3. Materials and Methods
3.1. General
All reagents and solvents were of commercial quality and were used without further purification, unless described otherwise. Unless otherwise stated, all reactions were carried out under an inert atmosphere of argon.
1H,
13C, and
31P-NMR spectra were collected on a Varian Mercury 400 MHz or Bruker Avance III 500 MHz spectrometer. All
1H and
13C NMR assignments are based on gCOSY, gHMBC, gHSQC, and DEPT−135 experiments. Abbreviations for splitting patterns are as follows: br, broad; s, singlet; d, doublet; t, triplet; m, multiplet. Coupling constants are given in hertz (Hz). High resolution time-of-flight mass spectra were obtained on a Bruker Daltonics micrOTOF mass spectrometer using electrospray ionization (ESI). The purity of new tested compounds was determined to be ≥95% by analytical HPLC (see
Supplementary Materials). Analytical HPLC analyses were carried out on a Waters 2695 Alliance module equipped with a Waters 2996 photodiode array detector (210–350 nm). The chromatographic system consisted of a Hichrom Guard column for HPLC and a Phenomenex Synergi 4 μm MAX-RP 80A column (150 mm × 4.60 mm), with elution at 1 mL/min with either (a) isocratic ion-pair buffer: 0.17% (
m/
v) cetrimide and 45% (
v/
v) phosphate buffer (pH 6.4) in MeOH or (b) a gradient of 0.05M Triethylammonium bicarbonate (TEAB):MeCN (95:5 → 35:65
v/
v). TEAB was prepared by bubbling CO
2 (g) through a 0.05M solution of triethylamine in MilliQ water to pH ≤ 8. “MilliQ” water refers to purified water from a MilliQ
® Reference Water Purification system, resistivity of 18.2 MΩ.cm (at 25 °C). Semi-preparative HPLC was performed on a Waters 2525 pump with manual FlexInject using a Phenomenex Gemini column (5u, C18, 110 Å, 250 × 10.00 mm), eluted at 5 mL min
−1 with a gradient of 0.05M TEAB:MeCN (95:5 → 35:65
v/
v). Synthetic phosphates were assayed and quantified by the Ames phosphate test [
45]. Non-phosphate final compounds were quantified by quantitative
1H-NMR. Note the use of 0.5% pyridine in chromatography systems with acid sensitive functional groups (e.g. phosphates protected with tert-butyl ethers) or free monophosphates. This system was used (rather than triethylamine) as it was less basic and therefore prevented decomposition of the analogues. Pyridine was evaporated from the TLC plate using a heat gun before visualization under UV light.
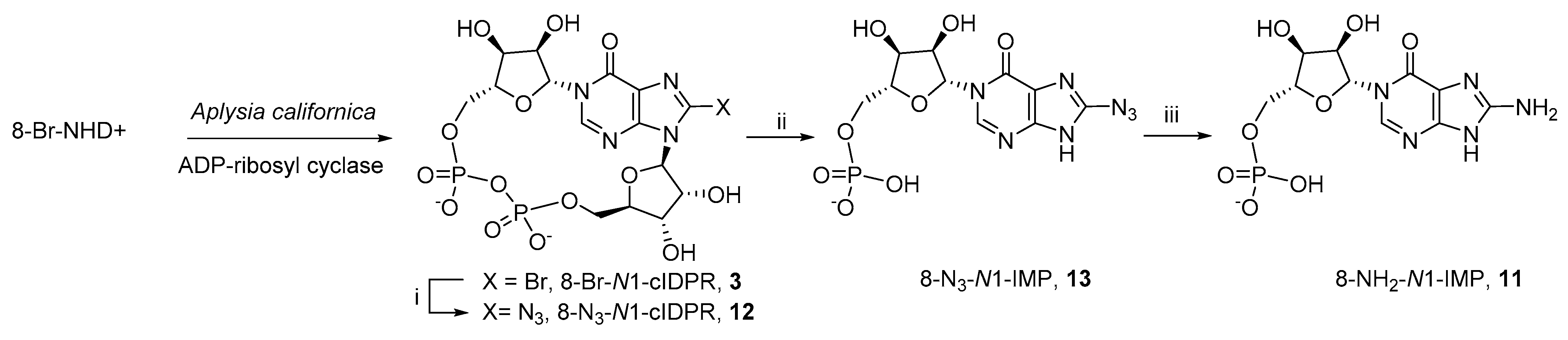
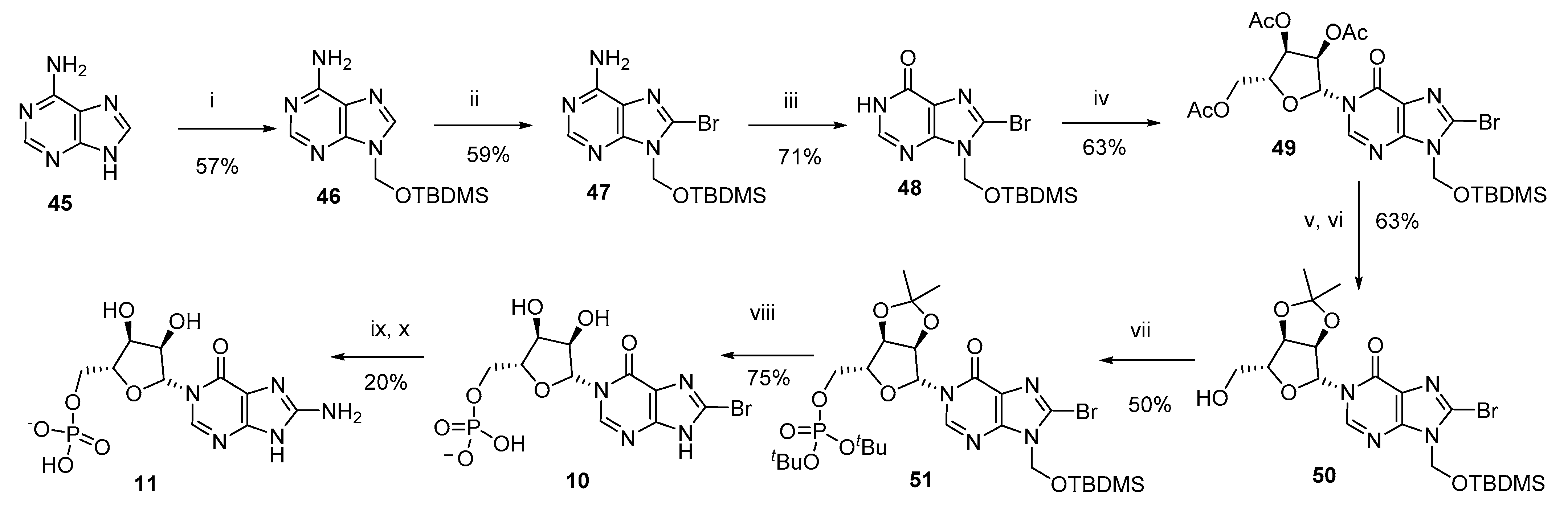
3.2. Total Synthesis of 8-NH2-N1-IMP (11)
N9-tert-Butyldimethylsilyloxymethyl-adenine (46)–Prepared from adenine following the method of Lang et al. [
43].
N9-tert-Butyldimethylsilyloxymethyl−8-bromoadenine (47)–To diisopropylamine (2.93 mL, 20.94 mmol) in THF (10 mL) at −78 °C was added n-butyllithium (8.55 mL, 2.5 M solution, 21.37 mmol), dropwise. After 1 h, a solution of N9-tert-Butyldimethylsilyloxymethyl-adenine (46, 1.17 g, 4.19 mmol) in THF (40 mL) was added dropwise and stirring continued for a further 1 h. Br2 (837 µL, 16.76 mmol) was added dropwise and the solution allowed to warm to rt and stirred for 16 h. The reaction was quenched by addition of NaOAc/AcOH (pH 4, 1N aq., 2 mL) and all solvents evaporated. The residue was taken up in DCM:H2O (1:1 v/v, 100 mL), the organic layer separated, washed with brine, dried (Na2SO4) and evaporated to dryness. The crude material was purified by column chromatography on silica gel eluting with DCM:Acetone (1:0 → 0:1 v/v) to afford the title compound (879 mg, 59%) as an amorphous cream solid; Rf = 0.59 (DCM/Acetone 1:1 v/v); 1H-NMR (400 MHz, CDCl3) δ 8.32 (s, 1H, H−2), 5.94 (br s, 2H, NH2), 5.71 (s, 2H, CH2OTBDMS), 0.86 (s, 9H) 0.10 (s, 6H) (15H, TBDMS); 13C-NMR (100 MHz, CDCl3) δ 154.1, 153.2, 151.03, 127.3, 119.7, 67.0, 25.5 (3C), 18.0 and −5.3 (2C); HRMS (ES+) calcd for C12H21N5OSi79Br 358.0693 (M+H)+ found 358.0699, calcd for C12H21N5OSi81Br 360.0678 (M+H)+ found 360.0691.
N9-tert-Butyldimethylsilyloxymethyl−8-bromohypoxanthine (48)–N9-tert-Butyldimethylsilyloxymethyl−8-bromoadenine (47, 600 mg, 1.67 mmol) was taken up in acetic acid-water (46 mL, 20:3 v/v). NaNO3 (1.39 g, 20.09 mmol) was added in one portion and the resulting solution stirred at 50 °C for 16 h. All solvents were evaporated and the residue partitioned between DCM and H2O, and the organic layer washed with NaHCO3 (sat. aq.), then brine, dried (Na2SO4) and evaporated to dryness. The residue was purified by column chromatography on silica gel eluting with DCM/Acetone (1:0 → 0:1 v/v) to afford the title compound (430 mg, 71%) as a pale orange amorphous solid; Rf = 0.57 (DCM/Acetone 1:1 v/v); 1H-NMR (400 MHz, CDCl3) δ 13.15 (br s, 1H, NH), 8.31 (s, 1H, H−2), 5.71 (s, 1H), 5.70 (s, 1H) (2H, CH2OTBDMS), 0.87 (s, 9H) 0.12 (s, 6H) (15H, TBDMS); 13C-NMR (100 MHz, CDCl3) δ 158.0, 150.2, 145.8, 126.4, 124.6, 67.5, 25.5 (3C), 18.0 and −5.3 (2C); HRMS (ES+) calcd for C12H20N4O2Si79Br 359.0539 (M+H)+ found 359.0544, calcd for C12H20N4O2Si81Br 361.0519 (M + H)+ found 361.0536.
N1-(2′,3′,5′-Tri-O-acetyl-β-D-ribofuranosyl)-N9-tert-butyldimethylsilyloxymethyl−8-bromohypoxanthine (49)–To N9-tert-butyldimethylsilyloxymethyl−8-bromohypoxanthine (48, 480 mg, 1.336 mmol) in DCM (5 mL) was added DBU (599 μL, 4.008 mmol). After 30 min, 1,2,3,5-tetra-O-acetyl-β-d-ribofuranose (468 mg, 1.47 mmol) was added and the solution cooled to −78 °C. Trimethylsilyl trifluoromethanesulfonate (967 μL, 5.344 mmol) was added dropwise and the solution stirred for a further 45 min before warming to rt. After 2 h, NaHCO3 (satd aq) was added and the crude material extracted into DCM (×3). The combined organic fractions were dried (Na2SO4), and solvent was evaporated under reduced pressure. The residue was purified by column chromatography on silica gel eluting with PE/EtOAc (1:0 → 0:1 v/v) to afford the title compound (517 mg, 63%) as a colourless glass; Rf = 0.64 (PE:EtOAc 1:3 v/v); 1H-NMR (400 MHz, CDCl3) δ 8.20 (s, 1H, H−2), 6.35 (d, J = 4.5 Hz, 1H, H−1′), 5.64 (d, J = 9.8 Hz, 1H), 5.62 (d, J = 9.8 Hz, 1H) (2H, CH2OTBDMS), 5.46−5.41 (m, 2H, H−2′, H−3′), 4.43−4.33 (m, 3H, H−4′, 2 × H−5′), 2.15 (s, 3H, AcetylCH3), 2.10 (s, 3H, AcetylCH3), 2.06 (s, 3H, AcetylCH3), 0.86 (s, 9H) 0.12 (s, 3H) and 0.11 (s, 3H) (15H, TBDMS); 13C-NMR (100 MHz, CDCl3) δ 170.2, 169.6 (2C), 154.7, 148.4, 144.5, 126.1, 123.9, 87.4, 80.4, 74.2, 70.1, 67.3, 62.9, 25.4 (3C), 20.7, 20.44, 20.38, 18.0, −5.2, −5.3; HRMS (ES+) calcd for C23H34N4O9Si79Br 617.1273 (M + H)+ found 617.1281, calcd for C23H34N4O9Si81Br 619.1252 (M + H)+ found 619.1241.
N1-(2′,3′-O-Isopropylidene-β-D-ribofuranosyl)-N9-tert-butyldimethylsilyloxymethyl−8-bromohypoxanthine (50)–N1-(2′,3′,5′-Tri-O-acetyl-β-d-ribofuranosyl)-N9-tert-butyldimethylsilyl oxymethyl−8-bromohypoxanthine (49, 370 mg, 0.599 mmol) was taken up in MeOH (5.0 mL) in a pressure tube. The solution was cooled to 0 °C in an ice-water bath and NH3 (g) bubbled through the solution to saturation. The tube was sealed and the resulting solution stirred at rt. When complete by TLC, the solvents were removed by evaporation under reduced pressure and the residue was purified by column chromatography on silica gel eluting with DCM/Acetone (1:0 → 0:1 v/v) to afford N1-(β-D-Ribofuranosyl)-N9-tert-butyldimethylsilyloxymethyl−8-bromohypoxanthine as an amorphous white solid, Rf = 0.21 (DCM:Acetone 1:1 v/v), which was used directly in the next step. HRMS (ES+) calcd for C17H27N4O6Si79BrNa 513.0775 (M+Na)+ found 513.0780, calcd for C17H27N4O6Si81BrNa 515.0755 (M + Na)+ found 515.0758.
