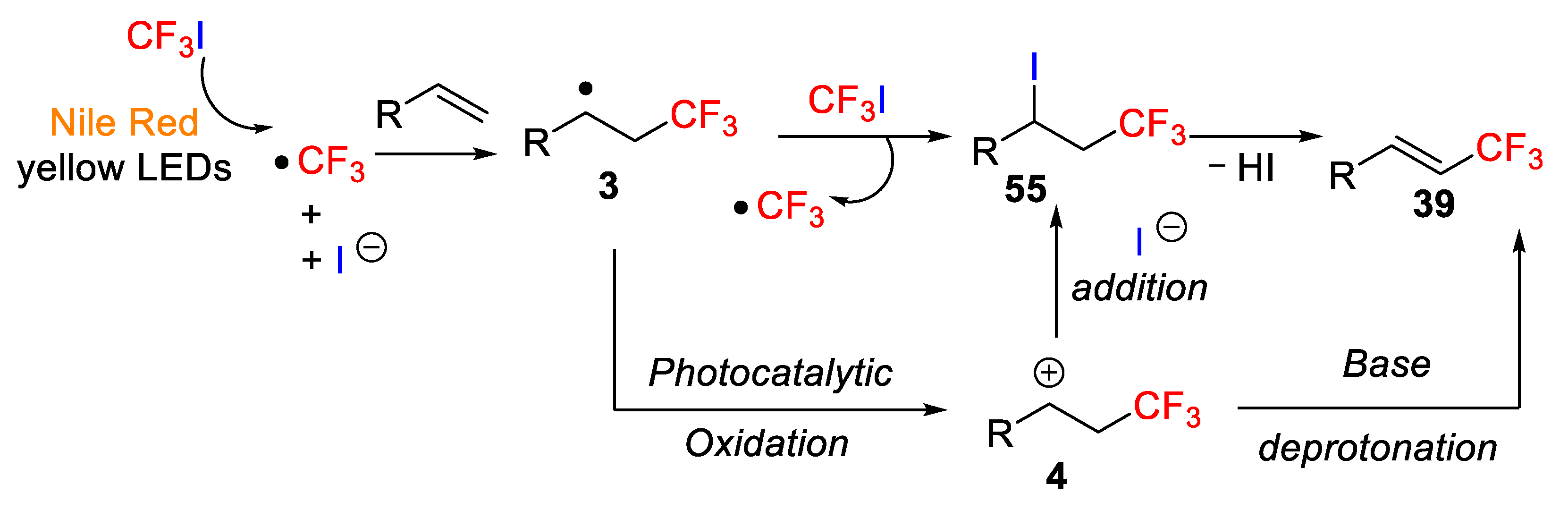{kind=link}
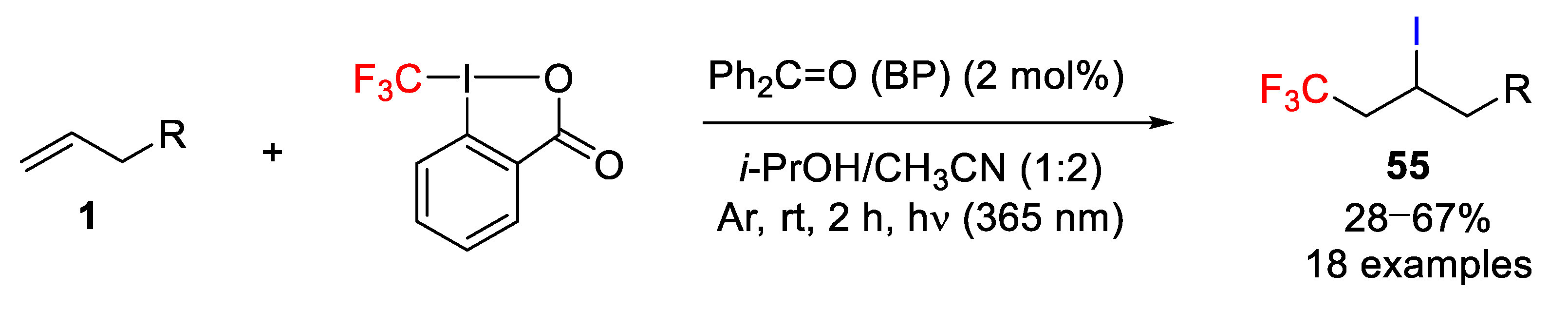{kind=link}
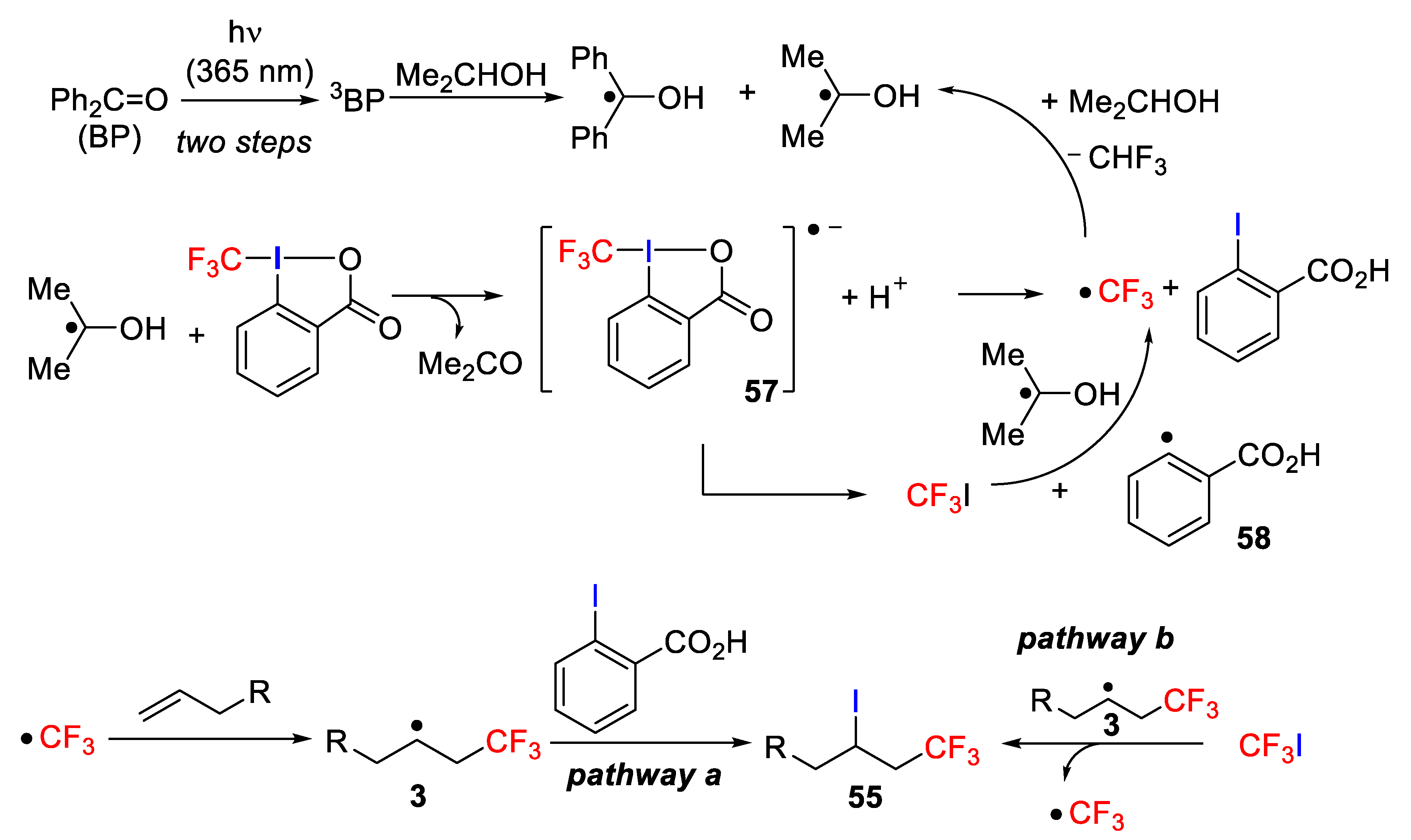{kind=link}
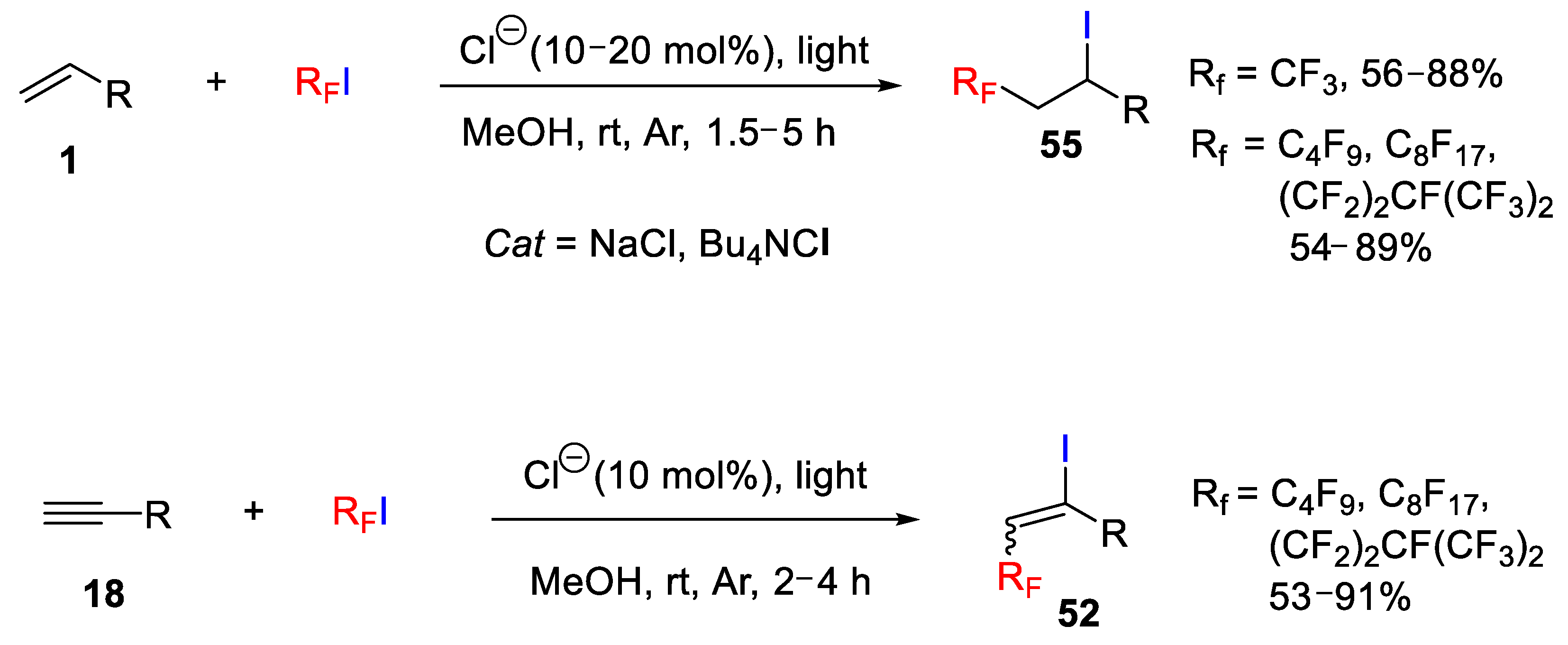{kind=link}
