
Synthetic Heterocyclic Derivatives as Kinase Inhibitors Tested for the Treatment of Neuroblastoma †

 ,
,  ,
,
Abstract
:1. Introduction
2. STK Inhibitors
2.1. CDK Inhibitors
2.1.1. Roniciclib
2.1.2. Ribociclib
2.1.3. Palbociclib
2.1.4. Dinaciclib
2.1.5. THZ1
2.2. Aurora Kinases (AURKs) Inhibitors
2.2.1. MLN8054 and Alisertib
2.2.2. Tozasertib
2.2.3. Barasertib
2.3. PLK Inhibitors
2.3.1. Volasertib
2.3.2. BI2536
2.3.3. GSK461364
2.3.4. UMB103
2.4. GSK-3 Inhibitors
2.4.1. AR-A014418
2.4.2. Tideglusib
2.4.3. 9-ING-41
2.4.4. LY2090314
2.5. Checkpoint Kinase 1 and 2 (CHEK1-2) Inhibitors
2.5.1. Prexasertib
2.5.2. PF-477736
2.6. PIM Inhibitors
AZD1208
2.7. DYRK Inhibitors
Harmine
2.8. Unc-51 Like Autophagy Kinase 1 (ULK1) Inhibitors
SBI-0206965
2.9. PAK4 Inhibitors
PF-3758309
2.10. PI3K/AKT/mTOR Inhibitors
2.10.1. Dactolisib
2.10.2. IBL-302
2.10.3. Alpelisib
2.10.4. LY294002 e SF1126
2.10.5. PI-103
2.10.6. Perifosine
2.10.7. Rapamycin and MK-066
2.10.8. Temsirolimus
2.10.9. VS-5584
3. MEK Inhibitors
3.1. Trametinib and CH5126766
3.2. Binimetinib
3.3. Cobimetinib
4. TK Inhibitors
4.1. ALK Inhibitors
4.1.1. Crizotinib
4.1.2. Alectinib
4.1.3. Ceritinib
4.1.4. Brigatinib
4.1.5. Lorlatinib
4.1.6. Entrectinib
4.1.7. TAE684
4.1.8. Other Studies on ALK Inhibitors in NB Models
4.2. TRK Inhibitors
4.2.1. AZ-23
4.2.2. GNF-4256
4.2.3. GZD2202
4.2.4. Other Compounds
4.3. FGFR Inhibitors
4.3.1. Erdafitinib
4.3.2. AZD4547
4.4. EGFR Inhibitors
Afatinib
4.5. AXL Inhibitors
4.6. FAK Inhibitors
4.7. Dual Src/Bcr-Abl Inhibitors
4.7.1. Dasatinib
4.7.2. Bosutinib
4.7.3. Ponatinib
4.7.4. Other Compounds
5. Multitargeted Inhibitors
5.1. Sorafenib
5.2. Cabozantinib
5.3. Agerafenib
5.4. Regorafenib
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Castleberry, R.P. Neuroblastoma. Eur. J. Cancer Part A 1997, 33, 1430–1437. [Google Scholar] [CrossRef]
- Mayor, R.; Theveneau, E. The neural crest. Development 2012, 140, 2247–2251. [Google Scholar] [CrossRef] [Green Version]
- Whittle, S.B.; Smith, V.; Doherty, E.; Zhao, S.; McCarty, S.; Zage, P.E. Overview and recent advances in the treatment of neuroblastoma. Expert Rev. Anticancer Ther. 2017, 17, 369–386. [Google Scholar] [CrossRef] [Green Version]
- Valter, K.; Zhivotovsky, B.; Gogvadze, V. Cell death-based treatment of neuroblastoma. Cell Death Dis. 2018, 9, 113. [Google Scholar] [CrossRef] [Green Version]
- Cohn, S.L.; Pearson, A.D.J.J.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG task force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Davidoff, A.M. Neuroblastoma. Semin. Pediatr. Surg. 2012, 21, 2–14. [Google Scholar] [CrossRef] [Green Version]
- Nakagawara, A.; Li, Y.; Izumi, H.; Muramori, K.; Inada, H.; Nishi, M. Neuroblastoma. Jpn. J. Clin. Oncol. 2018, 48, 214–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef] [PubMed]
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef]
- Fusco, P.; Mattiuzzo, E.; Frasson, C.; Viola, G.; Cimetta, E.; Esposito, M.R.; Tonini, G.P. Verteporfin induces apoptosis and reduces the stem cell-like properties in Neuroblastoma tumour-initiating cells through inhibition of the YAP/TAZ pathway. Eur. J. Pharmacol. 2021, 893, 173829. [Google Scholar] [CrossRef]
- Brodowska, K.; Al-Moujahed, A.; Marmalidou, A.; Meyer zu Horste, M.; Cichy, J.; Miller, J.W.; Gragoudas, E.; Vavvas, D.G. The clinically used photosensitizer Verteporfin (VP) inhibits YAP-TEAD and human retinoblastoma cell growth invitro without light activation. Exp. Eye Res. 2014, 124, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Coggins, G.E.; Farrel, A.; Rathi, K.S.; Hayes, C.M.; Scolaro, L.; Rokita, J.L.; Maris, J.M. YAP1 mediates resistance to MEK1/2 inhibition in neuroblastomas with hyperactivated Ras signaling. Cancer Res. 2019, 79, 6204–6214. [Google Scholar] [CrossRef] [Green Version]