To crude N1-(β-d-ribofuranosyl)-N9-tert-butyldimethylsilyloxymethyl−8-bromo-hypoxanthine in 2,2-dimethoxypropane-acetone (1:4 v/v, 5 mL) was added para-toluenesulfonic acid (114 mg, 0.599 mmol). After 1 h, DCM and NaHCO3 (satd. aq.) were added and the aqueous phase extracted with DCM (×3). The combined organic extracts were evaporated to dryness and purified by column chromatography on silica gel eluting with PE/EtOAc (1:0 → 0:1 v/v) to afford the title compound (200 mg, 63% over two steps) as a colourless glass. Rf = 0.65 (PE:EtOAc 1:3 v/v); 1H-NMR (400 MHz, CDCl3) δ 8.05 (s, 1H, H−2), 5.73 (d, J = 2.8 Hz, 1H, H−1′), 5.64 (d, J = 9.8 Hz, 1H), 5.62 (d, J = 9.8 Hz, 1H) (2H, CH2OTBDMS), 5.27 (dd, J = 6.5, 2.8 Hz, 1H, H−2′), 5.12 (dd, J = 6.5, 3.5 Hz, 1H, H−3′), 4.34 (ddd, J = 6.1, 5.6, 3.5 Hz, 1H, H−4′), 3.91 (ddd, J = 11.9, 5.6, 3.3 Hz, 1H, H−5′a), 3.83 (ddd, J = 11.9, 7.8, 6.1 Hz, 1H, H−5′b), 3.40 (dd, J = 7.8, 3.3 Hz, 1H, 5-OH), 1.57 (s, 3H, CHCH3), 1.34 (s, 3H, CHCH3), 0.86 (s, 9H) 0.11 (s, 3H) and 0.10 (s, 3H) (15H, TBDMS); 13C-NMR (100 MHz, CDCl3) δ 155.2, 148.8, 146.9, 126.5, 124.6, 114.2, 96.9, 88.0, 83.5, 80.6, 67.4, 62.8, 27.2, 25.4, 25.1, 17.9, −5.2 (2C); HRMS (ES+) calcd for C20H32N4O6Si79Br 531.1269 (M+H)+ found 531.1248, calcd for C20H32N4O6Si81Br 533.1249 (M+H)+ found 533.1246.
N1-[2′,3′-O-Isopropylidene−5′-O-(di-tert-butyl)-phosphoryl-β-d-ribofuranosyl]-N9-tert-butyldimethylsilyloxymethyl−8-bromohypoxanthine (51)–To N1-(2′,3′-O-isopropylidene-β-d-ribofuranosyl)-N9-tert-butyldimethylsilyloxymethyl−8-bromohypoxanthine (50, 80 mg, 0.151 mmol) in DCM (0.8 mL) was added 5-phenyl−1H-tetrazole (44 mg, 0.301 mmol) and the solution cooled to 0 °C. Di-tert-butyl N,N-diisopropylphosphoramidite (71 µL, 0.227 mmol) was added dropwise and the solution stirred at rt until phosphitylation was complete by TLC. After cooling to 0 °C, triethylamine (126 µL, 0.906 mmol) and H2O2 (30% in H2O, 33 µL, 0.375 mmol) were added and the solution stirred at rt until oxidation was complete. The reaction was diluted with DCM and washed with NaHCO3 (satd. aq.), dried over Na2SO4 and purified by column chromatography on silica gel eluting with PE/EtOAc (1:0 → 0:1 v/v + 0.5% pyridine in each solvent) to afford the title compound (55 mg, 50%) as a colourless glass; Rf = 0.46 (PE:EtOAc 1:3 v/v)–note there is no change in retention time compared to starting material (after oxidation); 1H-NMR (500 MHz, CDCl3) δ 8.00 (s, 1H, H−2), 5.90 (d, J = 1.8 Hz, 1H, H−1′), 5.54 (d, J = 10.4 Hz, 1H), 5.52 (d, J = 10.4 Hz, 1H) (2H, CH2OTBDMS), 4.96 (dd, J = 6.4, 1.8 Hz, 1H, H−2′), 4.88 (dd, J = 6.4, 4.1 Hz, 1H, H−3′), 4.32 (ddd, J = 6.5, 5.9, 4.1 Hz, 1H, H−4′), 4.14 (ddd, J = 11.2, 6.5, 4.2 Hz, 1H, H−5′a), 4.08 (ddd, J = 11.2, 6.8, 5.9 Hz, 1H, H−5′b), 1.47 (s, 3H, CHCH3), 1.354 (s, 9H, OtBu), 1.346 (s, 9H, OtBu), 1.23 (s, 3H, CHCH3), 0.76 (s, 9H) 0.01 (s, 3H) and 0.00 (s, 3H) (15H, TBDMS); 13C-NMR (125MHz, CDCl3) δ 154.8, 148.7, 146.2, 126.1, 124.4, 114.4, 94.1, 86.8 (d, J = 7.6 Hz), 85.1, 82.7 (d, J = 7.5 Hz), 81.3, 67.4, 66.4 (d, J = 6.3), 29.84 (3C, d, J = 4.1 Hz), 29.81 (3C, d, J = 3.7 Hz), 27.2, 25.5 (3C), 25.3, 18.0, −5.2 (2C); HRMS (ES+) calcd for C28H48N4O9Si79BrNa 745.2004 (M+Na)+ found 745.1990, calcd for C28H48N4O9Si 81BrNa 747.1983 (M+Na)+ found 747.1964.
N1-(5′-O-Phosphoryl-β-D-ribofuranosyl)−8-bromohypoxanthine (8-Br-N1-IMP, 10)–N1-[2′,3′-O-Isopropylidene−5′-O-(di-tert-butyl)-phosphoryl-β-d-ribofuranosyl]-N9-tert-butyldimethylsilyloxymethyl−8-bromohypoxanthine (51, 55 mg, 76 µmol) was treated with TFA (50% aq., 4 mL) for 16 h. All solvents were evaporated and the residue evaporated from MeOH × 3 to give the title compound (24 mg, 75%). 1H-NMR and MS data were as previously described39 and it was used in the next step without further purification.
N1-(5′-O-Phosphoryl-β-D-ribofuranosyl)−8-aminohypoxanthine (8-NH2-N1-IMP, 11)–To N1-(5′-O-phosphoryl-β-D-ribofuranosyl)−8-bromohypoxanthine (10, 24 mg, 56 µmol) in DMF (1 mL) was added TMSN3 (50 µL) and the solution stirred at 70 °C for 16 h. All solvents were evaporated and the crude N1-(5′-O-Phosphoryl-β-D-ribofuranosyl)−8-azidohypoxanthine (8-N3-N1-IMP) product used directly in the following step.
To crude N1-(5′-O-phosphoryl-β-D-ribofuranosyl)−8-azidohypoxanthine in TEAB (0.05 M, 5 mL) was added dithiothreitol (5 mg, 32 µmol) and the solution stirred at rt for 16 h before purification by semipreparative HPLC (1.1 cm × 25 cm C18 column), eluting with acetonitrile/0.1 M TEAB (1:19 → 13:7 v/v) over 25 min. Fractions were analyzed by analytical HPLC and appropriate fractions collected and evaporated under vacuum to give the title compound (5.3 mg, 20%). 1H-NMR (500 MHz, D2O) δ 8.42 (s, 1H, H−2), 6.32 (d, J = 3.3 Hz, 1H, H−1′), 4.31−4.35 (m, 2H, H−2′ and H−3′), 4.24 (brs, 1H, H−4′), 4.15−4.18 (m, 1H, H−5′a) and 4.07−4.03 (m, 1H, H−5′b). 13C-NMR (125 MHz, D2O) δ 161.4 (C−6), 155.4 (C−4), 152.7 (C−5), 143.7 (C−2), 112.2 (C−8), 89.1 (C−1′), 83.0 (d, J = 8.9 Hz, C−4′), 75.0 (C−2′), 69.0 (C−3′) and 63.6 (d, J = 4.5 Hz, C−5′). 31P-NMR (202 MHz, D2O) δ 0.36. HRMS (ES−) calcd for C10H13N5O8P 362.0507 (M − H)− found 362.0516.
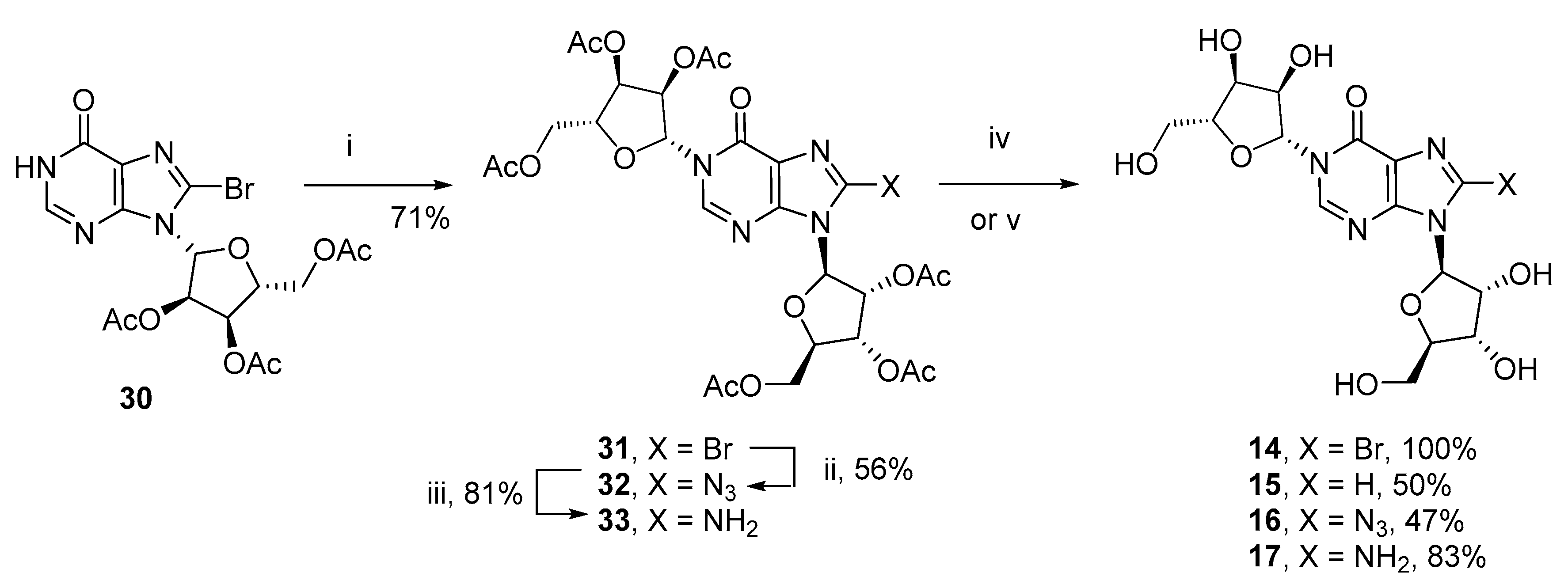
3.3. N1-Ribosyl-Inosine Analogues (14–17)
N1-(2″,3″,5″-Tri-O-acetyl-β-D-ribofuranosyl)−2′
,3′
,5′
-tri-O-acetyl−8-bromoinosine (31)–2′,3′,5′-tri-
O-acetyl−8-bromoinosine [
33] (
30, 615 mg, 1.30 mmol) was taken up in DCM (6 mL) and DBU (583 μL, 3.90 mmol) added. After 30 min, 1,2,3,5-tetra-
O-acetyl-β-
D-ribofuranose (455 mg, 1.43 mmol) was added and the solution cooled to −78 °C. Trimethylsilyl trifluoromethanesulfonate (941 μL, 5.20 mmol) was added dropwise and the solution stirred for a further 45 min before warming to rt. After 1 h, NaHCO
3 (satd aq) was added and the crude material extracted into DCM (×3). The combined organic fractions were dried (Na
2SO
4), and solvent was evaporated under reduced pressure. The residue was purified by column chromatography on silica gel eluting with DCM/Acetone (1:0 → 4:1
v/
v) to afford the title compound (678 mg, 71%) as a colourless glass:
Rf = 0.69 (DCM:Acetone 3:1
v/
v);
1H-NMR (400 MHz, CDCl
3) δ 8.28(s, 1H, 2-H), 6.38 (d,
J = 4.6 Hz, 1H), 6.20 (dd,
J = 6.0, 4.7, Hz, 1H), 6.10 (d,
J = 4.7 Hz, 1H), 5.79 (app. t,
J = 5.8 Hz, 1H), 5.51−5.44 (m, 2H), 4.48−4.33 (m, 6H), 2.20 (s, 3H), 2.16 (s, 3H), 2.14 (s, 3H), 2.13 (s, 3H), 2.11 (s, 3H), 2.07 (s, 3H);
13C-NMR (100 MHz, CDCl
3) δ 170.4, 170.3, 169.52, 169.47, 169.39, 169.29, 154.5, 148.1, 144.6, 126.3, 125.1, 88.7, 87.5, 80.3, 80.08, 74.2, 72.2, 70.3, 70.1, 63.1, 62.9, 20.8, 20.6, 20.48, 20.46, 20.39, 20.37; HRMS (ES
+) calcd for C
15H
20N
4O
979Br 479.0408 (M + H)
+ found 479.0401, calcd for C
15H
20N
4O
981Br 481.0388 (M + H)
+ found 481.0379.