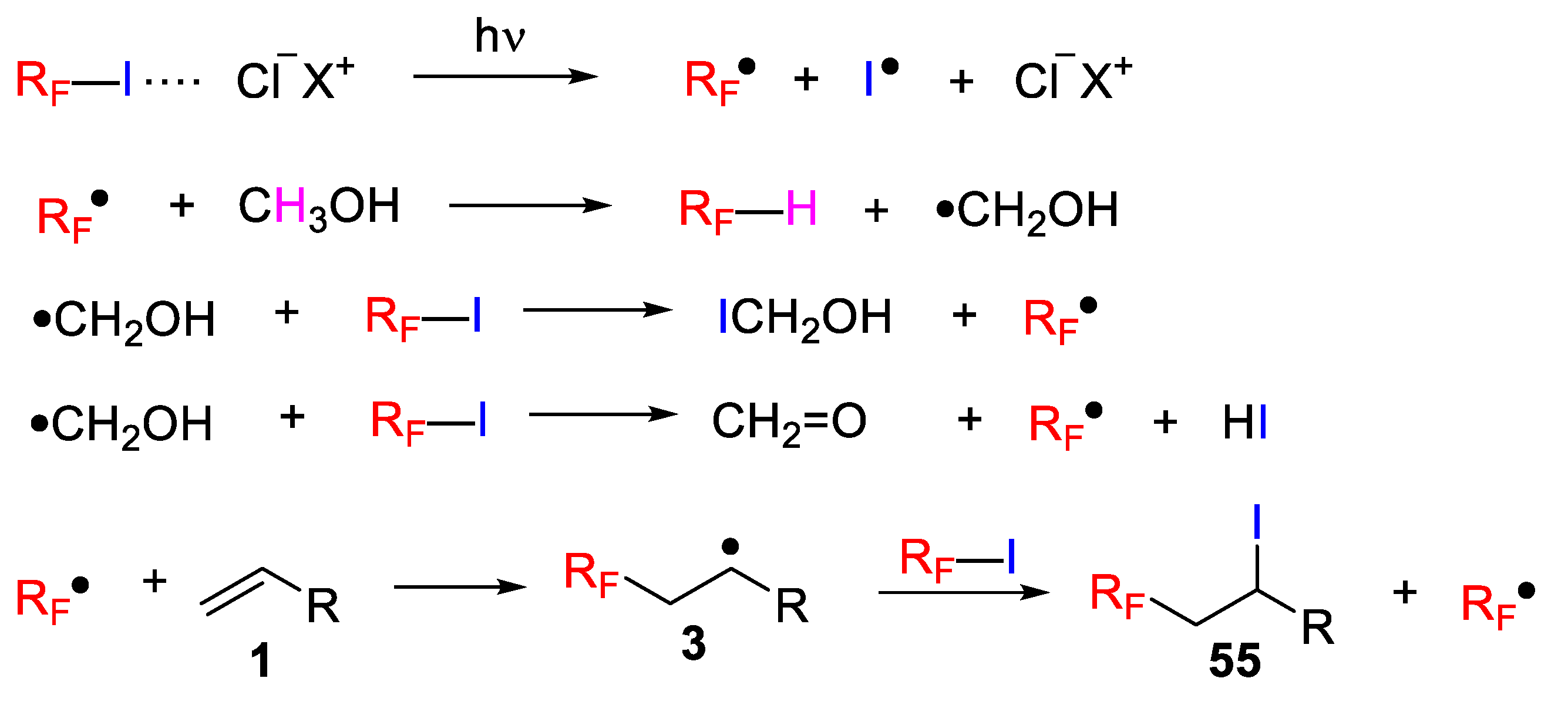{kind=link}
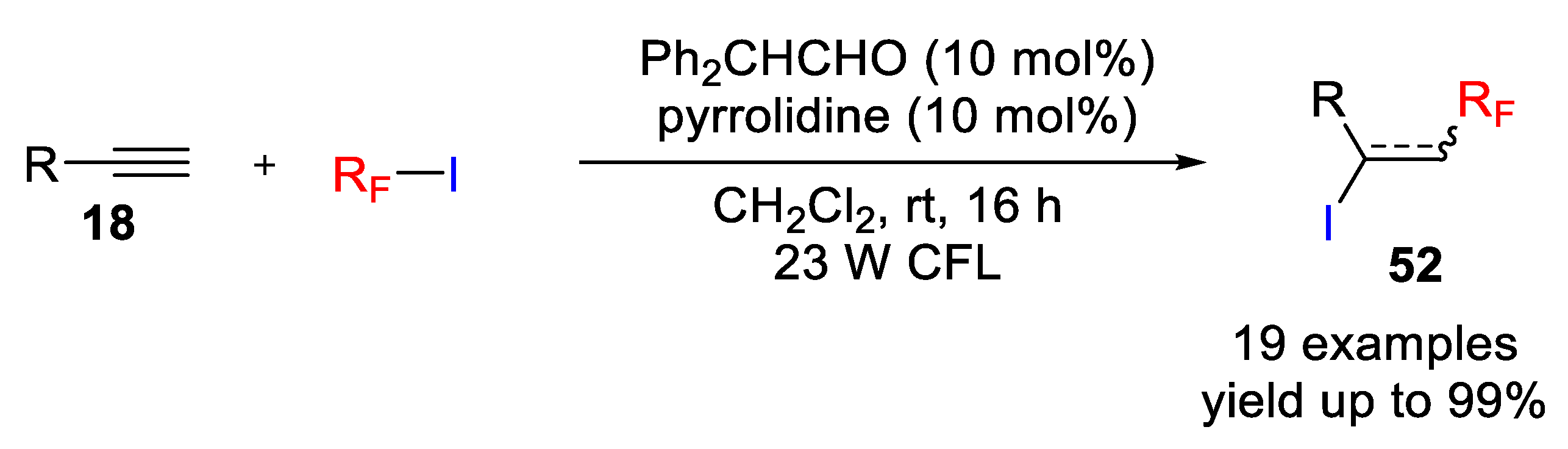{kind=link}
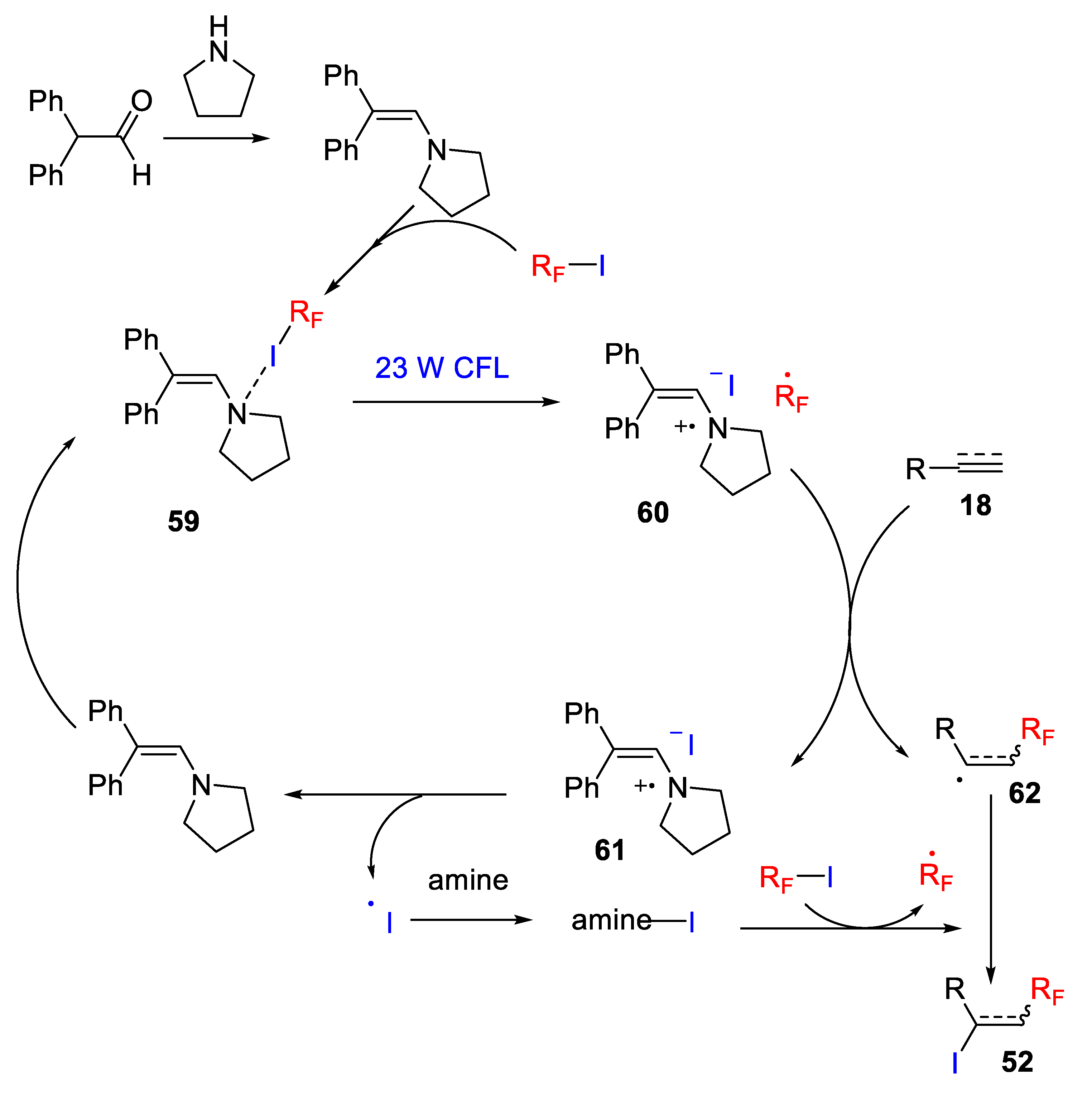{kind=link}
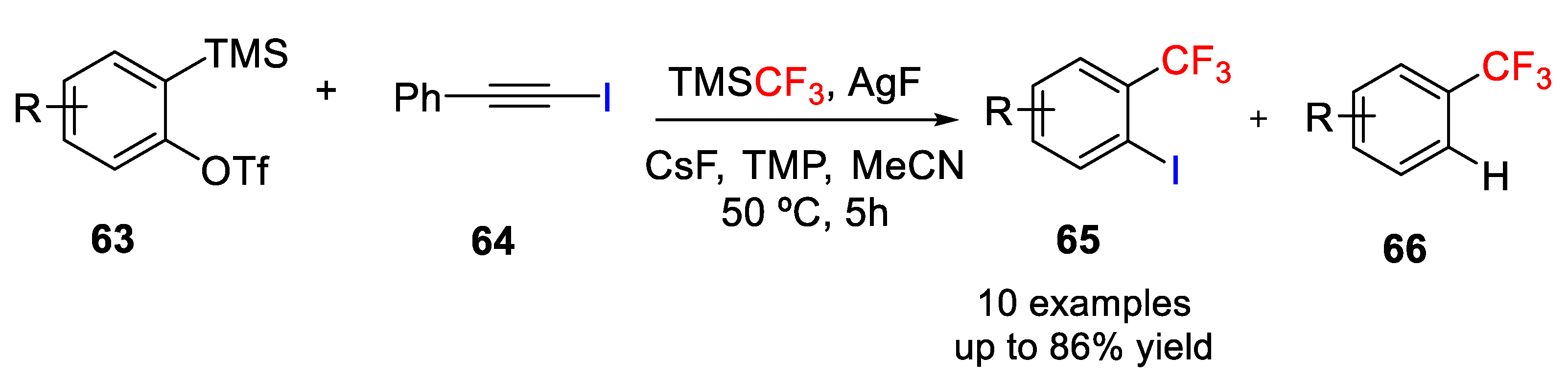{kind=link}