- Van Groningen, T.; Koster, J.; Valentijn, L.J.; Zwijnenburg, D.A.; Akogul, N.; Hasselt, N.E.; Broekmans, M.; Haneveld, F.; Nowakowska, N.E.; Bras, J.; et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat. Genet. 2017, 49, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.E.; Maldonado, N.V.; Khankaldyyan, V.; Shimada, H.; Song, M.M.; Maurer, B.J.; Reynolds, C.P. Reactive Oxygen Species Mediates the Synergistic Activity of Fenretinide Combined with the Microtubule Inhibitor ABT-751 against Multidrug-Resistant Recurrent Neuroblastoma Xenografts. Mol. Cancer Ther. 2016, 15, 2653–2664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, J.S.; El-Khoueiry, A.B.; Maurer, B.J.; Groshen, S.; Pinski, J.K.; Cobos, E.; Gandara, D.R.; Lenz, H.J.; Kang, M.H.; Reynolds, C.P.; et al. A phase I study of intravenous fenretinide (4-HPR) for patients with malignant solid tumors. Cancer Chemother. Pharmacol. 2021, 87, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Navid, F.; Armstrong, M.; Barfield, R.C. Immune therapies for neuroblastoma. Cancer Biol. Ther. 2009, 8, 874–882. [Google Scholar] [CrossRef] [Green Version]
- Cheresh, D.A.; Pierschbacher, M.D.; Herzig, M.A.; Mujoo, K. Disialogangliosides GD2 and GD3 are involved in the attachment of human melanoma and neuroblastoma cells to extracellular matrix proteins. J. Cell Biol. 1986, 102, 688–696. [Google Scholar] [CrossRef]
- FDA Grants Accelerated Approval to Naxitamab for High-Risk Neuroblastoma in Bone or Bone Marrow. Available online: https://ascopost.com/issues/december-10-2020/fda-grants-accelerated-approval-to-naxitamab-for-high-risk-neuroblastoma-in-bone-or-bone-marrow/ (accessed on 10 November 2021).
- Yu, A.L.L.; Gilman, A.L.L.; Ozkaynak, M.F.F.; London, W.B.B.; Kreissman, S.G.G.; Chen, H.X.X.; Smith, M.; Anderson, B.; Villablanca, J.G.G.; Matthay, K.K.K.; et al. Anti-GD2 Antibody with GM-CSF, Interleukin-2, and Isotretinoin for Neuroblastoma. N. Engl. J. Med. 2010, 363, 1324–1334. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Spetz, M.R.; Li, D.; Ho, M. Advances in immunotherapeutic targets for childhood cancers: A focus on glypican-2 and B7-H3. Pharmacol. Ther. 2021, 223, 107892. [Google Scholar] [CrossRef]
- Duong-Ly, K.C.; Peterson, J.R. The human kinome and kinase inhibition. Curr. Protoc. Pharmacol. 2013, 60, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Bellantoni, A.J.; Wagner, L.M. Pursuing Precision: Receptor Tyrosine Kinase Inhibitors for Treatment of Pediatric Solid Tumors. Cancers 2021, 13, 3531. [Google Scholar] [CrossRef] [PubMed]
- Greengard, E.G. Molecularly Targeted Therapy for Neuroblastoma. Children 2018, 5, 142. [Google Scholar] [CrossRef] [Green Version]
- Pacenta, H.L.; Macy, M.E. Entrectinib and other ALK/TRK inhibitors for the treatment of neuroblastoma. Drug Des. Dev. Ther. 2018, 12, 3549–3561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, S.S.; Kornev, A.P. Protein kinases: Evolution of dynamic regulatory proteins. Trends Biochem. Sci. 2011, 36, 65–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine kinase–role and significance in cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Manning, G.; Plowman, G.D.; Hunter, T.; Sudarsanam, S. Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci. 2002, 27, 514–520. [Google Scholar] [CrossRef]
- Musumeci, F.; Greco, C.; Grossi, G.; Molinari, A.; Schenone, S. Recent studies on ponatinib in cancers other than chronic myeloid leukemia. Cancers 2018, 10, 430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozharski, E.; Zhang, J.; Shapiro, P. ATP-Bound Form of the ERK2 Kinase. Available online: https://www.rcsb.org/structure/4GT3 (accessed on 10 November 2021).