N1-(β-D-Ribofuranosyl)−8-bromoinosine (8-Br-N1-ribosyl-IMP, 14)–N1-(2″,3″,5″-Tri-O-acetyl-β-D-ribofuranosyl)−2′,3′,5′-tri-O-acetyl−8-bromoinosine (31, 107 mg, 0.146 mmol) was taken up in MeOH (2.0 mL) in a pressure tube. The solution was cooled to 0 °C in an ice-water bath and NH3 (g) bubbled through the solution to saturation. The tube was sealed and the resulting solution stirred at rt. When complete by TLC, the precipitate was removed by filtration and dried under vacuum to yield the title compound (70 mg, 100%) as a white amorphous solid; 1H-NMR (400 MHz, CD3OD) δ 8.89 (s, 1H, 2-H), 6.26 (d, J = 2.8 Hz, 1H), 6.10 (d, J = 6.4 Hz, 1H), 5.11 (dd, J = 6.4, 5.7, Hz, 1H), 4.46 (dd, J = 5.4, 2.8 Hz, 1H), 4.35−4.30 (m, 2H), 4.20−4.16 (m, 2H), 4.01 (dd, J = 12.3, 2.5 Hz, 1H), 3.91 (dd, J = 12.4, 3.3 Hz, 1H), 3.86 (dd, J = 12.3, 2.8 Hz, 1H), 3.79 (dd, J = 12.4, 4.2 Hz, 1H); 13C-NMR (100 MHz, CD3OD) δ 156.9, 149.7, 147.0, 128.5, 125.8, 92.9, 91.6, 88.2, 86.3, 76.9, 74.0, 72.4, 70.5, 63.7, 61.6; HRMS (ES+) calcd for C15H20N4O979Br 479.0408 (M+H)+ found 479.0410, calcd for C15H20N4O981Br 481.0388 (M+H)+ found 481.0401.
N1-(β-D-Ribofuranosyl)-inosine (N1-ribosyl-IMP, 15)–N1-(2″,3″,5″-Tri-O-acetyl-β-D-ribofuranosyl)−2′,3′,5′-tri-O-acetyl−8-bromoinosine (31, 330 mg, 0.451 mmol), NaHCO3 (42 mg, 0.496 mmol) and Pd/C (45 mg, 45 µmol) were taken up in EtOH (6 mL) and the solution degassed with argon before being placed under an atmosphere of H2 for 12 h. The catalyst was removed by filtration and the resulting solution purified by column chromatography on silica gel eluting with DCM/Acetone (1:0 → 0:1 v/v) to afford the title compound (90 mg, 50%) as a clear glass; Rf = 0.27 (DCM:Acetone 4:1 v/v); 1H-NMR (500 MHz, CD3OD) δ 8.82 (s, 1H), 8.34 (s, 1H), 6.22 (d, J = 3.0 Hz, 1H), 6.00 (d, J = 5.7 Hz, 1H), 4.61 (dd, J = 5.7, 5.4 Hz, 1H), 4.31 (dd, J = 5.0, 3.7 Hz, 1H), 4.28–4.24 (m, 2H), 4.14−4.09 (m, 2H), 3.94 (dd, J = 12.3, 2.5 Hz, 1H), 3.85 (dd, J = 12.4, 3.0 Hz, 1H), 3.79 (dd, J = 12.4, 2.9 Hz, 1H), 3.74 (dd, J = 12.3, 3.3 Hz, 1H); 13C-NMR (125 MHz, CD3OD) δ 158.0, 148.7, 147.0, 141.4, 125.0, 91.4, 90.6, 87.6, 86.3, 76.9, 76.2, 72.1, 70.6, 63.0, 61.7; HRMS (ES+) calcd for C15H21N4O9 401.1303 (M + H)+ found 401.1297.
N1-(2″,3″,5″-Tri-O-acetyl-β-D-ribofuranosyl)−2′,3′,5′-tri-O-acetyl−8-azidoinosine (32)–To N1-(2″,3″,5″-tri-O-acetyl-β-d-ribofuranosyl)−2′,3′,5′-tri-O-acetyl−8-bromoinosine (31, 240 mg, 0.328 mmol) in DMF (2.4 mL) was added NaN3 (68 mg, 1.05 mmol) and the resulting solution stirred at 70 °C in the dark for 72 h. The solution was evaporated to dryness and the residue taken up in DCM, washed with H2O, dried over Na2SO4 and purified by column chromatography on silica gel eluting with PE/EtOAc (1:0 → 0:1 v/v) to afford the title compound (128 mg, 56%) as a clear glass; Rf = 0.53 (PE:EtOAc 1:3 v/v); 1H-NMR (400 MHz, CDCl3) δ 8.16 (s, 1H, 2-H), 6.30 (d, J = 4.5 Hz, 1H), 6.02 (dd, J = 5.8, 4.9 Hz, 1H), 5.90 (d, J = 4.9 Hz, 1H), 5.67 (app. t, J = 5.8 Hz, 1H), 5.47 (dd, J = 5.9, 4.5 Hz, 1H), 5.43 (dd, J = 5.8, 5.0 Hz, 1H), 4.42−4.22 (m, 6H), 2.14 (s, 3H), 2.10 (s, 3H), 2.09 (s, 3H), 2.064 (s, 3H), 2.061 (s, 3H), 2.03 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ 170.3, 170.2, 169.5, 169.4, 169.3, 169.2, 154.7, 146.9, 144.8, 143.8, 122.5, 87.9, 85.6, 80.3, 79.8, 74.2, 71.9, 70.2, 70.1, 63.0, 62.9, 20.7, 20.6, 20.4 (2C), 20.32, 20.31; HRMS (ES+) calcd for C27H31N7O15Na 716.1770 (M+Na)+ found 716.1758.
N1-(β-D-ribofuranosyl)−8-azidoinosine (8-N3-N1-ribosyl-IMP, 16)–N1-(2″,3″,5″-Tri-O-acetyl-β-D-ribofuranosyl)−2′,3′,5′-tri-O-acetyl−8-azidoinosine (32, 50 mg, 72 µmol) was taken up in MeOH (1.5 mL) in a pressure tube. The solution was cooled to 0 °C in an ice-water bath and NH3 (g) bubbled through the solution to saturation. The tube was sealed and the resulting solution stirred at rt. When complete by TLC, the solvents were removed by evaporation under reduced pressure and co-evaporation with MeOH (5 mL ×2). The residue was purified by column chromatography on silica gel eluting with DCM/MeOH (1:0 → 4:1 v/v + 0.5% pyridine) to afford the title compound (15 mg, 47%) as a clear glass; Rf = 0.23 (DCM:MeOH 8:2 v/v); 1H-NMR (500 MHz, D2O) δ 8.50 (s, 1H, 2-H), 6.08 (d, J = 3.0 Hz, 1H), 5.80 (d, J = 6.2 Hz, 1H), 4.83 (dd, J = 6.2, 5.8, Hz, 1H), 4.38 (dd, J = 5.8, 3.6 Hz, 1H), 4.34 (dd, J = 5.2, 3.0 Hz, 1H), 4.22 (dd, J = 7.0, 5.2 Hz, 1H), 4.14−4.10 (m, 2H), 3.93 (dd, J = 12.9, 2.7 Hz, 1H), 3.82−3.77 (m, 2H), 3.75 (dd, J = 12.7, 4.6 Hz, 1H); 13C-NMR (125 MHz, D2O) δ 156.1, 147.3, 146.4, 144.4, 121.4, 90.4, 87.7, 85.6, 83.9, 74.7, 72.4, 70.3, 68.7, 61.6, 60.2; HRMS (ES+) calcd for C15H20N7O9 442.1317 (M + H)+ found 442.1329.
N1-(2″,3″,5″-Tri-O-acetyl-β-D-ribofuranosyl)−2′,3′,5′-tri-O-acetyl−8-aminoinosine (33)–1-(2″,3″,5″-Tri-O-acetyl-β-D-ribofuranosyl)−2′,3′,5′-tri-O-acetyl−8-azidoinosine (32, 65 mg, 87 µmol) and Pd/C (9 mg, 9 µmol) were taken up in EtOH (2 mL) and the solution degassed with argon before being placed under an atmosphere of H2 for 12 h. The catalyst was removed by filtration and the resulting solution purified by column chromatography on silica gel eluting with DCM/Acetone (1:0 → 0:1 v/v) to afford the title compound (51 mg, 81%) as a clear glass; Rf = 0.48 (DCM:Acetone 1:1 v/v); 1H-NMR (400 MHz, CDCl3) δ 8.03 (s, 1H, 2-H), 6.29 (d, J = 4.3 Hz, 1H), 6.08 (d, J = 6.4 Hz, 1H), 5.84 (dd, J = 6.4, 6.2 Hz, 1H), 5.61 (br s, 2H, NH2), 5.53−5.49 (m, 2H), 5.45 (dd, J = 5.7, 5.5 Hz, 1H), 4.51 (dd, J = 12.1, 3.9 Hz, 1H), 4.44−4.28 (m, 5H), 2.15 (s, 3H), 2.12 (s, 3H), 2.11 (s, 3H), 2.10 (s, 3H), 2.09 (s, 3H), 2.06 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ 170.3, 170.2, 169.50, 169.47, 169.46, 169.41, 154.9, 151.0, 145.9, 141.7, 121.4, 88.0, 84.8, 80.5, 79.9, 74.2, 70.9, 70.2, 70.0, 63.0, 62.9, 20.7, 20.6, 20.5, 20.39, 20.38, 20.3; HRMS (ES+) calcd for C27H34N5O15 668.2046 (M+H)+ found 668.2021.
N1-(β-D-ribofuranosyl)−8-aminoinosine (8-NH2-N1-ribosyl-IMP, 17)–N1-(2″,3″,5″-Tri-O-acetyl-β-D-ribofuranosyl)−2′,3′,5′-tri-O-acetyl−8-aminoinosine (33, 50 mg, 69 µmol) was taken up in MeOH (2 mL) in a pressure tube. The solution was cooled to 0 °C in an ice-water bath and NH3 (g) bubbled through the solution to saturation. The tube was sealed and the resulting solution stirred at rt. When complete by TLC, the solvents were removed by evaporation under reduced pressure and co-evaporation with MeOH (5 mL ×2. The residue was purified by column chromatography on silica gel eluting with DCM/MeOH (1:0 → 7:3 v/v + 0.5% pyridine) to afford the title compound (24 mg, 83%) as a clear glass; Rf = 0.38 (DCM:MeOH 7:3 v/v + 0.5% pyridine); 1H-NMR (400 MHz, CD3OD) δ 8.67 (s, 1H, 2-H), 6.34 (d, J = 3.3 Hz, 1H), 6.09 (d, J = 7.3 Hz, 1H), 4.81 (dd, J = 7.3, 5.6, Hz, 1H), 4.38−4.30 (m, 3H), 4.20−4.15 (m, 2H), 4.00 (dd, J = 12.3, 2.5 Hz, 1H), 3.92−3.84 (m, 3H); 13C-NMR (100 MHz, CD3OD) δ 156.6, 153.5, 148.2, 144.1, 120.6, 91.8, 88.9, 87.7, 86.3, 76.9, 72.9, 72.6, 70.6, 62.9, 61.8; HRMS (ES+) calcd for C15H21N5O9Na 438.1231 (M+Na)+ found 438.1246.
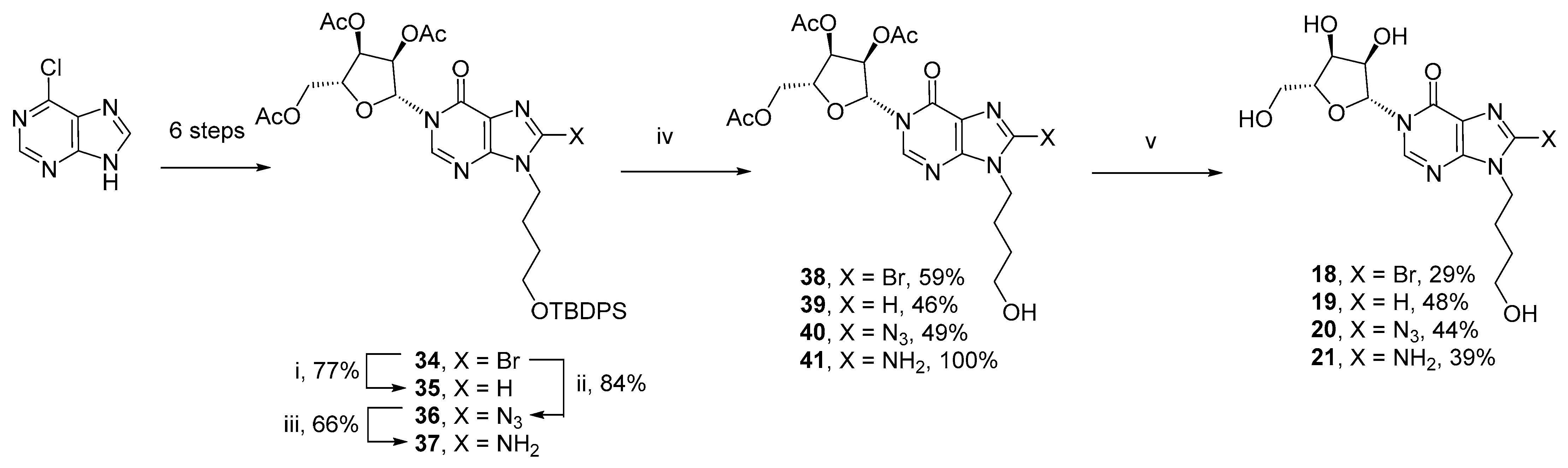
3.4. Total Synthesis of N9-(4-Hydroxybutyl)-N1-Inosine Analogues (18–21)
N1-(2′,3′,5′-Tri-
O-acetyl-β-
D-ribofuranosyl)-
N9-(4-(
tert-butyldiphenylsilyl)oxybutyl)−8-bromohypoxanthine (
34) was prepared as described previously [
36].