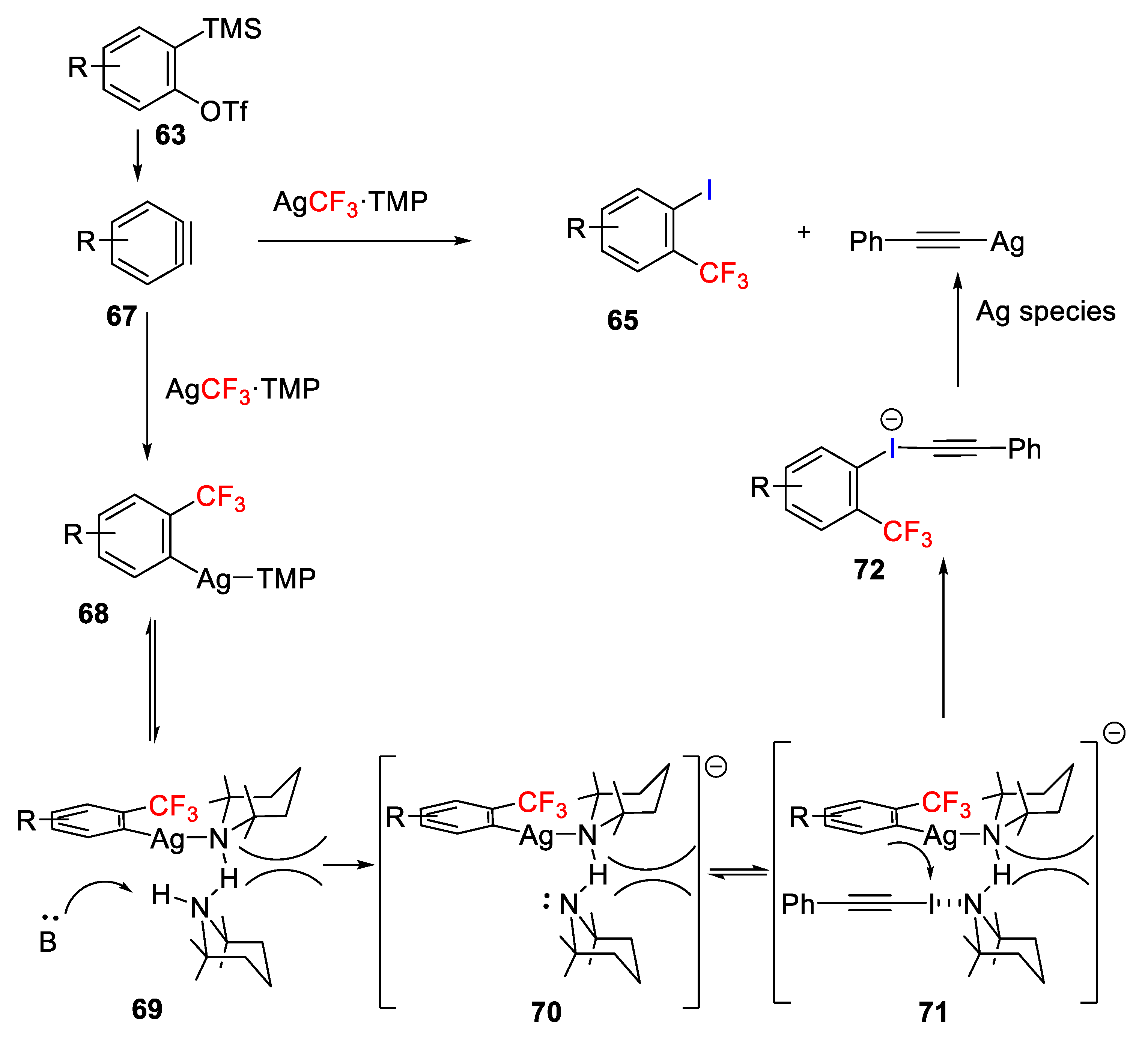{kind=link}
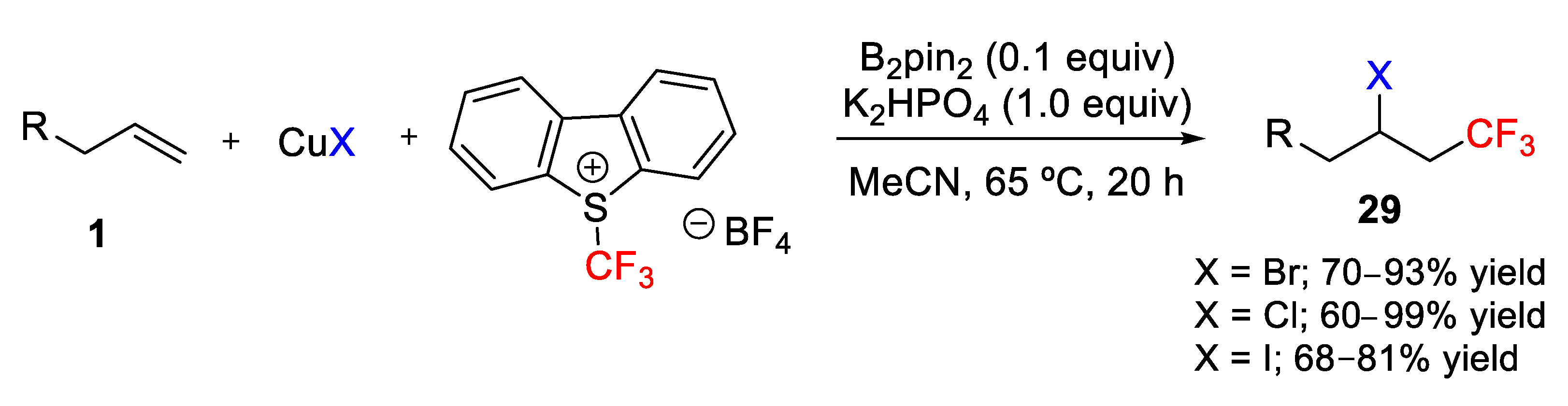{kind=link}
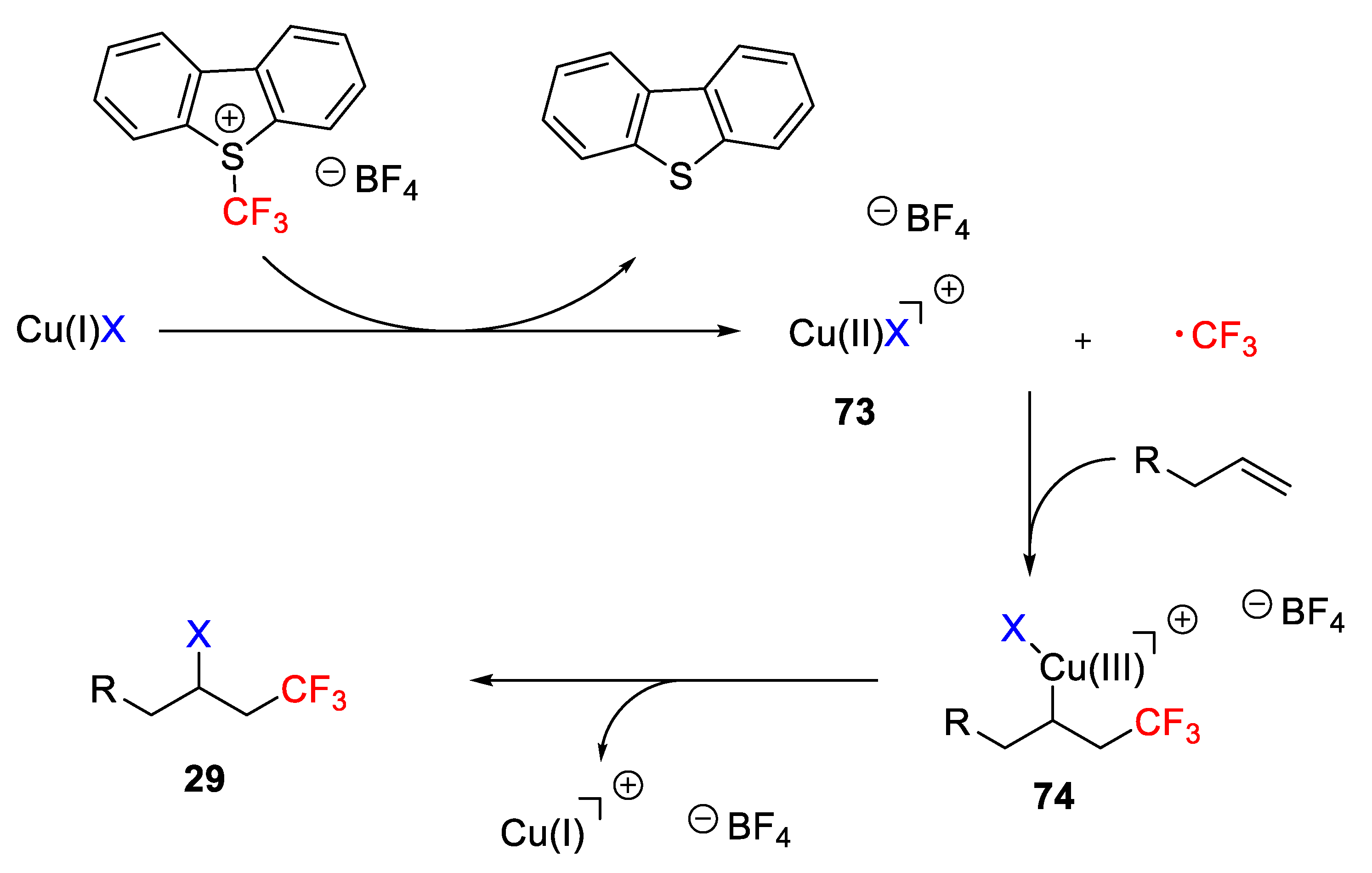{kind=link}
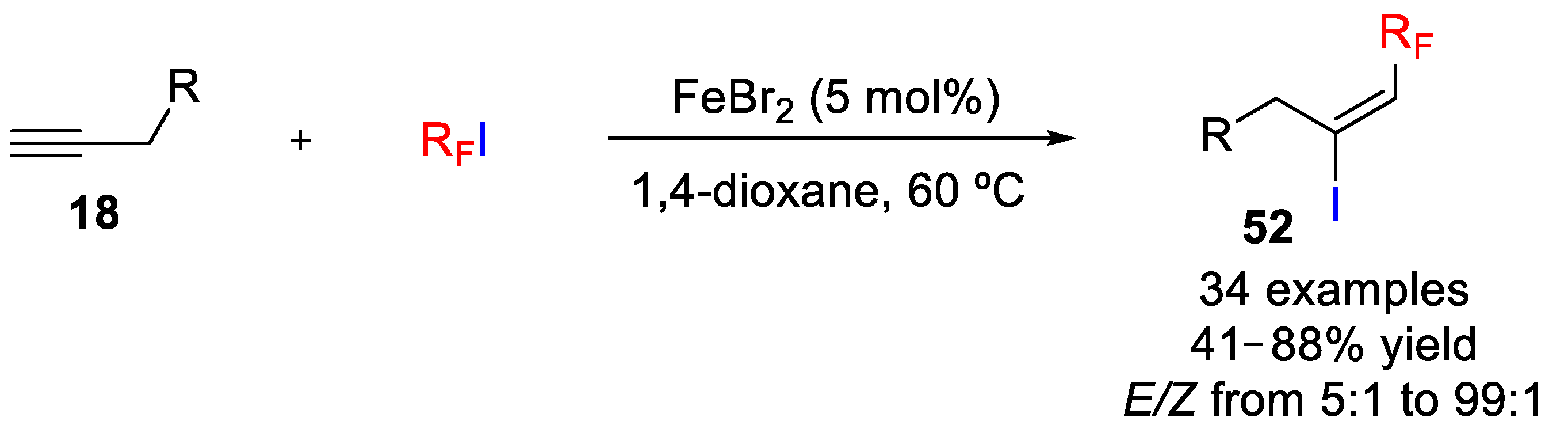{kind=link}