- The PyMOL Molecular Graphics System; Version 2.0; Schrödinger, Inc.: New York, NY, USA, 2017.
- Molenaar, J.J.; Ebus, M.E.; Koster, J.; Van Sluis, P.; Van Noesel, C.J.M.; Versteeg, R.; Caron, H.N. Cyclin D1 and CDK4 activity contribute to the undifferentiated phenotype in neuroblastoma. Cancer Res. 2008, 68, 2599–2609. [Google Scholar] [CrossRef] [Green Version]
- Swadi, R.R.; Sampat, K.; Herrmann, A.; Losty, P.D.; See, V.; Moss, D.J. CDK inhibitors reduce cell proliferation and reverse hypoxia-induced metastasis of neuroblastoma tumours in a chick embryo model. Sci. Rep. 2019, 9, 9136. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Wang, Z.; Pang, J.C.; Yu, Y.; Bieerkehazhi, S.; Lu, J.; Hu, T.; Zhao, Y.; Xu, X.; Zhang, H.; et al. Multiple CDK inhibitor dinaciclib suppresses neuroblastoma growth via inhibiting CDK2 and CDK9 activity. Sci. Rep. 2016, 6, 29090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayaz, P.; Andres, D.; Kwiatkowski, D.A.; Kolbe, C.-C.; Lienau, P.; Siemeister, G.; Lücking, U.; Stegmann, C.M. Conformational Adaption May Explain the Slow Dissociation Kinetics of Roniciclib (BAY 1000394), a Type I CDK Inhibitor with Kinetic Selectivity for CDK2 and CDK9. ACS Chem. Biol. 2016, 11, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Horn, L.; Novello, S.; Barlesi, F.; Albert, I.; Juhász, E.; Kowalski, D.; Robinet, G.; Cadranel, J.; Bidoli, P.; et al. Phase II Study of Roniciclib in Combination with Cisplatin/Etoposide or Carboplatin/Etoposide as First-Line Therapy in Patients with Extensive-Disease Small Cell Lung Cancer. J. Thorac. Oncol. 2019, 14, 701–711. [Google Scholar] [CrossRef]
- Yang, X.; Tang, F.; Shin, J.; Cunningham, J.M. A c-Myc-regulated stem celllike signature in high-risk neuroblastoma: A systematic discovery (target neuroblastoma ESC-like signature). Sci. Rep. 2017, 7, 41. [Google Scholar] [CrossRef]
- Ognibene, M.; Pezzolo, A. Roniciclib down-regulates stemness and inhibits cell growth by inducing nucleolar stress in neuroblastoma. Sci. Rep. 2020, 10, 12902. [Google Scholar] [CrossRef]
- Ayaz, P.; Andres, D.; Kwiatkowski, D.A.; Kolbe, C.; Lienau, P.; Siemeister, G.; Luecking, U.; Stegmann, C.M. Crystal Structure of BAY 1000394 (Roniciclib) Bound to CDK2. Available online: https://www.rcsb.org/structure/5IEV (accessed on 10 November 2021).