N1-(2′,3′,5′-Tri-O-acetyl-β-D-ribofuranosyl)-N9-(4-(tert-butyldiphenylsilyl)oxybutyl)-hypoxanthine (35) –N1-(2′,3′,5′-Tri-O-acetyl-β-D-ribofuranosyl)-N9-(4-(tert-butyldiphenylsilyl)oxybutyl)−8-bromohypo-xanthine (34, 140 mg, 0.179 mmol), NaHCO3 (75 mg, 0.895 mmol) and Pd/C (2 mg, 2 µmol) were taken up in EtOH (3 mL) and the solution degassed with argon before being placed under an atmosphere of H2 for 12 h. The catalyst was removed by filtration and the resulting solution purified by column chromatography on silica gel eluting with PE/EtOAc (1:0 → 0:1 v/v) to afford the title compound (95 mg, 77%) as a clear glass; Rf = 0.60 (PE:EtOAc 1:3 v/v); 1H-NMR (400 MHz, CDCl3) δ 8.15 (s, 1H), 7.68 (s, 1H), 7.63−7.61 (m, 4H), 7.42−7.33 (m, 6H)(10H, TBDPS), 6.39 (d, J = 4.7 Hz, 1H, H−1′), 5.51−5.45 (m, 2H, H−2′, H−3′), 4.44−4.36 (m, 3H, H−4′, 2 × H−5′), 4.13 (t, J = 7.2 Hz, 2H, CH2), 3.69 (t, J = 6.0 Hz, 2H, CH2), 2.14 (s, 3H), 2.11 (s, 3H), 2.06 (s, 3H)(3 × CH3), 2.02−1.93 (m, 2H, CH2), 1.64−1.52 (m, 2H, CH2), 1.03 (s, 9H, TBDPS); 13C-NMR (100 MHz, CDCl3) δ 170.2, 169.53, 169.51, 156.0, 147.4, 143.8, 140.0, 135.5 (4C), 133.6 (2C), 129.6 (2C), 127.6 (4C), 123.9, 87.3, 80.1, 74.2, 70.2, 63.0, 62.9, 43.9, 29.3, 26.83, 26.80 (3C), 20.7, 20.42, 20.37, 19.1; HRMS (ES+) calcd for C36H44N4O9SiNa 727.2770 (M+H)+ found 727.2741.
N1-(2′,3′,5′-Tri-O-acetyl-β-D-ribofuranosyl)-N9-(4-hydroxybutyl)-hypoxanthine (39)–To TBAF (128 mg, 0.404 mmol) in DMF (0.5 mL) was added AcOH (24 µL, 0.425 mmol). After 30 min, the solution was cooled to 0 °C and a solution of N1-(2′,3′,5′-Tri-O-acetyl-β-D-ribofuranosyl)-N9-(4-(tert-butyldiphenylsilyl)oxybutyl)-hypoxanthine (35, 95 mg, 0.135 mmol) in DMF (1.0 mL) added dropwise. After 6 h stirring at rt, NH4Cl (satd. aq.) was added and the solution extracted with DCM × 3. The combined organic layers were dried over Na2SO4 and purified by column chromatography on silica gel eluting with DCM/MeOH (1:0 → 4:1 v/v) to afford the title compound (29 mg, 46%) as a clear glass; Rf = 0.41 (DCM:MeOH 9:1 v/v); 1H-NMR (400 MHz, CDCl3) δ 8.17 (s, 1H), 7.76 (s, 1H), 6.36 (d, J = 4.6 Hz, 1H, H−1′), 5.49 (dd, J = 5.8, 4.6 Hz, 1H, H−2′), 5.46 (dd, J = 5.8, 5.1 Hz, 1H, H−3′), 4.43−4.35 (m, 3H, H−4′, 2 × H−5′), 4.21 (t, J = 7.2 Hz, 2H, CH2), 3.69 (t, J = 6.1 Hz, 2H, CH2), 2.16 (s, 3H), 2.09 (s, 3H), 2.07 (s, 3H)(3 × CH3), 2.02−1.91 (m, 2H, CH2) and 1.63−1.54 (m, 2H, CH2); 13C-NMR (125 MHz, CDCl3) δ 170.4, 169.70, 169.66, 156.0, 147.5, 144.0, 140.3, 123.9, 87.6, 80.1, 74.2, 70.2, 63.0, 61.9, 44.0, 29.2, 27.1, 20.9, 20.52, 20.49; HRMS (ES+) calcd for C20H27N4O9 467.1773 (M+H)+ found 467.1787.
N1-(β-D-Ribofuranosyl)-N9-(4-hydroxybutyl)-hypoxanthine (N9-hydroxybutyl-N1-Inosine, 19)–N1-(2′,3′,5′-Tri-O-acetyl-β-D-ribofuranosyl)-N9-(4-hydroxybutyl)-hypoxanthine (39, 25 mg, 54 µmol) was taken up in MeOH (2.0 mL) in a pressure tube. The solution was cooled to 0 °C in an ice-water bath and NH3 (g) bubbled through the solution to saturation. The tube was sealed and the resulting solution stirred at rt. When complete by TLC, the solvents were removed by evaporation under reduced pressure and co-evaporation with MeOH (5 mL ×2). The residue was purified by column chromatography on silica gel eluting with DCM/MeOH (1:0 → 3:1 v/v) to afford the title compound (8.7 mg, 48%) as a clear glass; Rf = 0.19 (DCM:MeOH 4:1 v/v); 1H-NMR (500 MHz, D2O) δ 8.48 (s, 1H), 7.96 (s, 1H), 6.04 (d, J = 3.3 Hz, 1H, H−1′), 4.28 (dd, J = 5.2, 3.3 Hz, 1H, H−2′), 4.17 (dd, J = 6.5, 5.2 Hz, 1H, H−3′), 4.11 (t, J = 7.1 Hz, 2H, CH2), 4.06 (ddd, J = 6.5, 4.1, 2.7 Hz, 1H, H−4′), 3.86 (dd, J = 12.9, 2.7 Hz, 1H, H−5′a), 3.72 (dd, J = 12.9, 2.7 Hz, 1H, H−5′b), 3.43 (t, J = 6.5 Hz, 2H, CH2), 1.76 (dt, J = 7.6, 7.1 Hz, 2H, CH2) and 1.37 (dt, J = 7.6, 6.5 Hz, 2H, CH2); 13C-NMR (125 MHz, D2O) δ 157.5, 147.7, 145.0, 142.5, 122.6, 90.1, 83.9, 74.7, 68.8, 60.9, 60.2, 44.0, 28.3, 25.9; HRMS (ES+) calcd for C14H20N4O6Na 363.1275 (M+Na)+ found 363.1276.
N1-(2′,3′,5′-Tri-O-acetyl-β-D-ribofuranosyl)-N9-(4-hydroxybutyl)−8-bromohypoxanthine (38)–To TBAF (121 mg, 0.384 mmol) in DMF (0.5 mL) was added AcOH (23 µL, 0.402 mmol). After 30 min, the solution was cooled to 0 °C and a solution of N1-(2′,3′,5′-Tri-O-acetyl-β-D-ribofuranosyl)-N9-(4-(tert-butyldiphenylsilyl)oxybutyl)−8-bromohypoxanthine (34, 100 mg, 0.128 mmol) in DMF (1.0 mL) added dropwise. After 6 h stirring at rt, NH4Cl (satd. aq.) was added and the solution extracted with DCM × 3. The combined organic layers were dried over Na2SO4 and purified by column chromatography on silica gel eluting with DCM/MeOH (1:0 → 4:1 v/v) to afford the title compound (41 mg, 59%) as a clear glass; Rf = 0.10 (PE:EtOAc 1:3 v/v); 1H-NMR (400 MHz, CDCl3) δ 8.16 (s, 1H), 6.35 (d, J = 4.6 Hz, 1H, H−1′), 5.46 (dd, J = 4.6, 2.4 Hz, 1H, H−2′), 5.43 (dd, J = 5.8, 2.4 Hz, 1H, H−3′), 4.43−4.34 (m, 3H, H−4′, 2 × H−5′), 4.21 (t, J = 7.2 Hz, 2H, CH2), 3.69 (t, J = 6.2 Hz, 2H, CH2), 2.15 (s, 3H), 2.10 (s, 3H), 2.07 (s, 3H)(3 × CH3), 1.96−1.88 (m, 2H, CH2) and 1.62−1.55 (m, 2H, CH2); 13C-NMR (100 MHz, CDCl3) δ 170.2, 169.6, 169.5, 154.7, 148.7, 144.2, 126.1, 124.0, 87.4, 80.1, 74.1, 70.1, 62.9, 62.0, 44.6, 29.1, 26.2, 20.7, 20.41, 20.35; HRMS (ES+) calcd for C20H25N4O9Na79Br 567.0697 (M+Na)+ found 567.0688; calcd for C20H25N4O9Na81Br 569.0677 (M+Na)+ found 569.0656.
N1-(β-D-Ribofuranosyl)-N9-(4-hydroxybutyl)−8-bromohypoxanthine (8-Br-N9-hydroxybutyl-N1-Inosine, 18)–N1-(2′,3′,5′-Tri-O-acetyl-β-D-ribofuranosyl)-N9-(4-hydroxybutyl)−8-bromohypoxanthine (38, 40 mg, 73 µmol) was taken up in MeOH (1.5 mL) in a pressure tube. The solution was cooled to 0 °C in an ice-water bath and NH3 (g) bubbled through the solution to saturation. The tube was sealed and the resulting solution stirred at rt. When complete by TLC, the solvents were removed by evaporation under reduced pressure and co-evaporation with MeOH (5 mL ×2). The residue was purified by column chromatography on silica gel eluting with DCM/MeOH (1:0 → 3:1 v/v) to afford the title compound (9 mg, 29%) as a clear glass; Rf = 0.12 (DCM:MeOH 9:1 v/v); 1H-NMR (400 MHz, CD3OD) δ 8.86 (s, 1H), 6.26 (d, J = 3.0 Hz, 1H, H−1′), 4.36−4.31 (m, 4H, H−2′, H−3′, CH2), 4.17 (ddd, J = 5.4, 2.9, 2.6 Hz, 1H, H−4′), 4.01 (dd, J = 12.3, 2.6 Hz, 1H, H−5′a), 3.87 (dd, J = 12.3, 2.9 Hz, 1H, H−5′b), 3.65 (t, J = 6.3 Hz, 2H, CH2), 1.98 (dt, J = 7.5, 7.1 Hz, 2H, CH2) and 1.62 (dt, J = 7.5, 6.3 Hz, 2H, CH2); 13C-NMR (100 MHz, CD3OD) δ 157.0, 150.6, 147.2, 127.7, 124.6, 91.5, 86.3, 76.9, 70.6, 62.2, 61.7, 45.8, 30.5, 27.2; HRMS (ES+) calcd for C14H19N4O6Na79Br 441.0380 (M+Na)+ found 441.0395; calcd for C14H19N4O6Na81Br 443.0360 (M+Na)+ found 443.0382.
N1-(2′,3′,5′-Tri-O-acetyl-β-D-ribofuranosyl)-N9-(4-(tert-butyldiphenylsilyl)oxybutyl)−8-azidohypoxan-thine (36)–To N1-(2′,3′,5′-tri-O-acetyl-β-D-ribofuranosyl)-N9-(4-(tert-butyldiphenylsilyl)oxybutyl)−8-bromohypoxanthine (34, 400 mg, 0.510 mmol) in DMF (4.0 mL) was added NaN3 (106 mg, 1.633 mmol) and the resulting solution stirred at 70 °C in the dark for 72 h. The solution was evaporated to dryness and the residue taken up in DCM, washed with H2O, dried over Na2SO4 and purified by column chromatography on silica gel eluting with PE/EtOAc (1:0 → 0:1 v/v) to afford the title compound (321 mg, 84%) as a clear glass; Rf = 0.73 (PE:EtOAc 1:3 v/v) – note there is no change in retention time compared to the starting material; 1H-NMR (400 MHz, CDCl3) δ 8.09 (s, 1H), 7.64−7.62 (m, 4H), 7.42−7.34 (m, 6H)(10H, TBDPS), 6.37 (d, J = 4.4 Hz, 1H, H−1′), 5.50−5.45 (m, 2H, H−2′, H−3′), 4.44−4.36 (m, 3H, H−4′, 2 × H−5′), 3.97 (t, J = 7.2 Hz, 2H, CH2), 3.68 (t, J = 6.1 Hz, 2H, CH2), 2.13 (s, 3H), 2.12 (s, 3H), 2.08 (s, 3H)(3 × CH3), 1.96−1.85 (m, 2H, CH2), 1.56−1.49 (m, 2H, CH2), 1.03 (s, 9H, TBDPS); 13C-NMR (100 MHz, CDCl3) δ 170.1, 169.53, 169.52, 154.9, 147.5, 145.0, 143.2, 135.5 (4C), 133.7 (2C), 129.6 (2C), 127.6 (4C), 121.7, 87.6, 80.2, 74.3, 70.3, 63.1, 62.8, 42.4, 29.2, 26.8 (3C), 25.9, 20.7, 20.43, 20.36, 19.1; HRMS (ES+) calcd for C36H43N7O9SiNa 768.2784 (M+H)+ found 768.2761.
N1-(2′,3′,5′-Tri-O-acetyl-β-D-ribofuranosyl)-N9-(4-hydroxybutyl)−8-azidohypoxanthine (40)–To TBAF (114 mg, 0.362 mmol) in DMF (0.5 mL) was added AcOH (22 µL, 0.380 mmol). After 30 min, the solution was cooled to 0 °C and a solution of N1-(2′,3′,5′-Tri-O-acetyl-β-D-ribofuranosyl)-N9-(4-(tert-butyldiphenylsilyl)oxybutyl)−8-azidohypoxanthine (36, 90 mg, 0.121 mmol) in DMF (1.0 mL) added dropwise. After 6 h stirring at rt, NH4Cl (satd. aq.) was added and the solution extracted with DCM × 3. The combined organic layers were dried over Na2SO4 and purified by column chromatography on silica gel eluting with DCM/MeOH (1:0 → 4:1 v/v) to afford the title compound (30 mg, 49%) as a clear glass; Rf = 0.41 (DCM:MeOH 9:1 v/v); 1H-NMR (400 MHz, CDCl3) δ 8.11 (s, 1H), 6.34 (d, J = 4.5 Hz, 1H, H−1′), 5.48 (dd, J = 5.9, 4.6 Hz, 1H, H−2′), 5.45 (dd, J = 5.9, 4.7 Hz, 1H, H−3′), 4.43−4.35 (m, 3H, H−4′, 2 × H−5′), 4.02 (t, J = 7.1 Hz, 2H, CH2), 3.67 (t, J = 6.3 Hz, 2H, CH2), 2.15 (s, 3H), 2.11 (s, 3H), 2.07 (s, 3H)(3 × CH3), 1.91−1.83 (m, 2H, CH2) and 1.58−1.51 (m, 2H, CH2); 13C-NMR (100 MHz, CDCl3) δ 170.2, 169.6, 169.5, 154.9, 147.5, 145.0, 143.4, 121.7, 87.7, 80.2, 74.2, 70.2, 63.1, 62.0, 42.4, 29.1, 26.0, 20.7, 20.42, 20.36; HRMS (ES+) calcd for C20H26N7O9 508.1787 (M+H)+ found 508.1799.