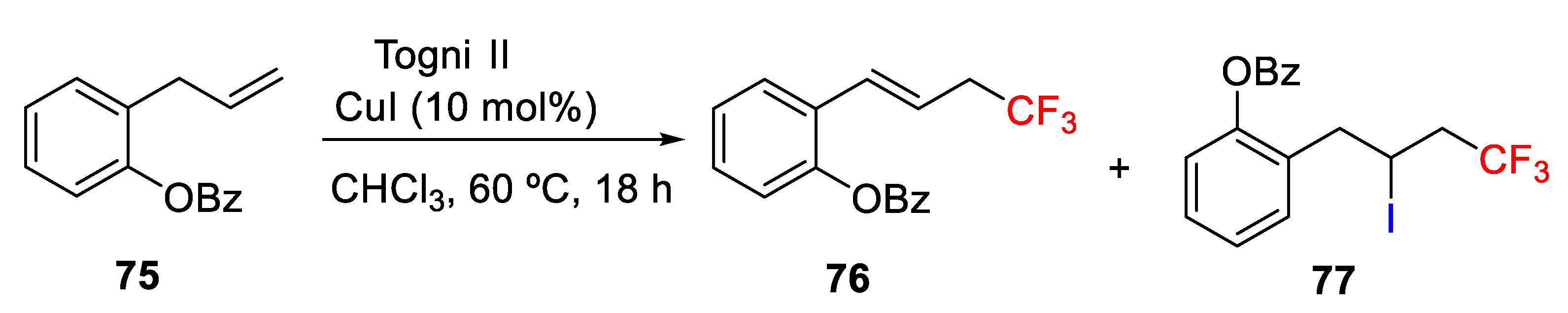{kind=link}
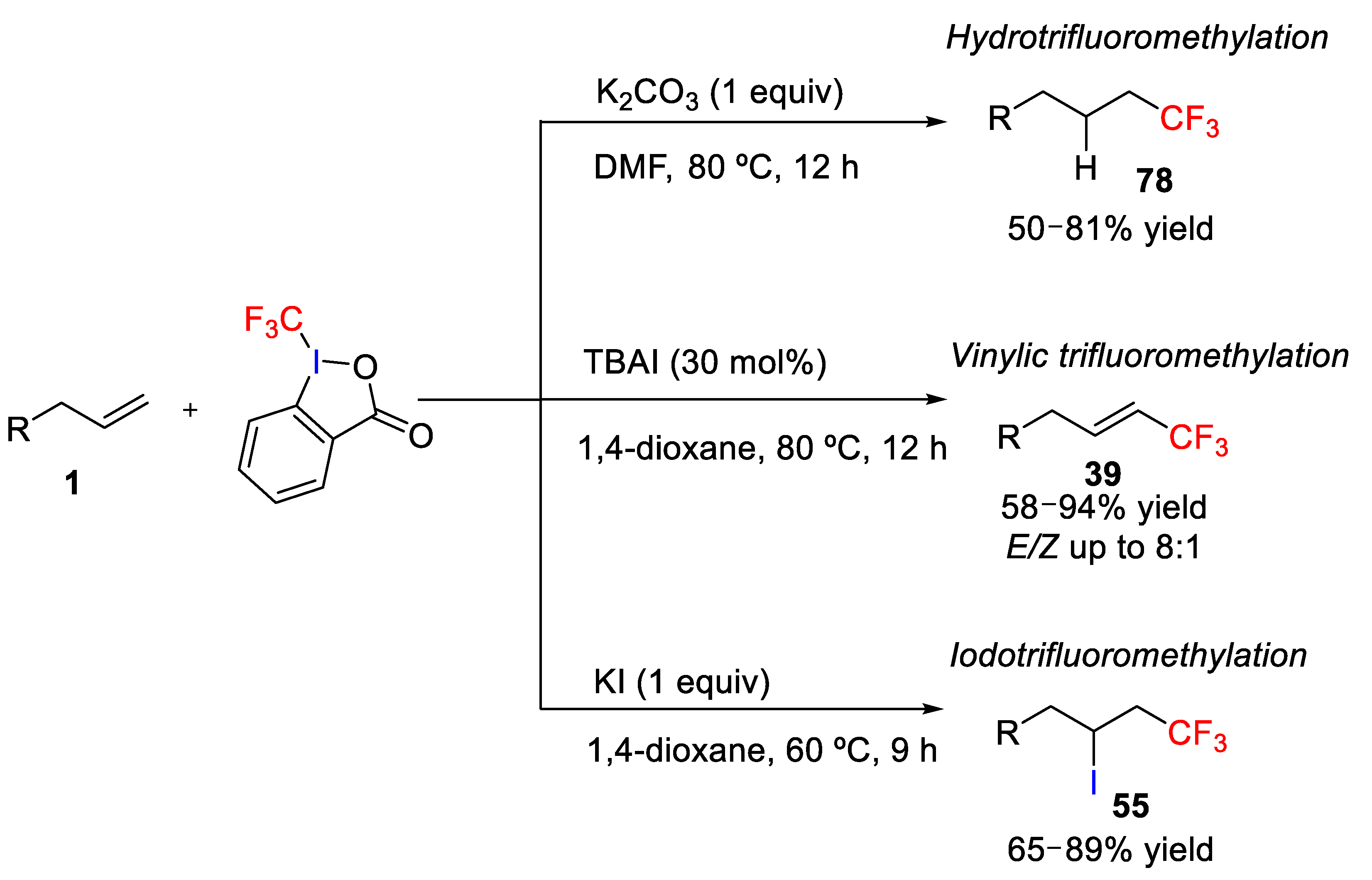{kind=link}
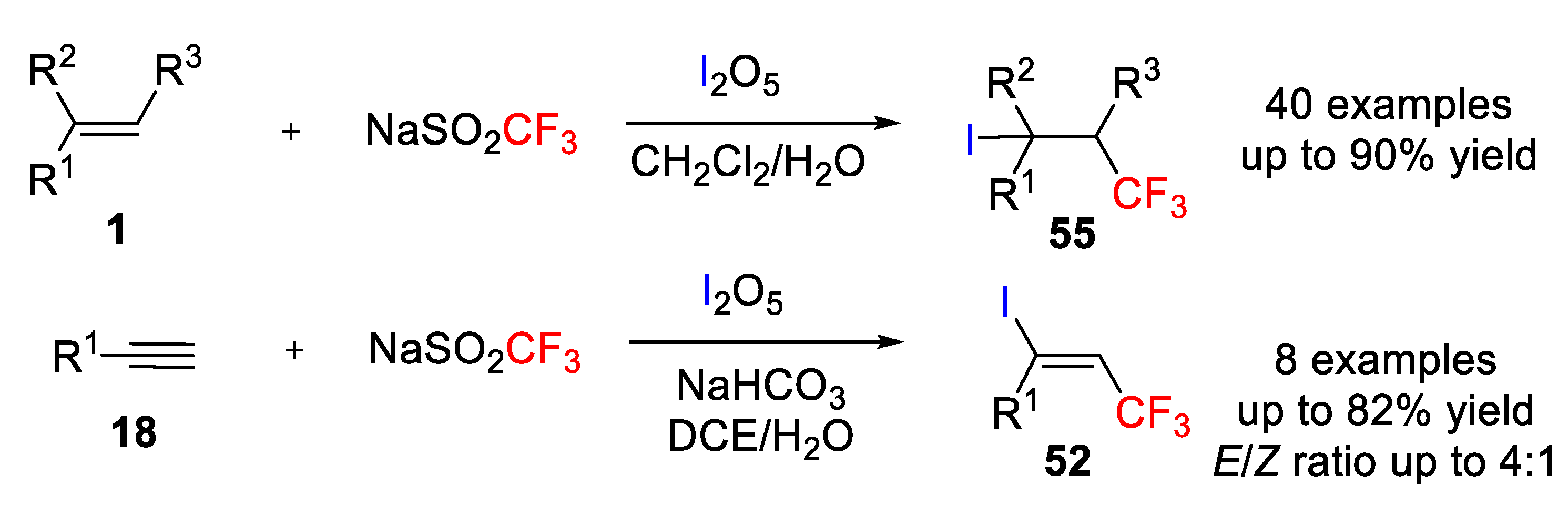{kind=link}
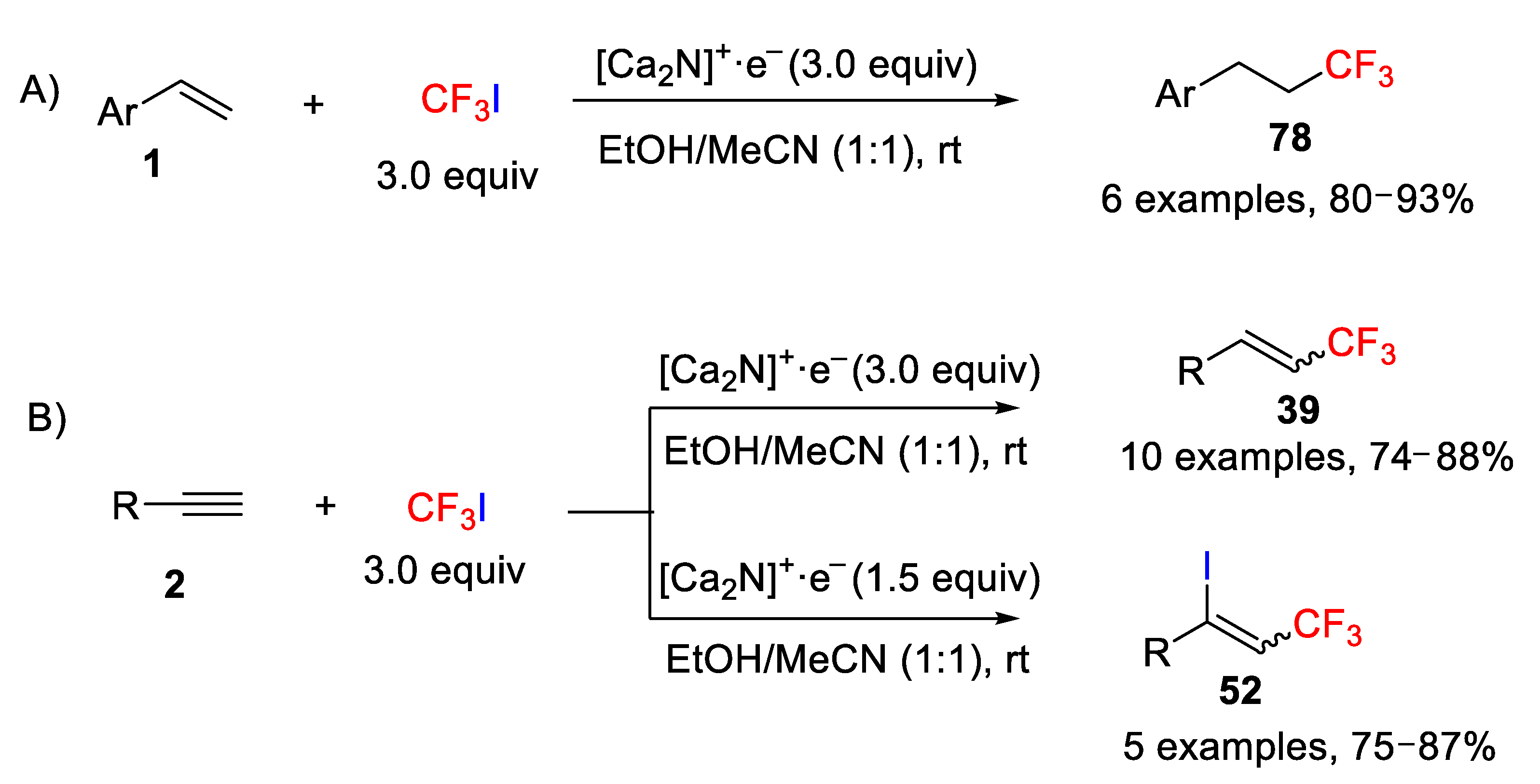{kind=link}