- Tripathy, D.; Bardia, A.; Sellers, W.R. Ribociclib (LEE011): Mechanism of Action and Clinical Impact of This Selective Cyclin-Dependent Kinase 4/6 Inhibitor in Various Solid Tumors. Clin. Cancer Res. 2017, 23, 3251–3262. [Google Scholar] [CrossRef] [Green Version]
- Besong, G.; Brain, C.T.; Brooks, C.A.; Congreve, M.S.; Dagostin, C.; He, G.; Hou, Y.; Howard, S.; Li, Y.; Lu, Y.; et al. Preparation of Pyrrolopyrimidine Compounds as CDK Inhibitors. International Application No. PCT/WO2010/020675A1, 25 February 2010. [Google Scholar]
- Rader, J.; Russell, M.R.; Hart, L.S.; Nakazawa, M.S.; Belcastro, L.T.; Martinez, D.; Li, Y.; Carpenter, E.L.; Attiyeh, E.F.; Diskin, S.J.; et al. Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma. Clin. Cancer Res. 2013, 19, 6173–6182. [Google Scholar] [CrossRef] [Green Version]
- Syed, Y.Y. Ribociclib: First Global Approval. Drugs 2017, 77, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Geoerger, B.; Bourdeaut, F.; DuBois, S.G.; Fischer, M.; Geller, J.I.; Gottardo, N.G.; Marabelle, A.; Pearson, A.D.J.; Modak, S.; Cash, T.; et al. A phase I study of the CDK4/6 inhibitor ribociclib (LEE011) in pediatric patients with malignant rhabdoid tumors, neuroblastoma, and other solid tumors. Clin. Cancer Res. 2017, 23, 2433–2441. [Google Scholar] [CrossRef] [Green Version]
- Hart, L.S.; Rader, J.A.; Raman, P.; Batra, V.; Russell, M.R.; Tsang, M.; Gagliardi, M.; Chen, L.; Martinez, D.; Li, Y.; et al. Preclinical therapeutic synergy of MEK1/2 and CDK4/6 inhibition in neuroblastoma. Clin. Cancer Res. 2017, 23, 1785–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, A.C.; Krytska, K.; Ryles, H.T.; Infarinato, N.R.; Sano, R.; Hansel, T.D.; Hart, L.S.; King, F.J.; Smith, T.R.; Ainscow, E.; et al. Dual ALK and CDK4/6 inhibition demonstrates synergy against neuroblastoma. Clin. Cancer Res. 2017, 23, 2856–2868. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Ferre, R.A.; Deihl, W.; Yu, X.; He, Y.-A. The X-ray Co-Crystal Structure of Human CDK6 and Ribociclib. Available online: https://www.wwpdb.org/pdb?id=pdb_00005l2t (accessed on 10 November 2021).
- Chen, P.; Lee, N.V.; Hu, W.; Xu, M.; Ferre, R.A.; Lam, H.; Bergqvist, S.; Solowiej, J.; Diehl, W.; He, Y.A.; et al. Spectrum and Degree of CDK Drug Interactions Predicts Clinical Performance. Mol. Cancer Ther. 2016, 15, 2273–2281. [Google Scholar] [CrossRef] [Green Version]
- Fry, D.W.; Harvey, P.J.; Keller, P.R.; Elliott, W.L.; Meade, M.; Trachet, E.; Albassam, M.; Zheng, X.; Leopold, W.R.; Pryer, N.K.; et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther. 2004, 3, 1427–1438. [Google Scholar] [PubMed]
- Rihani, A.; Vandesompele, J.; Speleman, F.; Van Maerken, T. Inhibition of CDK4/6 as a novel therapeutic option for neuroblastoma. Cancer Cell Int. 2015, 15, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, K.; Shimanuki, M.; Shikami, M.; Andreeff, M.; Nakakuma, H. Cyclin-dependent kinase 1 inhibitor RO-3306 enhances p53-mediated Bax activation and mitochondrial apoptosis in AML. Cancer Sci. 2009, 100, 1128–1136. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Ferre, R.A.; Deihl, W.; Yu, X.; He, Y.-A. The X-ray Co-Crystal Structure of Human CDK6 and Palbociclib. Available online: https://www.rcsb.org/structure/5l2i (accessed on 10 November 2021).
- Huang, M.; Zeki, J.; Sumarsono, N.; Coles, G.L.; Taylor, J.S.; Danzer, E.; Bruzoni, M.; Hazard, F.K.; Lacayo, N.J.; Sakamoto, K.M.; et al. Epigenetic targeting of TERT-associated gene expression signature in human neuroblastoma with TERT overexpression. Cancer Res. 2020, 80, 1024–1035. [Google Scholar] [CrossRef]
- Wood, L.; Huang, M.; Zeki, J.; Gong, M.; Taylor, J.; Shimada, H.; Chiu, B. Combining inhibitors of Brd4 and cyclin-dependent kinase can decrease tumor growth in neuroblastoma with MYCN amplification. J. Pediatr. Surg. 2021, 56, 1199–1202. [Google Scholar] [CrossRef]
- Martin, M.P.; Schonbrunn, E. CDK2 in Complex with Dinaciclib. Available online: https://www.ebi.ac.uk/pdbe/entry/pdb/4KD1 (accessed on 10 November 2021).