N1-(β-D-Ribofuranosyl)-N9-(4-hydroxybutyl)−8-azidohypoxanthine (8-N3-N9-hydroxybutyl-N1-inosine, 20)–N1-(2′,3′,5′-Tri-O-acetyl-β-D-ribofuranosyl)-N9-(4-hydroxybutyl)−8-azidohypoxanthine (40, 30 mg, 59 µmol) was taken up in MeOH (1.5 mL) in a pressure tube. The solution was cooled to 0 °C in an ice-water bath and NH3 (g) bubbled through the solution to saturation. The tube was sealed and the resulting solution stirred at rt. When complete by TLC, the solvents were removed by evaporation under reduced pressure and co-evaporation with MeOH (5 mL ×2). The residue was purified by column chromatography on silica gel eluting with DCM/MeOH (1:0 → 4:1 v/v) to afford the title compound (10 mg, 44%) as a clear glass; Rf = 0.11 (DCM:MeOH 9:1 v/v); 1H-NMR (400 MHz, CD3OD) δ 8.80 (s, 1H), 6.27 (d, J = 3.1 Hz, 1H, H−1′), 4.36−4.31 (m, 2H, H−2′, H−3′), 4.17 (ddd, J = 5.5, 2.9, 2.3 Hz, 1H, H−4′), 4.13 (t, J = 7.1 Hz, 2H, CH2), 4.01 (dd, J = 12.3, 2.6 Hz, 1H, H−5′a), 3.87 (dd, J = 12.3, 2.9 Hz, 1H, H−5′b), 3.63 (t, J = 6.4 Hz, 2H, CH2), 1.93 (dt, J = 7.3, 7.1 Hz, 2H, CH2) and 1.58 (dt, J = 7.3, 6.4 Hz, 2H, CH2); 13C-NMR (125 MHz, CD3OD) δ 157.3, 149.5, 146.7, 146.2, 122.4, 91.6, 86.3, 76.9, 70.6, 62.1, 61.7, 43.5, 30.5, 26.9; HRMS (ES+) calcd for C14H19N7O6Na 404.1289 (M+Na)+ found 404.1293.
N1-(2′,3′,5′-Tri-O-acetyl-β-D-ribofuranosyl)-N9-(4-(tert-butyldiphenylsilyl)oxybutyl)−8-aminohypoxan-thine (37)–N1-(2′,3′,5′-Tri-O-acetyl-β-D-ribofuranosyl)-N9-(4-(tert-butyldiphenylsilyl)oxybutyl)−8-azidohypoxanthine (36, 110 mg, 0.147 mmol) and Pd/C (9 mg, 9 µmol) were taken up in EtOH (3 mL) and the solution degassed with argon before being placed under an atmosphere of H2 for 12 h. The catalyst was removed by filtration and the resulting solution purified by column chromatography on silica gel eluting with DCM/Acetone (1:0 → 0:1 v/v) to afford the title compound (70 mg, 66%) as a clear glass; Rf = 0.60 (DCM:Acetone 1:1 v/v); 1H-NMR (500 MHz, CDCl3) δ 8.03 (s, 1H), 7.65−7.63 (m, 4H), 7.44−7.36 (m, 6H)(10H, TBDPS), 6.40 (d, J = 4.7 Hz, 1H, H−1′), 5.50 (dd, 1H, J = 5.7, 4.7, H−2′), 5.47−5.45 (m, 1H, H−3′), 4.84 (brs, 2H, NH2), 4.44−4.36 (m, 3H, H−4′, 2 × H−5′), 3.98 (dt, J = 7.1, 1.5 Hz, 2H, CH2), 3.68 (m, 2H, CH2), 2.15 (s, 3H), 2.11 (s, 3H), 2.09 (s, 3H)(3 × CH3), 1.92−1.88 (m, 2H, CH2), 1.61−1.56 (m, 2H, CH2), 1.05 (s, 9H, TBDPS); 13C-NMR (125 MHz, CDCl3) δ 170.3, 169.63, 169.61, 155.1, 150.7, 146.7, 141.4, 135.5 (4C), 133.5 (2C), 129.8 (2C), 127.7 (4C), 120.6, 87.3, 80.0, 74.3, 70.2, 64.3, 63.3, 63.1, 41.7, 29.0, 26.9 (3C), 25.9, 20.8, 20.52, 20.49, 19.2; HRMS (ES+) calcd for C36H45N5O9SiNa 742.2879 (M+Na)+ found 742.2850.
N1-(2′,3′,5′-Tri-O-acetyl-β-D-ribofuranosyl)-N9-(4-hydroxybutyl)−8-aminohypoxanthine (41)–To TBAF (92 mg, 0.292 mmol) in DMF (0.5 mL) was added AcOH (18 µL, 0.306 mmol). After 30 min, the solution was cooled to 0 °C and a solution of N1-(2′,3′,5′-Tri-O-acetyl-β-D-ribofuranosyl)-N9-(4-(tert-butyldiphenylsilyl)oxybutyl)−8-aminohypoxanthine (37, 70 mg, 0.0972 mmol) in DMF (0.5 mL) added dropwise. After 6 h stirring at rt, NH4Cl (satd. aq.) was added and the solution extracted with DCM × 3. The combined organic layers were dried over Na2SO4 and purified by column chromatography on silica gel eluting with DCM/MeOH (1:0 → 4:1 v/v) to afford the title compound (47 mg, 100%) as a clear glass; Rf = 0.47 (DCM:MeOH 9:1 v/v); 1H-NMR (400 MHz, CDCl3) δ 7.99 (s, 1H), 6.24 (d, J = 4.0 Hz, 1H, H−1′), 6.16 (br s, 2H, NH2), 5.55 (dd, J = 5.8, 4.0 Hz, 1H, H−2′), 5.45 (dd, J = 5.8, 5.7 Hz, 1H, H−3′), 4.43−4.31 (m, 3H, H−4′, 2 × H−5′), 4.03 (t, J = 7.0 Hz, 2H, CH2), 3.69 (t, J = 5.8 Hz, 2H, CH2), 2.12 (s, 3H), 2.08 (s, 6H)(3 × CH3), 1.91−1.82 (m, 2H, CH2) and 1.58−1.51 (m, 2H, CH2); 13C-NMR (100 MHz, CDCl3) δ 170.4, 169.61, 169.59, 154.9, 152.4, 146.8, 141.1, 120.5, 88.4, 79.7, 74.1, 70.0, 62.9, 62.0, 41.5, 28.4, 26.1, 20.7, 20.41 (2C); HRMS (ES+) calcd for C20H28N5O9 482.1882 (M+H)+ found 482.1891.
N1-(β-D-Ribofuranosyl)-N9-(4-hydroxybutyl)−8-aminohypoxanthine (8-NH2-N9-hydroxybutyl-N1-inosine, 21)–N1-(2′,3′,5′-Tri-O-acetyl-β-D-ribofuranosyl)-N9-(4-hydroxybutyl)−8-aminohypoxanthine (41, 45 mg, 93 µmol) was taken up in MeOH (1.5 mL) in a pressure tube. The solution was cooled to 0 °C in an ice-water bath and NH3 (g) bubbled through the solution to saturation. The tube was sealed and the resulting solution stirred at rt. When complete by TLC, the solvents were removed by evaporation under reduced pressure and co-evaporation with MeOH (5 mL ×2). The residue was purified by column chromatography on silica gel eluting with DCM/MeOH (1:0 → 4:1 v/v + 0.5% pyridine) to afford the title compound (13 mg, 39%) as a clear glass; Rf = 0.47 (DCM:MeOH 7:3 v/v); 1H-NMR (400 MHz, CD3OD) δ 8.63 (s, 1H), 6.25 (d, J = 3.4 Hz, 1H, H−1′), 4.37 (dd, J = 5.2, 3.4 Hz, 1H, H−2′), 4.33 (dd, J = 5.7, 5.2 Hz, 1H, H−3′), 4.16 (ddd, J = 5.7, 3.0, 2.5 Hz, 1H, H−4′), 4.12 (t, J = 7.2 Hz, 2H, CH2), 4.00 (dd, J = 12.3, 2.5 Hz, 1H, H−5′a), 3.86 (dd, J = 12.3, 3.0 Hz, 1H, H−5′b), 3.65 (t, J = 6.4 Hz, 2H, CH2), 1.90 (dt, J = 7.5, 7.2 Hz, 2H, CH2) and 1.61 (dt, J = 7.5, 6.4 Hz, 2H, CH2); 13C-NMR (125 MHz, CD3OD) δ 156.9, 154.1, 148.6, 143.8, 121.2, 91.7, 86.2, 76.8, 70.7, 62.4, 61.9, 42.4, 30.4, 26.5; HRMS (ES+) calcd for C14H22N5O6 356.1565 (M+H)+ found 356.1561.
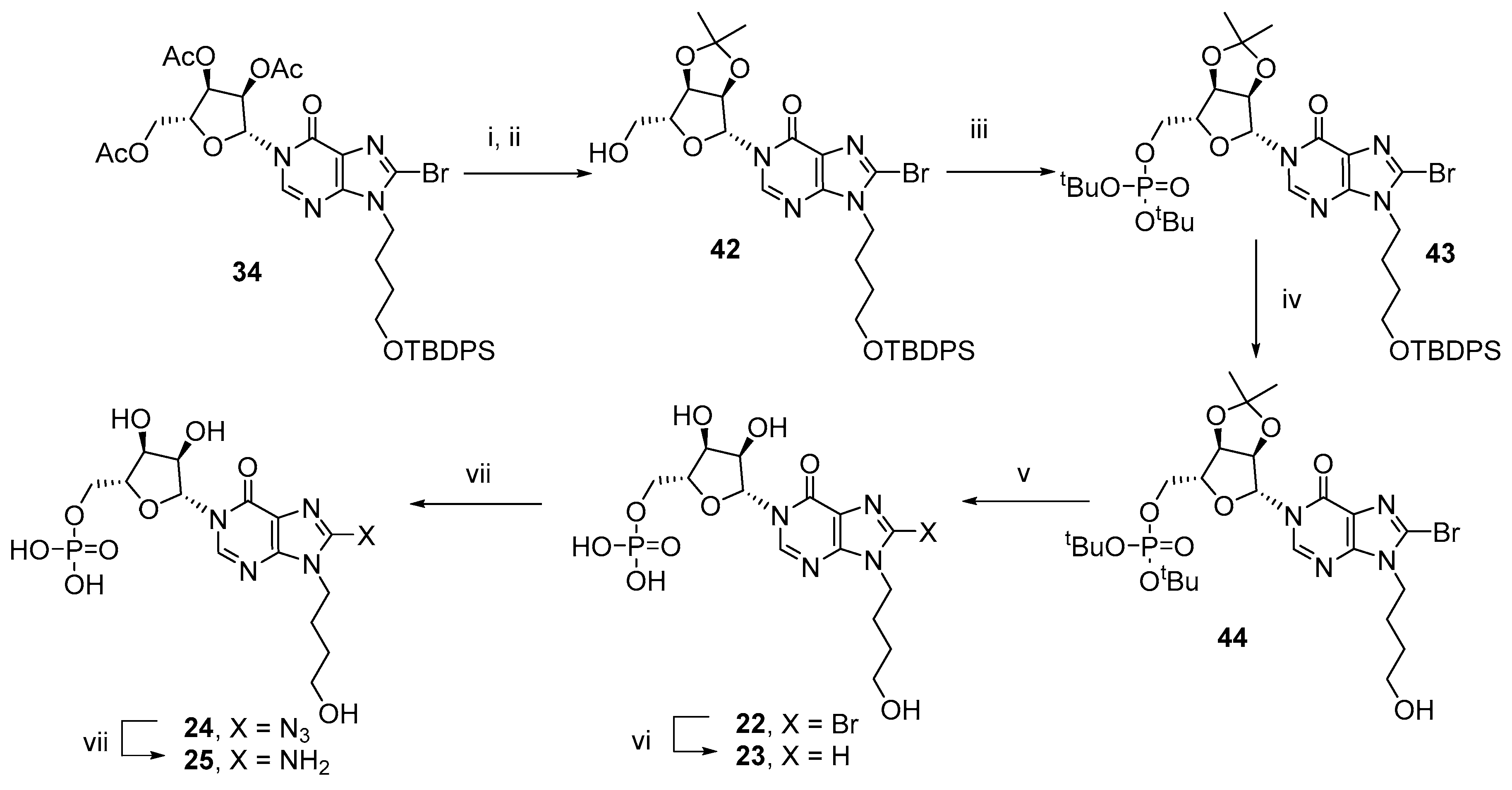
3.5. Total Synthesis of N9-(4-Hydroxybutyl)-N1-IMP Analogues (22–25)
N1-[2′,3′-O-Isopropylidine−5′-O-(di-tert-butyl)-phosphoryl-β-D-ribofuranosyl]-N9-(4-hydroxybutyl)−8-bromohypoxanthine (44) was prepared in 10 steps from 6-chloropurine as described previously [
36].