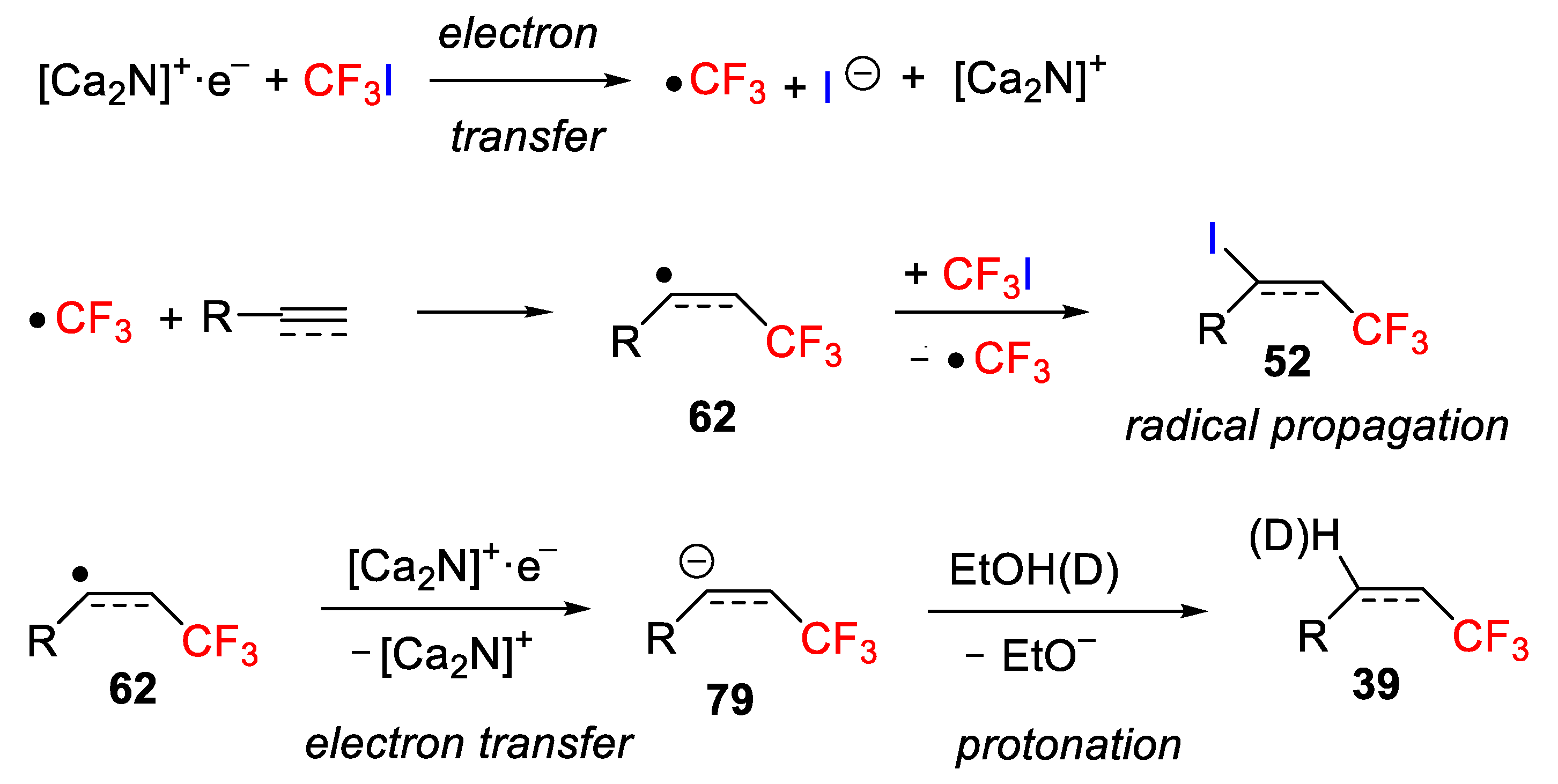{kind=link}
{kind=link}
{kind=link}
{kind=link}
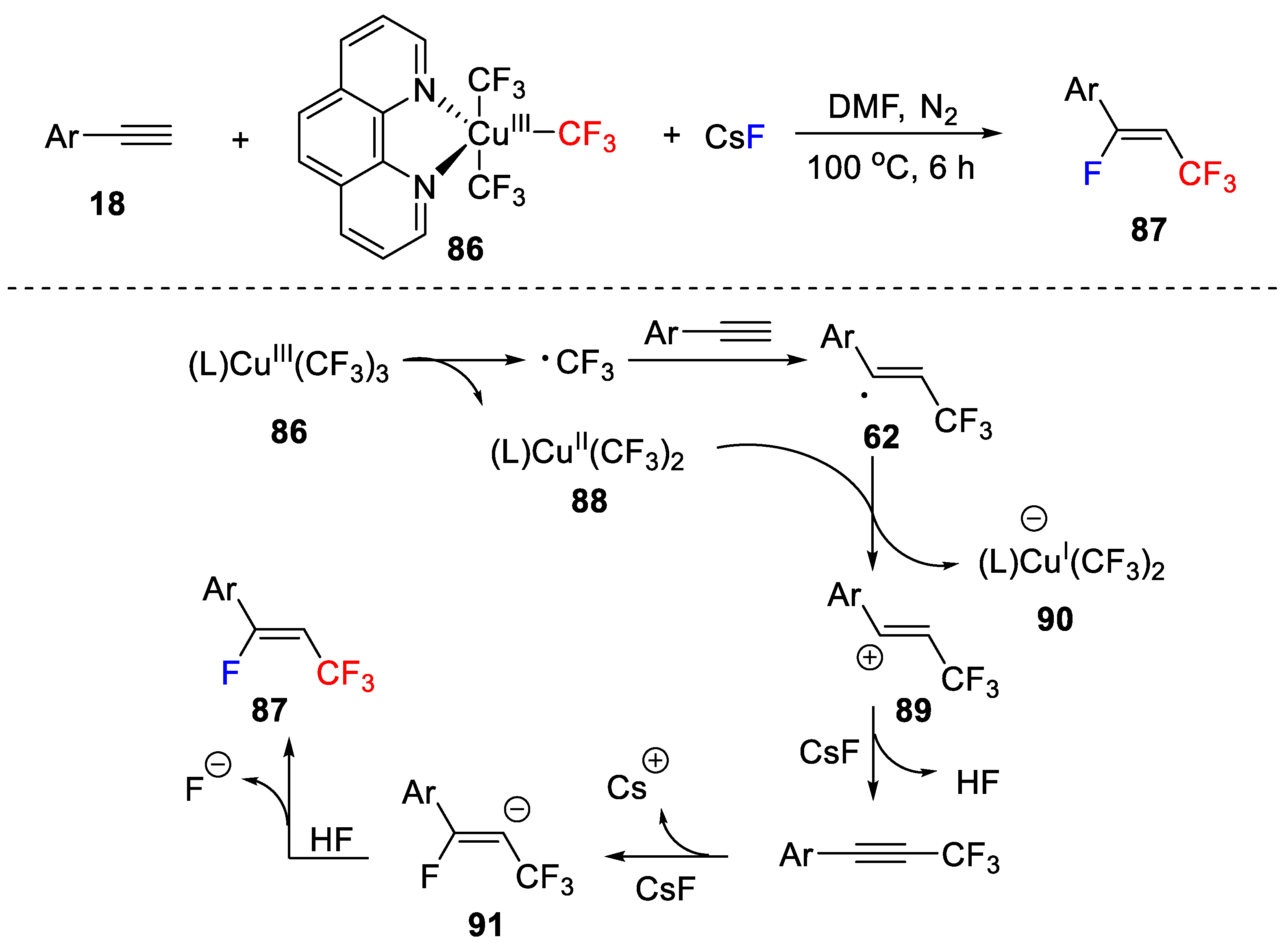{kind=link}
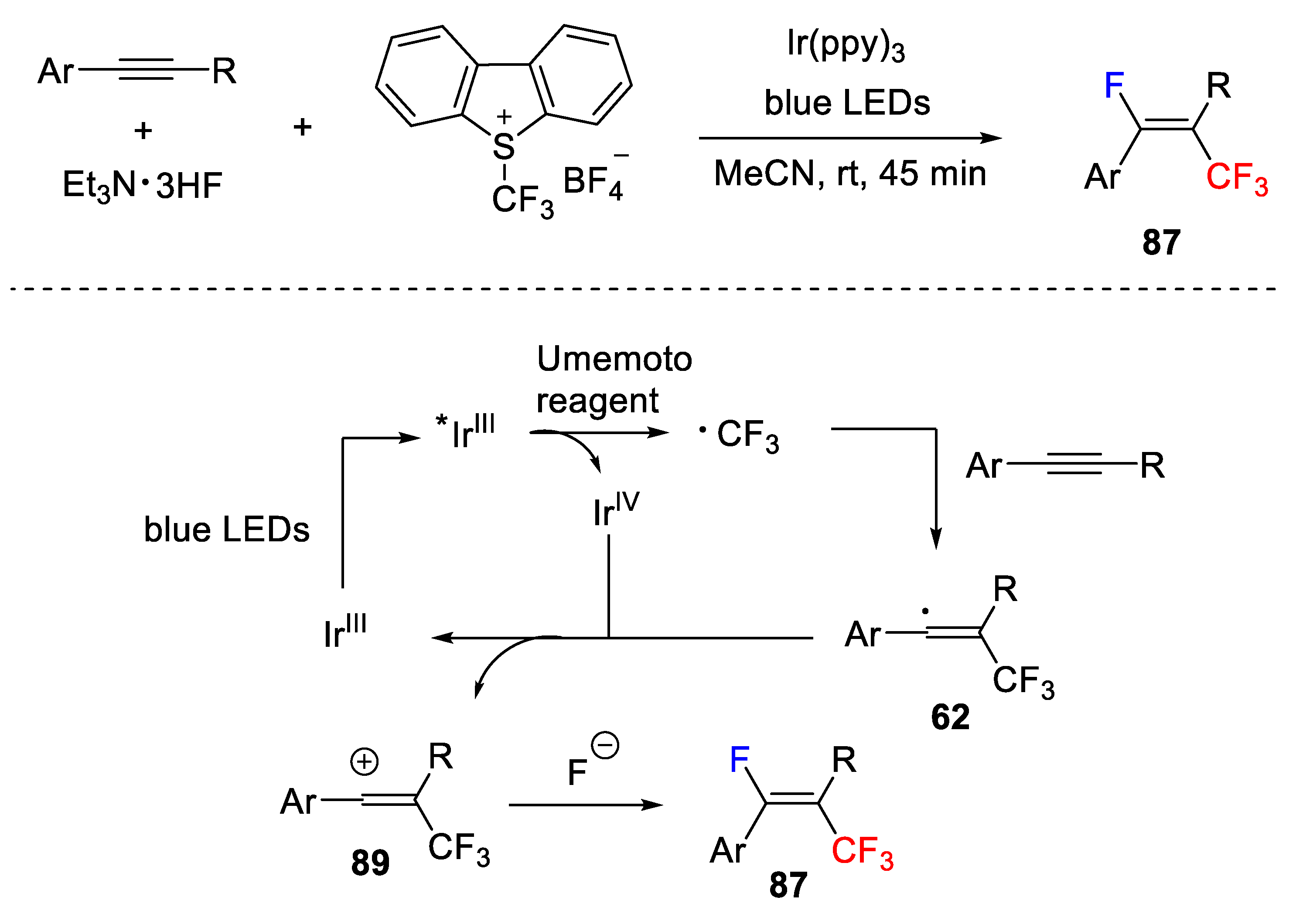{kind=link}
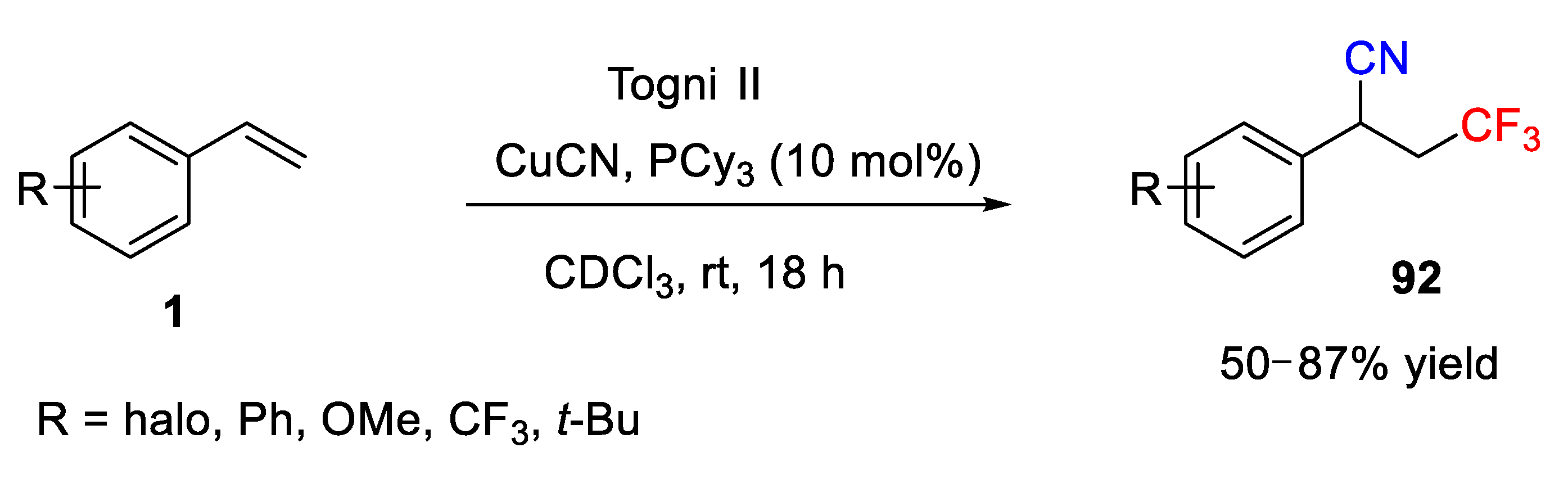{kind=link}