- Martin, M.P.; Olesen, S.H.; Georg, G.I.; Schönbrunn, E. Cyclin-dependent kinase inhibitor dinaciclib interacts with the acetyl-lysine recognition site of bromodomains. ACS Chem. Biol. 2013, 8, 2360–2365. [Google Scholar] [CrossRef]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef] [Green Version]
- Tee, A.E.; Ciampa, O.C.; Wong, M.; Fletcher, J.I.; Kamili, A.; Chen, J.; Ho, N.; Sun, Y.; Carter, D.R.; Cheung, B.B.; et al. Combination therapy with the CDK7 inhibitor and the tyrosine kinase inhibitor exerts synergistic anticancer effects against MYCN-amplified neuroblastoma. Int. J. Cancer 2020, 147, 1928–1938. [Google Scholar] [CrossRef]
- Greber, B.J.; Perez-Bertoldi, J.M.; Lim, K.; Iavarone, A.T.; Toso, D.B.; Nogales, E. The cryoelectron microscopy structure of the human CDK-activating kinase. Proc. Natl. Acad. Sci. USA 2020, 117, 22849–22857. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Kuang, J.; Zhong, L.; Kuo, W.L.; Gray, J.W.; Sahin, A.; Brinkley, B.R.; Sen, S. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat. Genet. 1998, 20, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.; Horn, S.; Brockmann, M.; Eilers, U.; Schüttrumpf, L.; Popov, N.; Kenney, A.M.; Schulte, J.H.; Beijersbergen, R.; Christiansen, H.; et al. Stabilization of N-Myc Is a Critical Function of Aurora A in Human Neuroblastoma. Cancer Cell 2009, 15, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Richards, M.W.; Burgess, S.G.; Poon, E.; Carstensen, A.; Eilers, M.; Chesler, L.; Bayliss, R. Structural basis of N-Myc binding by Aurora-A and its destabilization by kinase inhibitors. Proc. Natl. Acad. Sci. USA 2016, 113, 13726–13731. [Google Scholar] [CrossRef] [Green Version]
- Shang, X.; Burlingame, S.M.; Okcu, M.F.; Ge, N.; Russell, H.V.; Egler, R.A.; David, R.D.; Vasudevan, S.A.; Yang, J.; Nuchtern, J.G. Aurora A is a negative prognostic factor and a new therapeutic target in human neuroblastoma. Mol. Cancer Ther. 2009, 8, 2461–2469. [Google Scholar] [CrossRef] [Green Version]
- Manfredi, M.G.; Ecsedy, J.A.; Meetze, K.A.; Balani, S.K.; Burenkova, O.; Chen, W.; Galvin, K.M.; Hoar, K.M.; Huck, J.J.; LeRoy, P.J.; et al. Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora a kinase. Proc. Natl. Acad. Sci. USA 2007, 104, 4106–4111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brockmann, M.; Poon, E.; Berry, T.; Carstensen, A.; Deubzer, H.E.; Rycak, L.; Jamin, Y.; Thway, K.; Robinson, S.P.; Roels, F.; et al. Small molecule inhibitors of aurora-a induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer Cell 2013, 24, 75–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matulonis, U.A.; Sharma, S.; Ghamande, S.; Gordon, M.S.; Del Prete, S.A.; Ray-Coquard, I.; Kutarska, E.; Liu, H.; Fingert, H.; Zhou, X.; et al. Phase II study of MLN8237 (alisertib), an investigational Aurora A kinase inhibitor, in patients with platinum-resistant or -refractory epithelial ovarian, fallopian tube, or primary peritoneal carcinoma. Gynecol. Oncol. 2012, 127, 63–69. [Google Scholar] [CrossRef]
- Carol, H.; Boehm, I.; Reynolds, C.P.; Kang, M.H.; Maris, J.M.; Morton, C.L.; Gorlick, R.; Kolb, E.A.; Keir, S.T.; Wu, J.; et al. Efficacy and pharmacokinetic/pharmacodynamic evaluation of the Aurora kinase A inhibitor MLN8237 against preclinical models of pediatric cancer. Cancer Chemother. Pharmacol. 2011, 68, 1291–1304. [Google Scholar] [CrossRef] [Green Version]