N1-(5′-O-Phosphoryl-β-D-ribofuranosyl)-N9-(4-hydroxybutyl)−8-bromohypoxanthine (22)–N1-[2′,3′-O-Isopropylidine−5′-O-(di-tert-butyl)-phosphoryl-β-D-ribofuranosyl]-N9-(4-hydroxybutyl)−8-bromohypoxanthine (44, 50 mg, 77 µmol) was stirred at 0 °C in TFA (50% aq., 2 mL). Solvents were evaporated to dryness and the residue evaporated with MeOH (× 3) before purification by semipreparative HPLC (1.1 cm × 25 cm C18 column), eluting with acetonitrile/0.1 M TEAB (1:19 → 13:7 v/v) over 25 min. Fractions were analyzed by analytical HPLC and appropriate fractions collected and evaporated under vacuum to give the title compound (33 mg, 87%); UV (H2O, pH 7), λmax = 257 nm (ε = 14 700); 1H-NMR (500 MHz, D2O) δ 8.64 (s, 1H, H−2), 6.10 (d, J = 2.4 Hz, 1H, H−1′), 4.31−4.27 (m, 2H, H−2′ and H−3′), 4.21−4.18 (m, 1H, H−4′), 4.11−4.06 (m, 3H, H−5′a, CH2), 3.95−3.92 (m, 1H, H−5′b), 3.45 (t, J = 6.5 Hz, 2H, CH2), 1.72 (dt, J = 7.6, 7.3 Hz, 2H, CH2) and 1.41 (dt, J = 8.3, 6.5 Hz, 2H, CH2); 13C-NMR (125 MHz, D2O) δ 156.1, 148.7, 145.3, 128.2, 122.8, 89.1 (C−1′), 83.4 (d, J = 8.9 Hz, C−4′), 75.2 (C−2′), 68.8 (C−3′), 62.7 (d, J = 3.8 Hz, C−5′), 60.9, 44.8, 28.3, 25.3 (4 × CH2); 31P-NMR (202 MHz, D2O) δ 2.49. HRMS (ES−) calcd for C14H19N4O9P79Br 497.0079 (M−H)− found 497.0073, calcd for C14H19N4O9P81Br 499.0058 (M−H)− found 499.0072.
N1-(5′-O-Phosphoryl-β-D-ribofuranosyl)-N9-(4-hydroxybutyl)hypoxanthine (23)–N1-(5′-O-Phosphoryl-β-D-ribofuranosyl)-N9-(4-hydroxybutyl)−8-bromohypoxanthine (22, 38 mg, 77 µmol), NaHCO3 (20 mg, 0.23 mmol) and Pd/C (5 mg) were taken up in MilliQ-EtOH (2:1 v/v, 3 mL) and the solution degassed with argon before being placed under an atmosphere of H2 for 12 h. The catalyst was removed by filtration and the resulting solution purified by semipreparative HPLC (1.1 cm × 25 cm C18 column), eluting with acetonitrile/0.1 M TEAB (1:19 → 13:7 v/v) over 25 min. Fractions were analyzed by analytical HPLC and appropriate fractions collected and evaporated under vacuum to give the title compound (25 mg, 78%); UV (H2O, pH 7), λmax = 252 nm (ε = 10 300); 1H-NMR (500 MHz, D2O) δ 8.52 (s, 1H, H−2), 7.96 (s, 1H, H−8), 6.11 (d, J = 2.3 Hz, 1H, H−1′), 4.30−4.27 (m, 2H, H−2′ and H−3′), 4.22−4.19 (m, 1H, H−4′), 4.13−4.16 (m, 1H, H−5′a), 4.10 (t, J = 7.1 Hz, 2H, CH2), 4.02−3.98 (m, 1H, H−5′b), 3.44 (t, J = 6.5 Hz, 2H, CH2), 1.76 (dt, J = 7.6, 7.1 Hz, 2H, CH2) and 1.38 (dt, J = 8.1, 6.5 Hz, 2H, CH2); 13C-NMR (125 MHz, D2O) δ 157.4, 147.5, 144.7, 142.4, 122.3, 89.1 (C−1′), 82.9 (d, J = 8.8 Hz, C−4′), 75.1 (C−2′), 68.8 (C−3′), 63.4 (d, J = 4.1 Hz, C−5′), 60.9, 44.0, 28.3, 25.9 (4 × CH2); 31P-NMR (202 MHz, D2O) δ 0.49. HRMS (ES−) calcd for C14H20N4O9P 419.0973 (M−H)− found 419.0958.
N1-(5′-O-Phosphoryl-β-D-ribofuranosyl)-N9-(4-hydroxybutyl)−8-azidohypoxanthine (24)–N1-(5′-O-phosphoryl-β-D-ribofuranosyl)-N9-(4-hydroxybutyl)−8-bromohypoxanthine (22, 20 mg, 40 µmol) was evaporated from DMF (2 × 2 mL), taken up in DMF (1 mL) and TMSN3 (53 µL, 401 µL) added. After 16 h at 70 °C in the dark, all solvents were evaporated and the residue purified by semi preparative HPLC (1.1 cm × 25 cm C18 column), eluting with acetonitrile/0.1 M TEAB (1:19 → 13:7 v/v) over 25 min. Fractions were analyzed by analytical HPLC and appropriate fractions collected and evaporated under vacuum to give the title compound (5.2 mg, 28%); UV (H2O, pH 7), λmax = 277 nm (ε = 15,600); 1H-NMR (500 MHz, D2O) δ 8.59 (s, 1H, H−2), 6.18 (d, J = 2.2 Hz, 1H, H−1′), 4.35−4.31 (m, 2H, H−2′ and H−3′), 4.27−4.24 (m, 1H, H−4′), 4.19−4.15 (m, 1H, H−5′a), 4.06−4.03 (m, 1H, H−5′b), 3.96 (t, J = 7.0 Hz, 2H, CH2), 3.50 (t, J = 6.6 Hz, 2H, CH2), 1.74 (dt, J = 7.5, 7.0 Hz, 2H, CH2) and 1.43 (dt, J = 8.1, 6.6 Hz, 2H, CH2); 13C-NMR (125 MHz, D2O) δ 156.2, 147.7, 146.6, 144.2, 120.5, 89.2 (C−1′), 83.0 (d, J = 8.9 Hz, C−4′), 75.2 (C−2′), 68.7 (C−3′), 63.2 (d, J = 4.6 Hz, C−5′), 60.9, 42.4, 28.3, 25.0 (4 × CH2); 31P-NMR (202 MHz, D2O) δ 0.97. HRMS (ES−) calcd for C14H19N7O9P 460.0987 (M−H)− found 460.0969.
N1-(5′-O-Phosphoryl-β-D-ribofuranosyl)-N9-(4-hydroxybutyl)−8-aminohypoxanthine (25)–To N1-(5′-O-phosphoryl-β-D-ribofuranosyl)-N9-(4-hydroxybutyl)−8-azidohypoxanthine (24, 18 mg, 39 µmol) in TEAB (0.05 M, 5 mL) was added dithiothreitol (5 mg, 32 µmol). After 16 h, the reaction was complete by HPLC (λmax 277 → 262) and was purified by semipreparative HPLC (1.1 cm × 25 cm C18 column), eluting with acetonitrile/0.1 M TEAB (1:19 → 13:7 v/v) over 25 min. Fractions were analyzed by analytical HPLC and appropriate fractions collected and evaporated under vacuum to give the title compound (9.6 mg, 57%); UV (H2O, pH 7), λmax = 265 nm (ε = 25,400); H-NMR (500 MHz, D2O) δ 8.46 (s, 1H, H−2), 6.12 (d, J = 2.4 Hz, 1H, H−1′), 4.29−4.26 (m, 2H, H−2′ and H−3′), 4.18−4.16 (m, 1H, H−4′), 4.09−4.06 (m, 1H, H−5′a), 3.96−3.93 (m, 1H, H−5′b), 3.87 (t, J = 7.0 Hz, 2H, CH2), 3.42 (t, J = 6.4 Hz, 2H, CH2), 1.65 (dt, J = 7.5, 7.0 Hz, 2H, CH2) and 1.38 (dt, J = 8.1, 6.4 Hz, 2H, CH2); 13C-NMR (125 MHz, D2O) δ 155.8, 152.9, 147.3, 142.3, 119.3, 89.1 (C−1′), 83.1 (d, J = 8.8 Hz, C−4′), 75.2 (C−2′), 68.8 (C−3′), 62.8 (d, J = 4.3 Hz, C−5′), 61.1, 41.5, 28.4, 24.5 (4 × CH2); 31P-NMR (202 MHz, D2O) δ 2.03; HRMS (ES−) calcd for C14H21N5O9P 434.1082 (M−H)− found 434.1063.
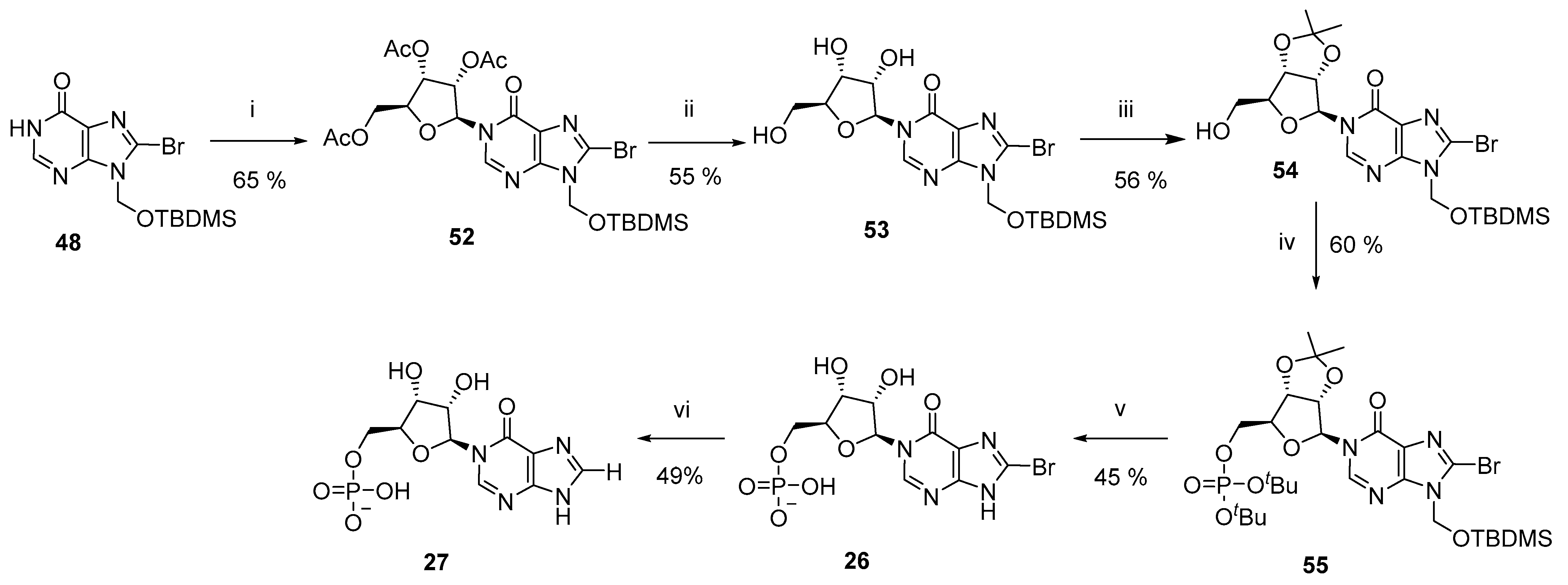
3.6. Total Synthesis of L-N1-IMP Analogues (26–27)
N1-(2′,3′,5′-Tri-O-acetyl-β-L-ribofuranosyl)-N9-tert-butyldimethylsilyloxymethyl−8-bromohypoxanthine (52)–To N9-tert-butyldimethylsilyloxymethyl−8-bromohypoxanthine (48, 430 mg, 1.197 mmol) in DCM (4.5 mL) was added DBU (537 μL, 3.590 mmol). After 30 min, 1,2,3,5-tetra-O-acetyl-β-L-ribofuranose (419 mg, 1.317 mmol) was added and the solution cooled to −78 °C. Trimethylsilyl trifluoromethanesulfonate (867 μL, 4.788 mmol) was added dropwise and the solution stirred for a further 45 min before warming to rt. After 2 h, NaHCO3 (satd aq) was added and the crude material extracted into DCM (×3). The combined organic fractions were dried (Na2SO4), and solvent was evaporated under reduced pressure. The residue was purified by column chromatography on silica gel eluting with PE/EtOAc (1:0 → 0:1 v/v) to afford the title compound (481 mg, 65%) as acolourless glass; Rf = 0.48 (PE:EtOAc 1:3 v/v); 1H-NMR (400 MHz, CDCl3) δ 8.20 (s, 1H, H−2), 6.35 (d, J = 4.6 Hz, 1H, H−1′), 5.64 (d, J = 9.8 Hz, 1H), 5.61 (d, J = 9.8 Hz, 1H) (2H, CH2OTBDMS), 5.46−5.40 (m, 2H, H−2′, H−3′), 4.43−4.32 (m, 3H, H−4′, 2 × H−5′), 2.13 (s, 3H, AcetylCH3), 2.09 (s, 3H, AcetylCH3), 2.05 (s, 3H, AcetylCH3), 0.85 (s, 9H) 0.11 (s, 3H) and 0.10 (s, 3H) (15H, TBDMS); 13C-NMR (100 MHz, CDCl3) δ 170.2, 169.5 (2C), 154.7, 148.4, 144.5, 126.1, 123.9, 87.4, 80.1, 74.2, 70.1, 67.3, 62.9, 25.5 (3C), 20.7, 20.43, 20.37, 18.0, −5.26 and −5.27; HRMS (ES+) calcd for C23H34N4O9Si79Br 617.1273 (M+H)+ found 617.1298, calcd for C23H34N4O9Si81Br 619.1252 (M+H)+ found 619.1298.