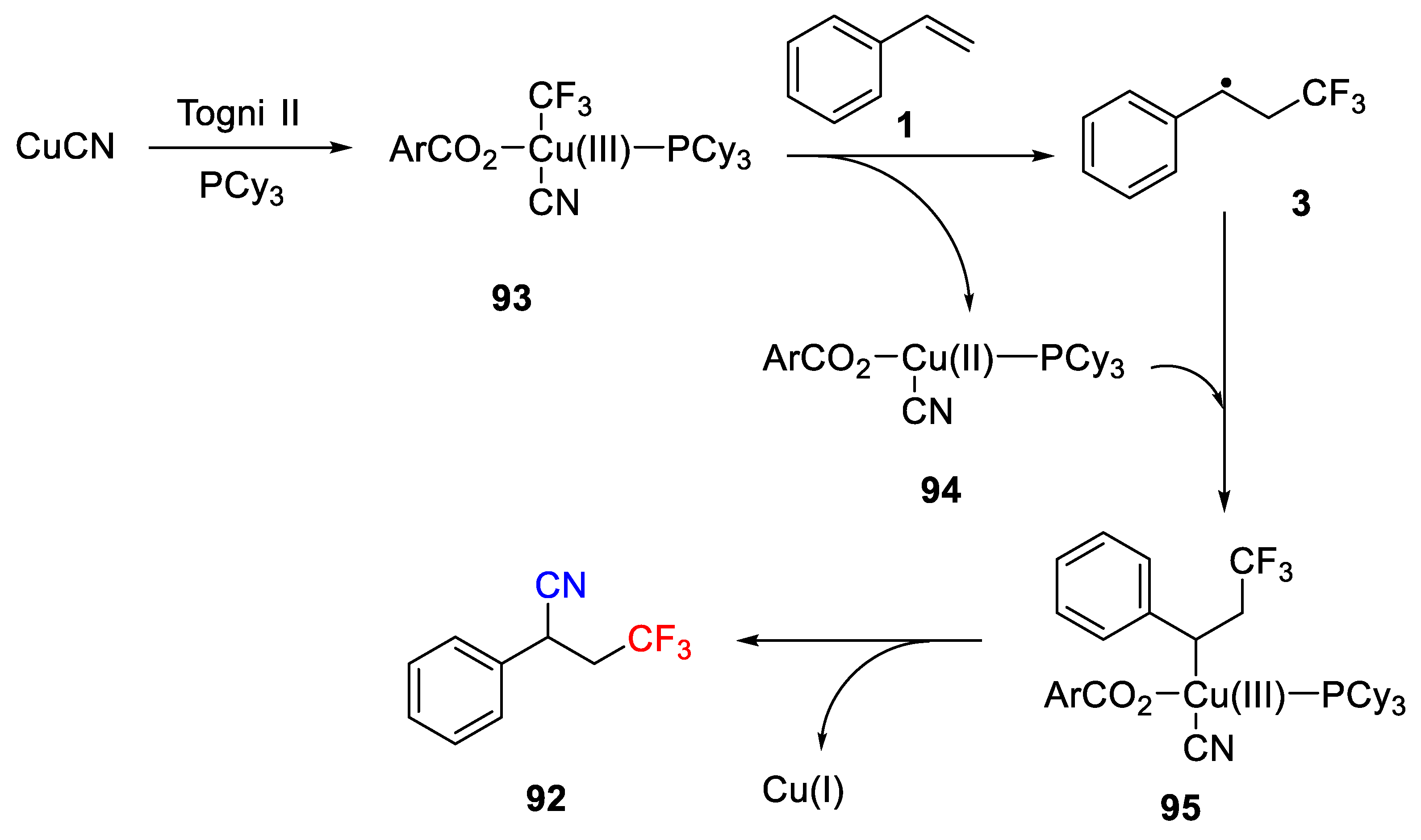{kind=link}
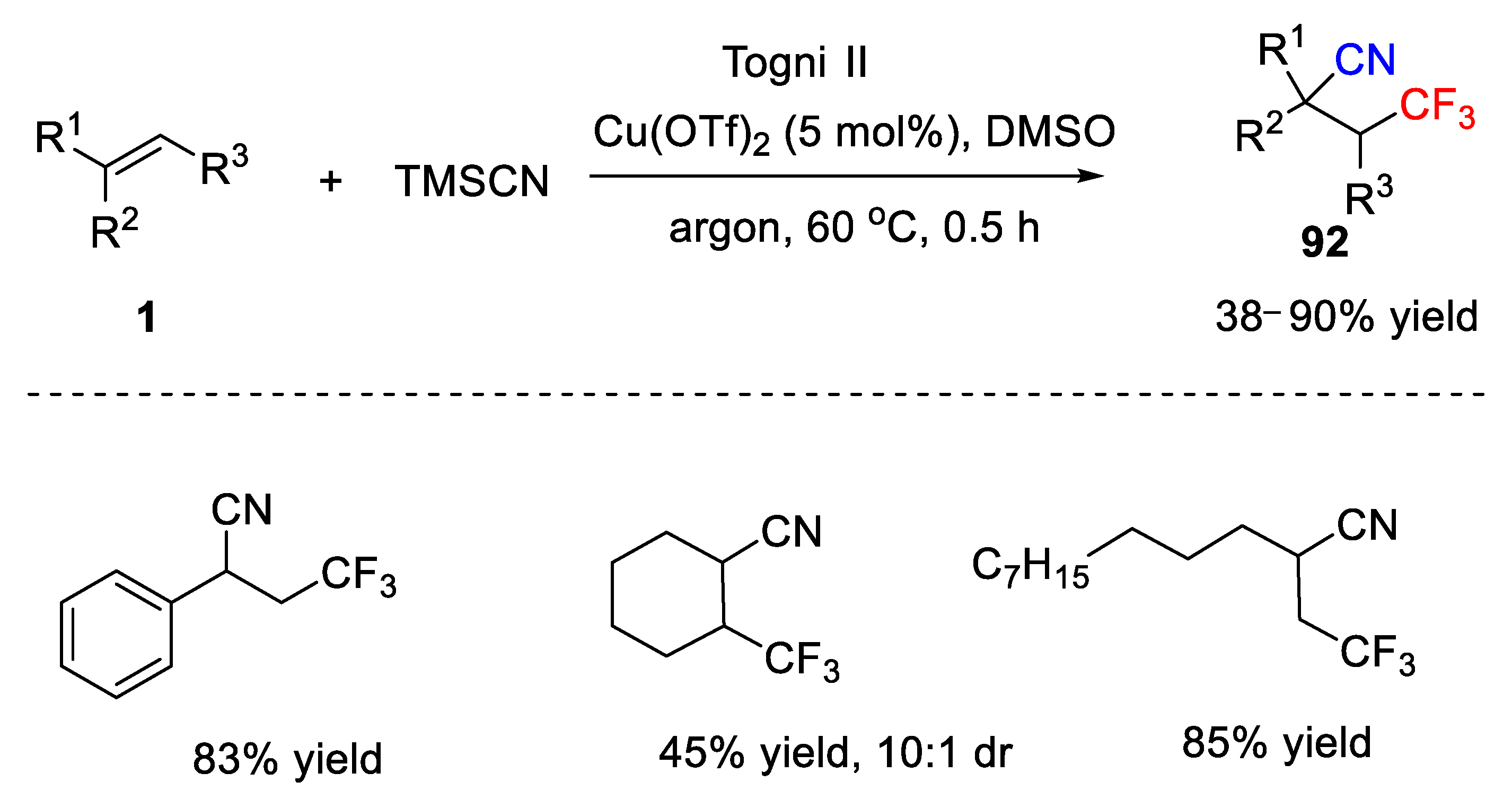{kind=link}
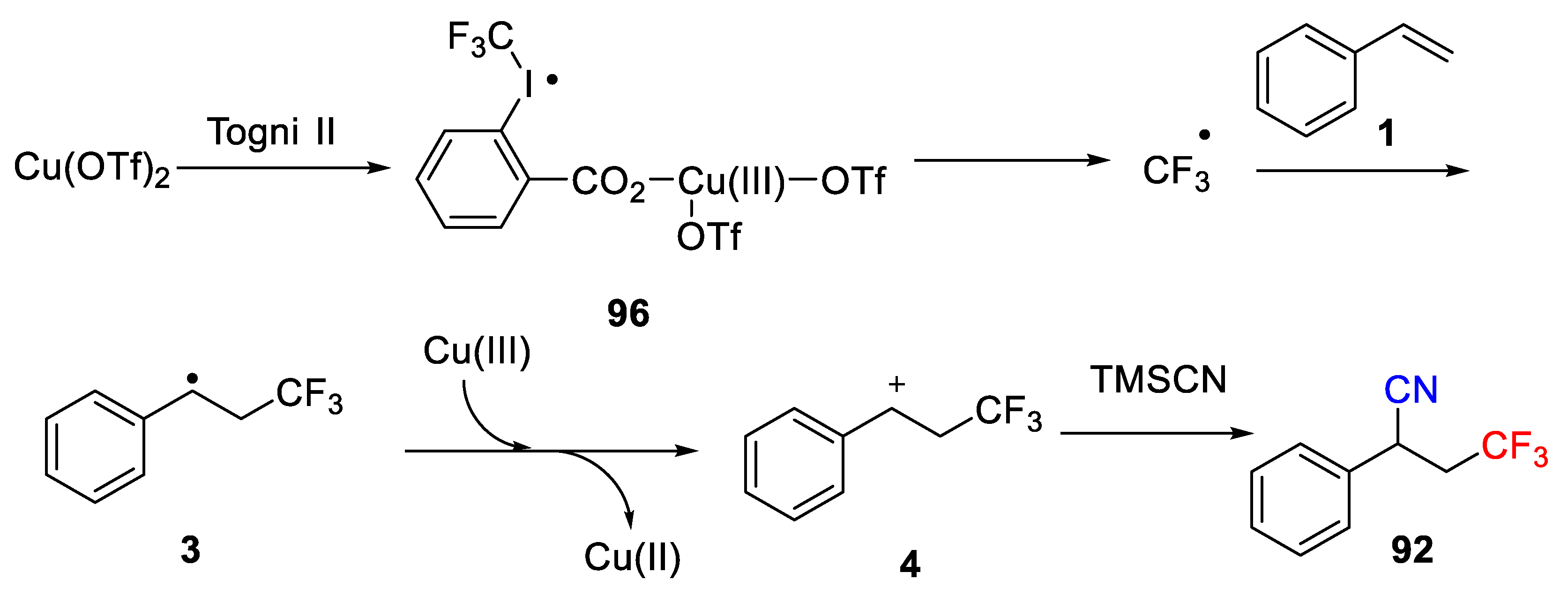{kind=link}
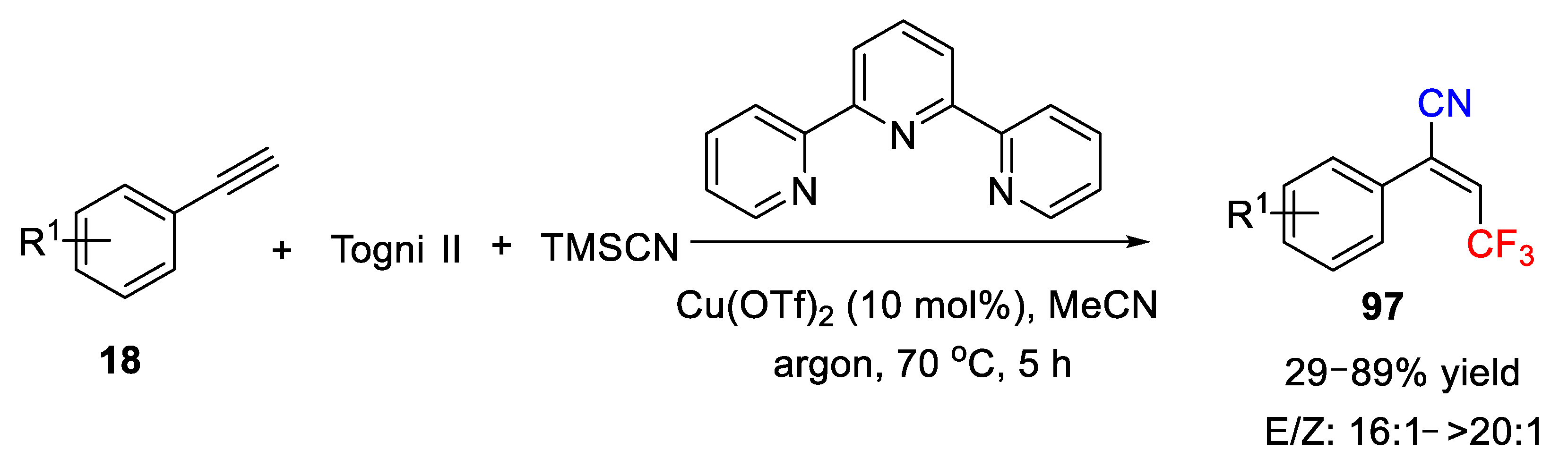{kind=link}