N1-(2′,3′-O-Isopropylidene-β-L-ribofuranosyl)-N9-tert-butyldimethylsilyloxymethyl−8-bromohypoxanthine (54)–N1-(2′,3′,5′-Tri-O-acetyl-β-L-ribofuranosyl)-N9-tert-butyldimethylsilyloxy-methyl−8-bromohypoxanthine (52, 470 mg, 0.761 mmol) was taken up in MeOH (5.0 mL) in a pressure tube. The solution was cooled to 0 °C in an ice-water bath and NH3 (g) bubbled through the solution to saturation. The tube was sealed and the resulting solution stirred at rt. When complete by TLC, the solvents were removed by evaporation under reduced pressure and the residue was purified by column chromatography on silica gel eluting with DCM/Acetone (1:0 → 0:1 v/v) to afford the deacetylated intermediate 53 (205 mg, 55%) as an amorphous white solid, Rf = 0.34 (EtOAc), which was used directly in the next step.
To N1-(β-L-ribofuranosyl)-N9-tert-butyldimethylsilyloxymethyl−8-bromohypoxanthine (53, 170 mg, 0.346 mmol) in 2,2-dimethoxypropane-acetone (1:4 v/v, 10 mL) was added para-toluenesulfonic acid (66 mg, 0.346 mmol). After 1 h, DCM and NaHCO3 (satd. aq.) were added and the aqueous phase extracted with DCM (×3). The combined organic extracts were evaporated to dryness, taken up in MeOH (5 mL) and treated with pre-washed DOWEX® H+ resin (50WX8, 100−200 mesh, ion-exchange resin) for 30 min. Note - the Dowex® H+ resin was used as a source of cations to remove the unwanted partial 5′-O-[2-methoxypropyl] protecting group, while leaving the desired 2′,3′-O-isopropylidene in tact. The resin was removed by filtration and the filtrate purified by column chromatography on silica gel eluting with PE/EtOAc (1:0 → 0:1 v/v) to afford the title compound (103 mg, 56%) as acolourless glass; Rf = 0.71 (EtOAc); 1H-NMR (500 MHz, CDCl3) δ 8.03 (s, 1H, 2-H), 5.71 (d, J = 2.9 Hz, 1H, H−1′), 5.66 (d, J = 9.9 Hz, 1H), 5.63 (d, J = 9.9 Hz, 1H) (2H, CH2OTBDMS), 5.31 (dd, J = 6.5, 2.9 Hz, 1H, H−2′), 5.15 (dd, J = 6.5, 3.6 Hz, 1H, H−3′), 4.36 (ddd, J = 6.0, 3.7, 3.6 Hz, 1H, H−4′), 3.93 (ddd, J = 12.0, 6.0, 3.1 Hz, 1H, H−5′a), 3.84 (ddd, J = 12.0, 8.2, 3.7 Hz, 1H, H−5′b), 3.31 (dd, J = 8.2, 3.1, 5′-OH), 1.59 (s, 3H, CCH3), 1.36 (s, 3H, CCH3), 0.87 (s, 9H), 0.12 (s, 3H), 0.11 (s, 3H) (15H, TBDMS); 13C-NMR (125 MHz, CDCl3) δ 155.3 (C−6), 148.9 (C−4), 146.9 (C−2), 126.6 (C−8), 124.8 (C−5), 114.3 (C(CH3)2), 97.2 (C−1′), 88.1 (C−4′), 83.5 (C−2′), 80.7 (C−3′), 67.5 (CH2OTBDMS), 62.9 (C−5′), 27.3 (CCH3), 25.5 (3C, SiC(CH3)3), 25.2 (CCH3), 18.1 (SiC(CH3)3), −5.2 (2C, Si(CH3)2); HRMS (ES+) calcd for C20H32N4O679BrSi 531.1275 (M+H)+ found 531.1288, calcd for C20H32N4O679BrSi 533.1254 (M+H)+ found 533.1249.
N1-[2′,3′-O-Isopropylidene−5′-O-(di-tert-butyl)-phosphoryl-β-L-ribofuranosyl]-N9-tert-butyldimethyl-silyloxymethyl−8-bromohypoxanthine (55)–To N1-(2′,3′-O-isopropylidene-β-L-ribofuranosyl)-N9-tert-butyldimethylsilyloxymethyl−8-bromohypoxanthine (54, 100 mg, 0.188 mmol) in DCM (1.0 mL) was added 5-phenyl−1H-tetrazole (55 mg, 0.376 mmol) and the solution cooled to 0 °C. Di-tert-butyl N,N-diisopropylphosphoramidite (89 µL, 0.282 mmol) was added dropwise and the solution stirred at rt until phosphitylation was complete by TLC. After cooling to 0 °C, triethylamine (157 µL, 1.128 mmol) and H2O2 (30% in H2O, 48 µL, 0.470 mmol) were added and the solution stirred at rt until oxidation was complete. The reaction was diluted with DCM and washed with NaHCO3 (satd. aq.), dried over Na2SO4 and purified by column chromatography on silica gel eluting with PE/EtOAc (1:0 → 0:1 v/v + 0.5% pyridine in each solvent) to afford the title compound (82 mg, 60%) as a colourless glass; Rf = 0.37 (PE:EtOAc 1:3 v/v); 1H-NMR (500 MHz, CDCl3) δ 8.10 (s, 1H, 2-H), 6.01 (d, J = 2.0 Hz, 1H, H−1′), 5.65 (d, J = 9.9 Hz, 1H), 5.63 (d, J = 9.9 Hz, 1H) (2H, CH2OTBDMS), 5.07 (dd, J = 6.4, 2.0 Hz, 1H, H−2′), 4.99 (dd, J = 6.4, 4.0 Hz, 1H, H−3′), 4.42 (ddd, J = 5.8, 4.1, 4.0 Hz, 1H, H−4′), 4.25 (ddd, J = 12.9, 6.5, 4.1 Hz, 1H, H−5′a), 4.18 (ddd, J = 12.9, 7.2, 5.8 Hz, 1H, H−5′b), 1.58 (s, 3H, CCH3), 1.47 (s, 9H, OtBu), 1.46 (s, 9H, OtBu), 1.34 (s, 3H, CCH3), 0.87 (s, 9H), 0.12 (s, 3H), 0.11 (s, 3H) (15H, TBDMS); 13C-NMR (125 MHz, CDCl3) δ 154.7, 148.7, 146.2, 126.1, 124.4, 114.4 (C(CH3)2), 94.1 (C−1′), 86.8 (d, J = 7.8 Hz, C−4′), 85.1 (C−2′), 82.7 (d, J = 7.1 Hz, 2C, 2 × POC(CH3)3), 81.3 (C−3′), 67.3 (CH2OTBDMS), 66.4 (d, J = 6.3 Hz, C−5′), 29.81 (d, J = 4.3 Hz, 3C, POC(CH3)3), 29.78 (d, J = 4.2 Hz, 3C, POC(CH3)3), 27.1 (CCH3), 25.5 (3C, SiC(CH3)3), 25.3 (CCH3), 18.0 (SiC(CH3)3), −5.2 (2C, Si(CH3)2); 31P-NMR (202 MHz, CDCl3) δ −10.10; HRMS (ES+) calcd for C28H48N4O9P79BrSiNa 745.2010 (M+Na)+ found 745.2040, calcd for C28H48N4O9P81BrSiNa 747.1989 (M+Na)+ found 747.2045.
N1-(5′-O-Phosphoryl-β-L-ribofuranosyl)−8-bromohypoxanthine (8-Br-L-N1-IMP, 26)–N1-[2′,3′-O-Isopropylidene−5′-O-(di-tert-butyl)-phosphoryl-β-L-ribofuranosyl]-N9-tert-butyldimethylsilyloxymethyl−8-bromohypoxanthine (55, 55 mg, 76 µmol) was treated with TFA (50% aq., 4 mL) for 16 h. All solvents were evaporated and the residue evaporated from MeOH × 3 before purification by semipreparative HPLC (1.1 cm × 25 cm C18 column), eluting with acetonitrile/0.1 M TEAB (1:19 → 13:7 v/v) over 25 min. Fractions were analyzed by analytical HPLC and appropriate fractions collected and evaporated under vacuum to give the title compound (14.4 mg, 45%); UV (H2O, pH 7), λmax = 261 nm (ε = 15,900); 1H-NMR (500 MHz, D2O) δ 8.49 (s, 1H, 2-H), 6.22 (d, J = 4.2 Hz, 1H, H−1′), 4.39 (dd, J = 5.3, 4.4 Hz, 1H, H−2′), 4.35 (dd, J = 5.3, 5,2 Hz, 1H, H−3′), 4.27−4.21 (m, 1H, H−4′), 4.07−4.03 (m, 1H, H−5′a) and 3.99−3.94 (m, 1H, H−5′b); 13C-NMR (125 MHz, D2O) δ 157.3, 156.4, 142.5, 136.3, 123.8, 88.4 (C−1′), 83.5 (d, J = 8.5 Hz, C−4′), 75.0 (C−2′), 69.4 (C−3′) and 63.1 (d, J = 4.4 Hz, C−5′); 31P-NMR (202 MHz, D2O) δ 2.82; HRMS (ES−) calcd for C10H11N4O8P79Br 424.9503 (M−H)− found 424.9524, calcd for C10H11N4O8P81Br 426.9478 (M−H)− found 426.9503.
N1-(5′-O-Phosphoryl-β-L-ribofuranosyl)-hypoxanthine (L-N1-IMP, 27)–N1-(5′-O-Phosphoryl-β-L-ribofuranosyl)−8-bromohypoxanthine (26, 5.3 mg, 14 µmol), NaHCO3 (12 mg, 0.14 mmol) and Pd/C (5 mg) were taken up in MilliQ-EtOH (2:1 v/v, 3 mL) and the solution degassed with argon before being placed under an atmosphere of H2 for 16 h. The catalyst was removed by filtration through celite and the resulting solution purified by semipreparative HPLC (1.1 cm × 25 cm C18 column), eluting with acetonitrile/0.1 M TEAB (1:19 → 13:7 v/v) over 25 min. Fractions were analyzed by analytical HPLC and appropriate fractions collected and evaporated under vacuum to give the title compound (2.1 mg, 49%); UV (H2O, pH 7), λmax = 251 nm (ε = 11,500); 1H-NMR (500 MHz, D2O) δ 8.65 (s, 1H), 8.10 (s, 1H), 6.22 (d, J = 3.8 Hz, 1H, H−1′), 4.39 (dd, J = 5.3, 3.8 Hz, 1H, H−2′), 4.36 (dd, J = 5.5, 5,3 Hz, 1H, H−3′), 4.27−4.23 (m, 1H, H−4′), 4.16−4.10 (m, 1H, H−5′a) and 4.04−4.00 (m, 1H, H−5′b); 13C-NMR (125 MHz, D2O) δ 156.3, (147), 144.7, 141.8, (122)*, 88.9 (C−1′), 83.2 (d, J = 8.4 Hz, C−4′), 75.0 (C−2′), 68.9 (C−3′) and 63.0 (d, J = 3.4 Hz, C−5′); 31P-NMR (202 MHz, D2O) δ 1.71. HRMS (ES−) calcd for C10H12N4O8P 347.0398 (M−H)− found 347.0409. * Only three of 5 hypoxanthine C visible in 13C, the values in parenthesis are estimates.
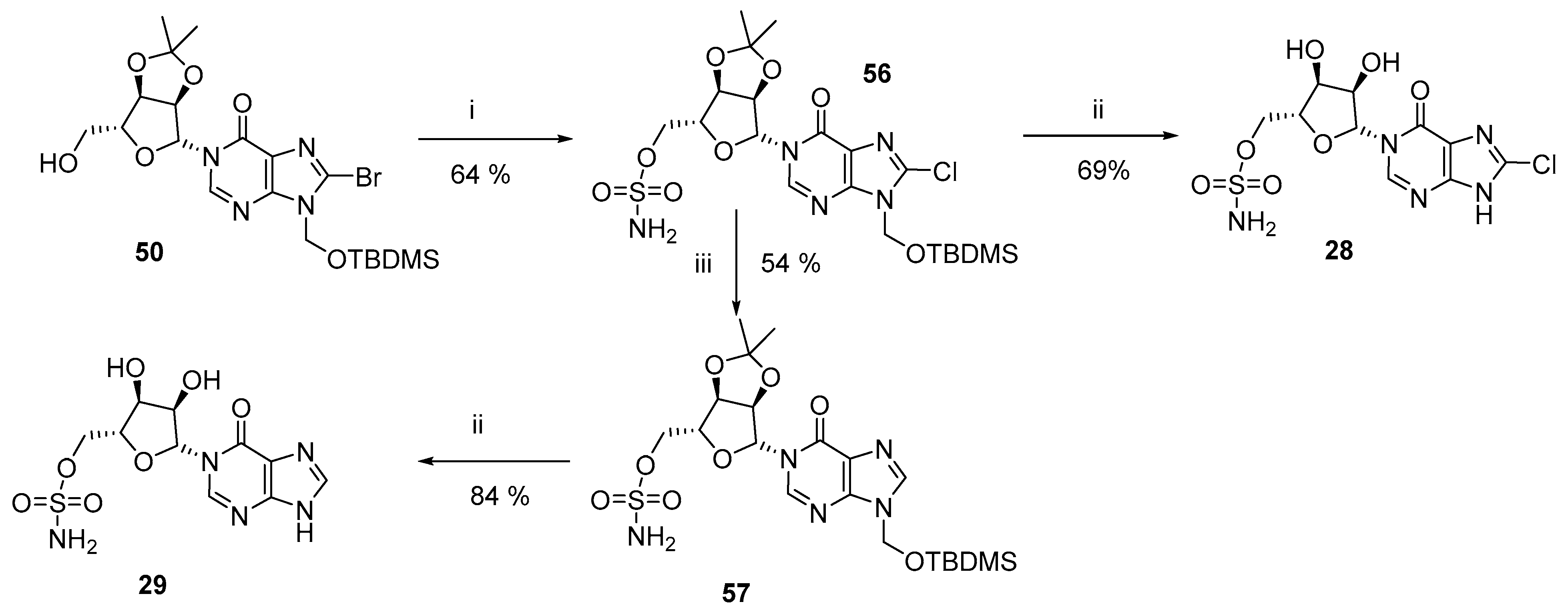
3.7. Total Synthesis of Pyrophosphate Bioisostere Analogues (28–29)
N1-(2′,3′-O-Isopropylidene−5′-O-sulfonamide-β-D-ribofuranosyl)-N9-tert-butyldimethylsilyloxymethyl−8-chlorohypoxanthine (56)–N1-(2′,3′-O-Isopropylidene-β-D-ribofuranosyl)-N9-tert-butyldimethylsilyl-oxymethyl−8-bromohypoxanthine (50, 150 mg, 0.28 mmol) was taken up in DCM (1.5 mL) and cooled to 0 °C. Triethylamine (47 µL, 0.34 mmol) was added and the solution stirred for 30 min before dropwise addition of sulfamoyl chloride in toluene (1.28 mL, 0.56 mmol, 0.44 M solution). After 16 h at rt, MeOH (1 mL) was added and all solvent evaporated under reduced pressure. The residue was purified by column chromatography on silica gel eluting with PE/EtOAc (1:0 → 0:1 v/v) to afford the title compound (102 mg, 64%) as acolourless glass; Rf = 0.68 (PE:EtOAc 1:3 v/v); 1H-NMR (400 MHz, CDCl3) δ 8.02 (s, 1H, 2-H), 5.77 (d, J = 1.4 Hz, 1H, H−1′), 5.67 (d, J = 9.8 Hz, 1H), 5.64 (d, J = 9.8 Hz, 1H) (2H, CH2OTBDMS), 5.40 (br s, 2H, NH2), 5.25 (dd, J = 6.5, 1.4 Hz, 1H, H−2′), 5.08−5.05 (m, 1H, H−3′), 4.51−4.44 (m, 3H, H−4′, 2 × H−5′), 1.56 (s, 3H, CCH3), 1.34 (s, 3H, CCH3), 0.88 (s, 9H), 0.14 (s, 3H), 0.12 (s, 3H) (15H, TBDMS); 13C-NMR (100 MHz, CDCl3) δ 155.2, 148.4, 147.0, 138.2, 123.0, 114.5 (C(CH3)2), 96.7 (C−1′), 86.7 (C−4′), 84.5 (C−2′), 81.8 (C−3′), 69.8 (C−5′), 66.8 (CH2OTBDMS), 27.0 (CCH3), 25.5 (3C, SiC(CH3)3), 25.1 (CCH3), 18.0 (SiC(CH3)3), −5.3 (2C, Si(CH3)2); HRMS (ES+) calcd for C20H32N5O8SSi35ClNa 588.1322 (M+Na)+ found 588.1317, calcd for C20H32N5O8Ssi37ClNa 590.1294 (M+Na)+ found 590.1347.
N1-(5′-O-sulfonamide-β-D-ribofuranosyl)−8-chlorohypoxanthine (8-Cl-N1-IMS, 28)–N1-(2′,3′-O-Isopropylidene−5′-O-sulfonamide-β-D-ribofuranosyl)-N9-tert-butyldimethylsilyloxymethyl−8-chlorohypo-xanthine (56, 15 mg, 25 µmol) was cooled to 0 °C and H2O (1 mL) then TFA (1 mL) added. The solution was allowed to warm to rt and stirred for 3 h. All solvent was evaporated and the residue co-evaporated with MeOH (×3). The residue was purified by column chromatography on silica gel eluting with DCM/MeOH (1:0 → 4:1 v/v) to afford the title compound (7 mg, 69%) as a colourless glass. The final compound was further purified by semipreparative HPLC (1.1 cm × 25 cm C18 column), eluting with acetonitrile/0.1 M TEAB (1:19 → 13:7 v/v) over 25 min. Fractions were analyzed by analytical HPLC and appropriate fractions collected and evaporated under vacuum; Rf = 0.59 (DCM:MeOH 9:1 v/v); 1H-NMR (500 MHz, D2O) δ 8.13 (s, 1H, H−2), 6.05 (d, J = 2.2 Hz, 1H, H−1′), 4.47−4.44 (m, 1H, H−2′), 4.36−4.33 (m, 2H, H−3′, H−4′) and 4.29−4.26 (m, 2H, 2 × H−5); 13C-NMR (125 MHz, D2O) δ 156.2, 150.1, 142.3, 142.1, 123.4, 90.2, 80.9, 74.2, 68.9 and 68.5; HRMS (ES−) calcd for C10H1135Cl N5O7S 380.0062 (M−H)− found 380.0099, calcd for C10H1137Cl N5O7S 382.0033 (M−H)− found 382.0076.
N1-(2′,3′-O-Isopropylidene−5′-O-sulfonamide-β-D-ribofuranosyl)-N9-tert-butyldimethylsilyloxymethyl-hypoxanthine (57)–N1-(2′,3′-O-Isopropylidene−5′-O-sulfonamide-β-D-ribofuranosyl)-N9-tert-butyldimethylsilyloxymethyl−8-chlorohypoxanthine (56, 15 mg, 25 µmol) was taken up in EtOH (1 mL). Pd/C (<1 mg, 10 mol %) and NaHCO3 (11 mg, 0.125 mmol) were added and the flask evacuated and purged with Argon (×3) before placing under an atmosphere of H2. After stirring for 16 h, the suspension was filtered through cotton wool to remove the catalyst and all solvent evaporated under reduced pressure. The residue was purified by column chromatography on silica gel eluting with DCM/MeOH (1:0 → 4:1 v/v) to afford the title compound (7 mg, 54%) as a colourless glass; Rf = 0.32 (PE:EtOAc 1:3 v/v); 1H-NMR (400 MHz, CDCl3) δ 8.00 (s, 1H, 2-H), 7.93 (s, 1H, 8-H), 5.78 (d, J = 1.3 Hz, 1H, H−1′), 5.69 (d, J = 9.6 Hz, 1H), 5.66 (d, J = 9.6 Hz, 1H) (2H, CH2OTBDMS), 5.43 (br s, 2H, NH2), 5.29 (dd, J = 6.4, 1.3 Hz, 1H, H−2′), 5.10 (dd, J = 6.4, 2.8 Hz, 1H, H−3′), 4.53−4.46 (m, 3H, H−4′, 2 × H−5′), 1.57 (s, 3H, CCH3), 1.35 (s, 3H, CCH3), 0.88 (s, 9H), 0.12 (s, 3H), 0.11 (s, 3H) (15H, TBDMS); 13C-NMR (125 MHz, CDCl3) δ 156.4, 147.4, 146.8, 140.4, 124.2, 114.5 (C(CH3)2), 96.8 (C−1′), 86.8 (C−4′), 84.6 (C−2′), 81.9 (C−3′), 69.9 (C−5′), 67.7 (CH2OTBDMS), 27.0 (CCH3), 25.5 (3C, SiC(CH3)3), 25.2 (CCH3), 18.0 (SiC(CH3)3), −5.2 (2C, Si(CH3)2); HRMS (ES+) calcd for C20H34N5O8SSi 532.1892 (M+Na)+ found 532.1909.
N1-(5′-O-sulfonamide-β-D-ribofuranosyl)-hypoxanthine (N1-IMS, 29)–N1-(2′,3′-O-Isopropylidene−5′-O-sulfonamide-β-D-ribofuranosyl)-N9-tert-butyldimethylsilyloxy-methylhypoxanthine (57, 5.5 mg, 10 µmol) was cooled to 0 °C and H2O (1 mL) then TFA (1 mL) added. The solution was allowed to warm to rt and stirred for 3 h. All solvent was evaporated and the residue co-evaporated with MeOH (×3). The residue was purified by column chromatography on silica gel eluting with DCM/MeOH (1:0 → 4:1 v/v) to afford the title compound (3 mg, 84%) as a colourless glass; 1H-NMR (500 MHz, D2O) δ 8.32 (s, 1H), 8.18 (br s, 1H), 6.06 (d, J = 3.3 Hz, 1H), 4.48 (dd, J = 11.4, 1.9 Hz, 1H), 4.36 (dd, J = 11.4, 3.3 Hz, 1H), 4.33 (dd, J = 4.6, 3.5 Hz, 1H) and 4.29−4.25 (m, 2H); 13C-NMR (125 MHz, D2O) δ 155.7, 151.2, 145.0, 141.9, 117.7, 90.4, 81.1, 74.2, 68.8 and 68.3; HRMS (ES−) calcd for C10H13N5O7SNa 370.0428 (M−H)− found 370.0437.
3.8. Enzymatic Assay for cADPR Hydrolysis
The inhibition of cADPR hydrolysis by various concentrations of inhibitor (0–1 mM) was determined by incubating 1 µM cADPR with 1 µg/mL of CD38 for 10 min at 20–24°C in 25 mM sodium acetate, pH 4.5. The reaction was stopped by the addition of 150 mM HCl. The precipitated protein was filtered, and the pH was neutralized with Tris base. After diluting the mixture 20-fold, the concentration of the unhydrolyzed cADPR present in the diluted reaction mixture was assayed by the fluorimetric cycling assay as previously described [
46].
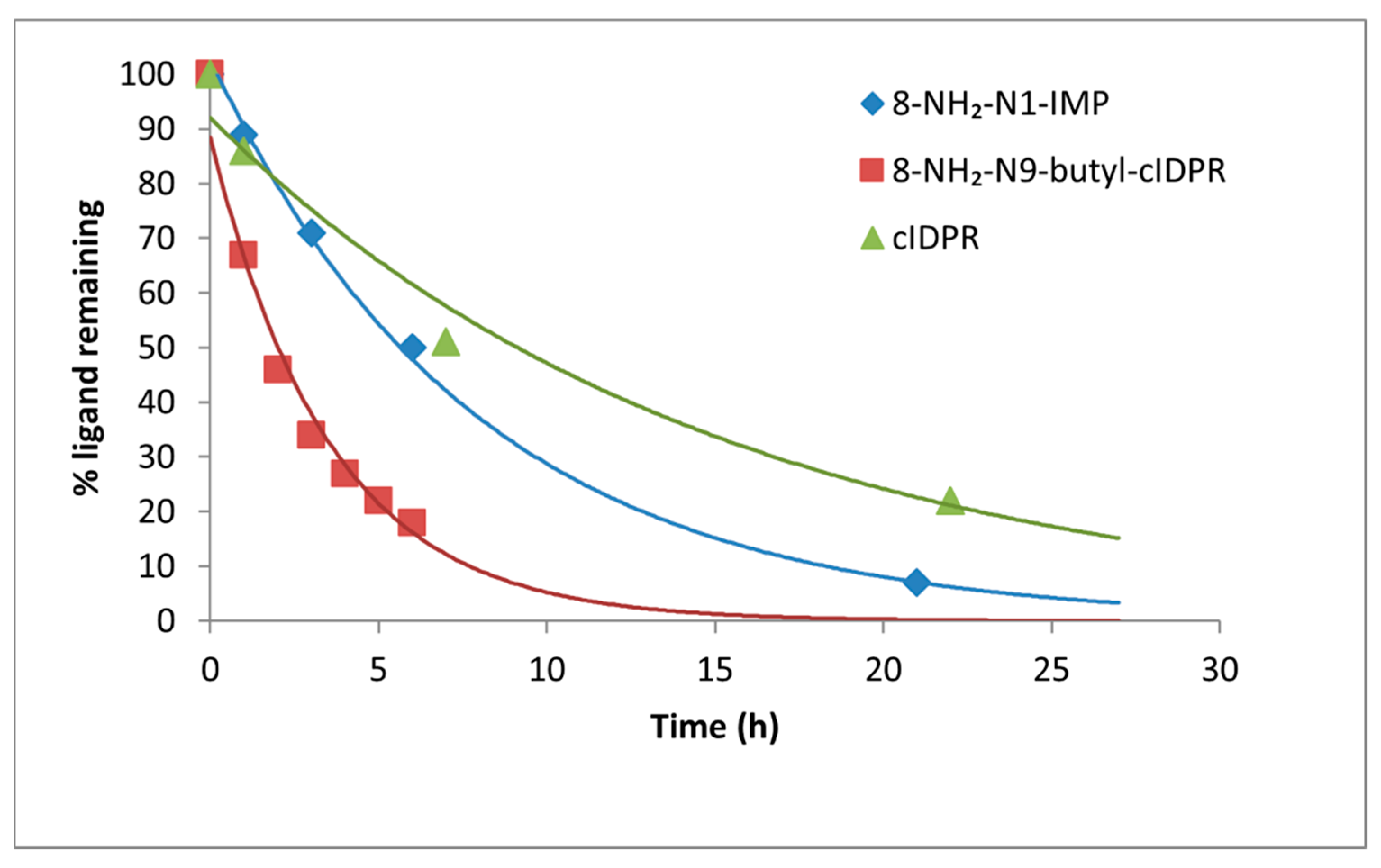
3.9. HPLC Studies
HPLC studies were carried out as previously described [
36]. Briefly, the solution containing shCD38 was adjusted to the desired concentration (4 mg/mL) using Tris-HCl buffer (20 mM, pH 8) and 50 µL was added to the inhibitor (0.05 µmole in MilliQ (2 mL)–1 mM final concentration) in an Eppendorf tube at room temperature (T = 0). At a given time point, a sample of 5 µL was removed and diluted with 95 µL MilliQ water. 10 µL Of this sample was injected directly into the analytical HPLC system (see General Experimental), eluting at 1 mL/min with an isocratic ion-pair buffer: 0.17% (m/v) cetrimide and 45% (
v/
v) phosphate buffer (pH 6.4) in MeOH.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}