{kind=link}
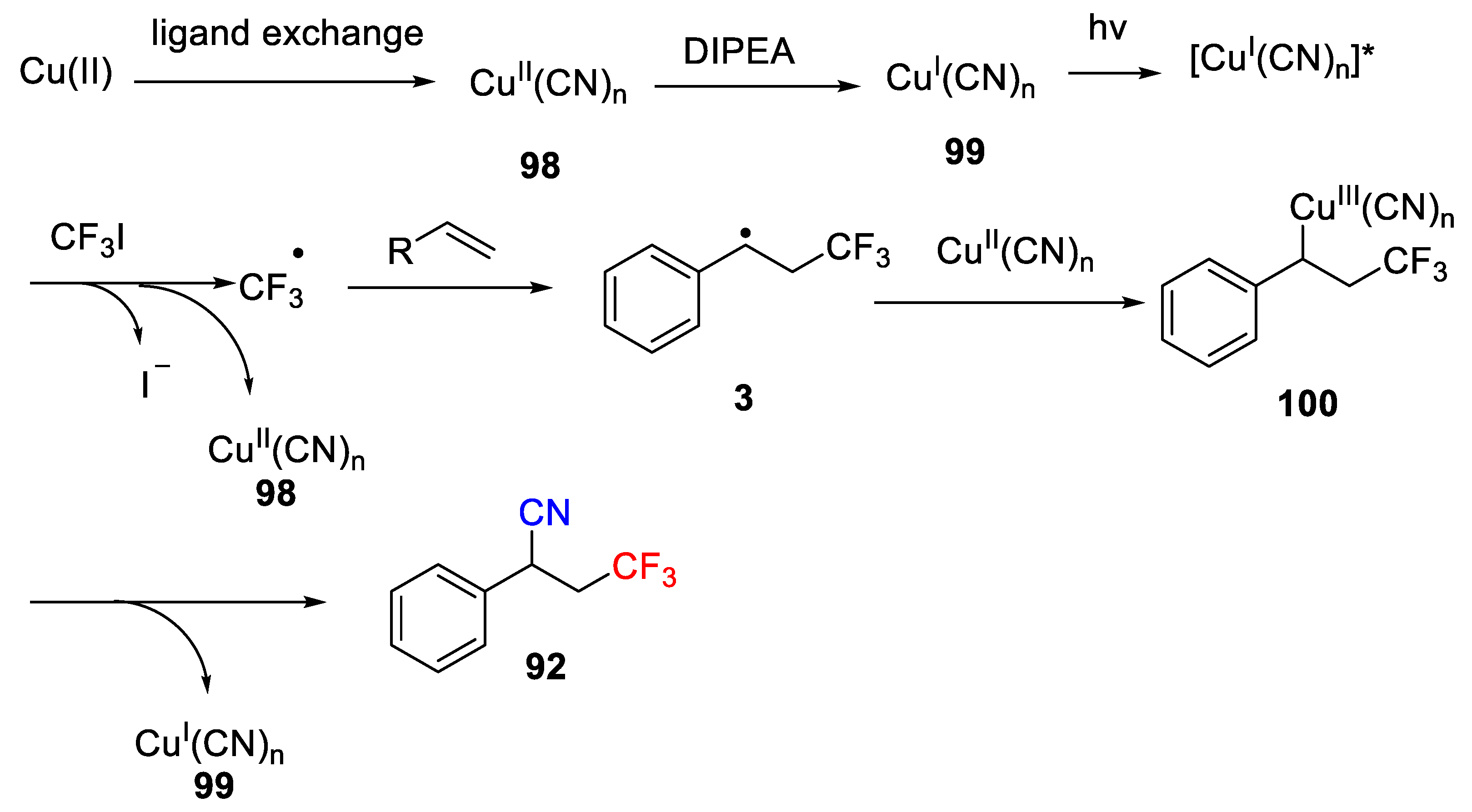{kind=link}
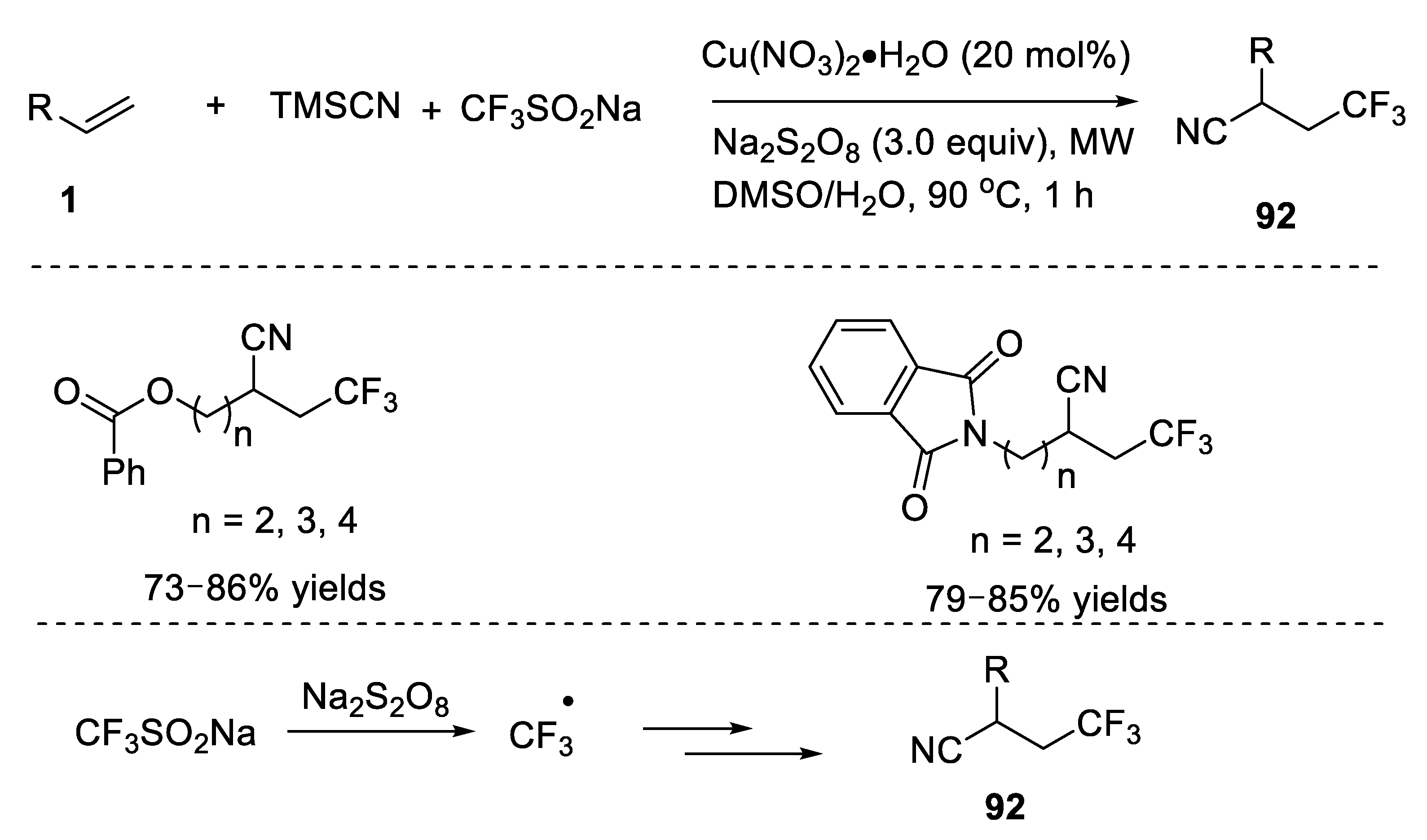{kind=link}
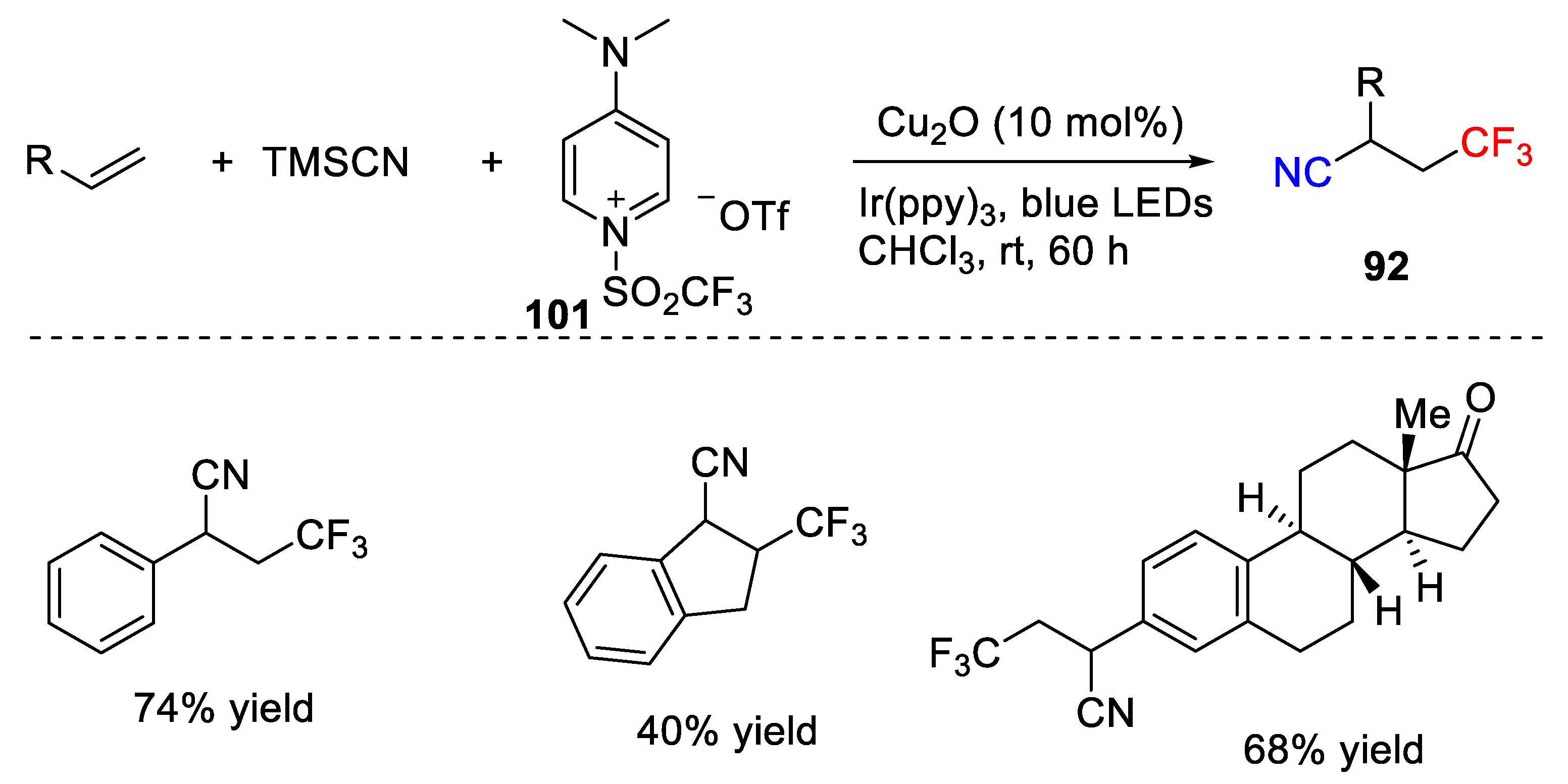{kind=link}
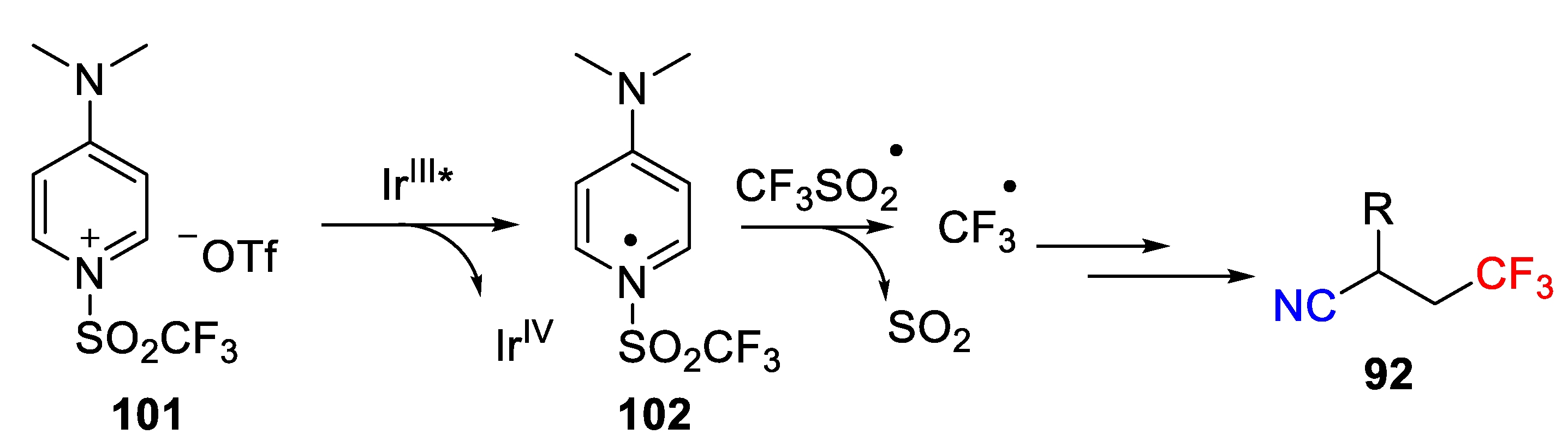{kind=link}
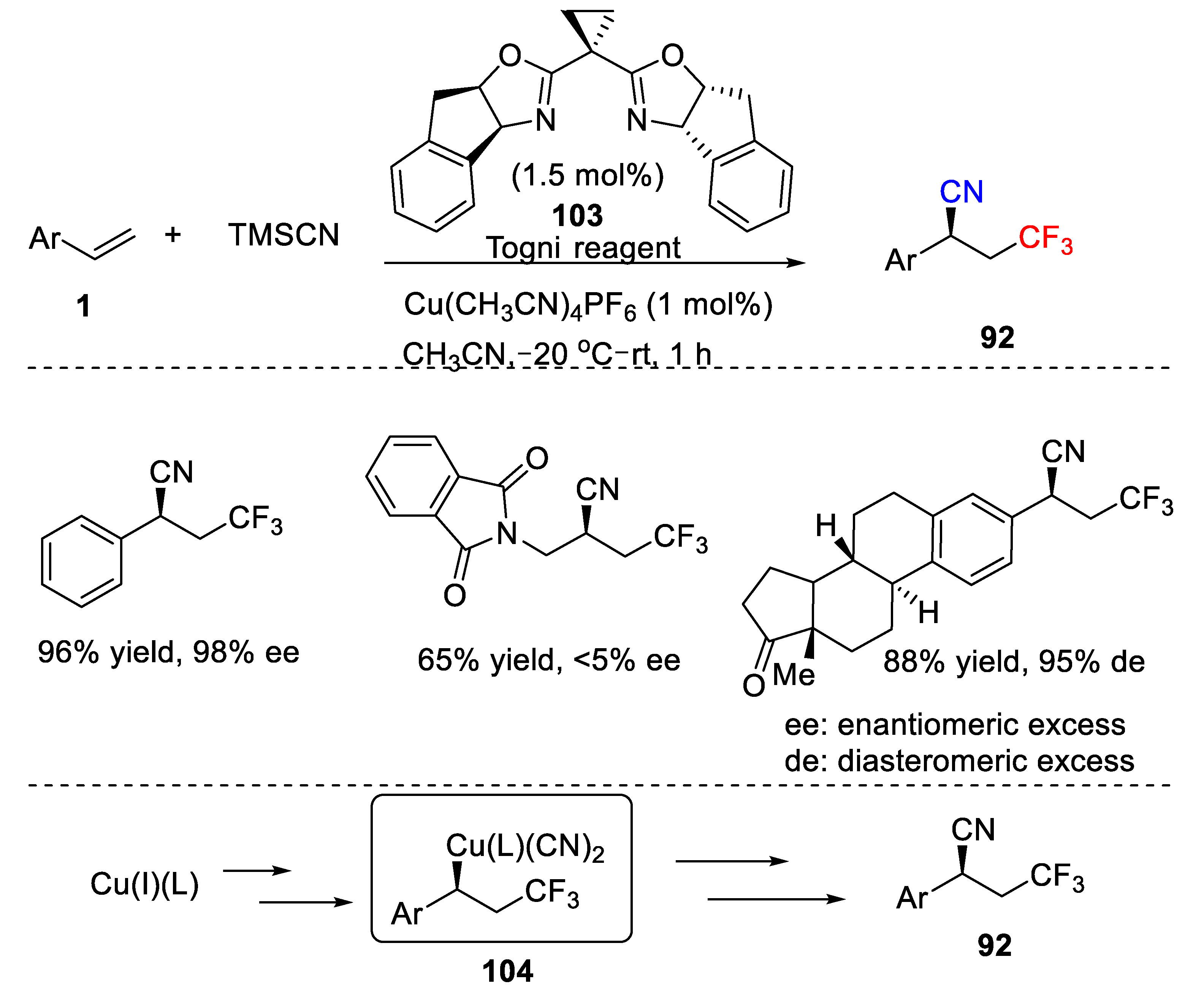{kind=link}
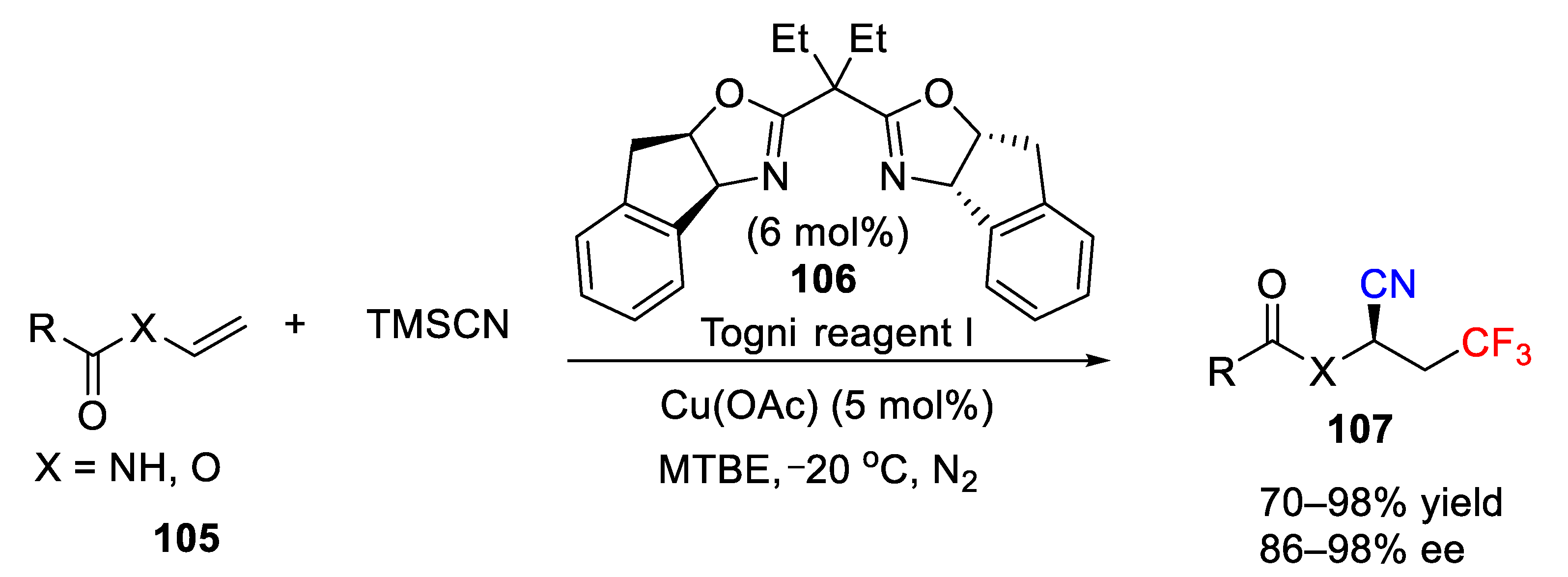{kind=link}















































