- DuBois, S.G.; Marachelian, A.; Fox, E.; Kudgus, R.A.; Reid, J.M.; Groshen, S.; Malvar, J.; Bagatell, R.; Wagner, L.; Maris, J.M.; et al. Phase I study of the aurora A kinase inhibitor alisertib in combination with irinotecan and temozolomide for patients with relapsed or refractory neuroblastoma: A nant (new approaches to neuroblastoma therapy) trial. J. Clin. Oncol. 2016, 34, 1368–1375. [Google Scholar] [CrossRef] [Green Version]
- DuBois, S.G.; Mosse, Y.P.; Fox, E.; Kudgus, R.A.; Reid, J.M.; McGovern, R.; Groshen, S.; Bagatell, R.; Maris, J.M.; Twist, C.J.; et al. Phase II trial of alisertib in combination with irinotecan and temozolomide for patients with relapsed or refractory neuroblastoma. Clin. Cancer Res. 2018, 24, 6142–6149. [Google Scholar] [CrossRef] [Green Version]
- Kollareddy, M.; Sherrard, A.; Park, J.H.; Szemes, M.; Gallacher, K.; Melegh, Z.; Oltean, S.; Michaelis, M.; Cinatl, J.; Kaidi, A.; et al. The small molecule inhibitor YK-4-279 disrupts mitotic progression of neuroblastoma cells, overcomes drug resistance and synergizes with inhibitors of mitosis. Cancer Lett. 2017, 403, 74–85. [Google Scholar] [CrossRef]
- Currier, M.A.; Sprague, L.; Rizvi, T.A.; Nartker, B.; Chen, C.Y.; Wang, P.Y.; Hutzen, B.J.; Franczek, M.R.; Patel, A.V.; Chaney, K.E.; et al. Aurora A kinase inhibition enhances oncolytic herpes virotherapy through cytotoxic synergy and innate cellular immune modulation. Oncotarget 2017, 8, 17412–17427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felgenhauer, J.; Tomino, L.; Selich-Anderson, J.; Bopp, E.; Shah, N. Dual BRD4 and AURKA Inhibition Is Synergistic against MYCN-Amplified and Nonamplified Neuroblastoma. Neoplasia 2018, 20, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ding, L.; Zhou, Q.; Fen, L.; Cao, Y.; Sun, J.; Zhou, X.; Liu, A. Silencing of AURKA augments the antitumor efficacy of the AURKA inhibitor MLN8237 on neuroblastoma cells. Cancer Cell Int. 2020, 20, 9. [Google Scholar] [CrossRef]
- Savory, W.; Mueller, I.; Mason, C.S.; Lamers, M.; Williams, D.H.; Eyers, P.A. Structure of Aurora A in Complex with MLN8054. Available online: https://www.rcsb.org/structure/2X81 (accessed on 10 November 2021).
- Sloane, D.A.; Trikic, M.Z.; Chu, M.L.H.; Lamers, M.B.A.C.; Mason, C.S.; Mueller, I.; Savory, W.J.; Williams, D.H.; Eyers, P.A. Drug-resistant aurora A mutants for cellular target validation of the small molecule kinase inhibitors MLN8054 and MLN8237. ACS Chem. Biol. 2010, 5, 563–576. [Google Scholar] [CrossRef]
- Michaelis, M.; Selt, F.; Rothweiler, F.; Löschmann, N.; Nüsse, B.; Dirks, W.G.; Zehner, R.; Cinatl, J. Aurora kinases as targets in drug-resistant neuroblastoma cells. PLoS ONE 2014, 9, e108758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, C.H.; Cheung, C.H.Y.; Liu, Y.L.; Hou, C.L.; Hsu, C.L.; Huang, C.T.; Yang, T.S.; Chen, S.F.; Chen, C.N.; Hsu, W.M.; et al. Quantitative proteomics of Th-MYCN transgenic mice reveals Aurora kinase inhibitor altered metabolic pathways and enhanced ACADM to suppress neuroblastoma progression. J. Proteome Res. 2019, 18, 3850–3866. [Google Scholar] [CrossRef]
- Zhao, B.; Smallwood, A.; Lai, Z. Crystal Structure of Aurora A in Complex with VX-680 and TPX2. Available online: https://www.rcsb.org/structure/3e5a (accessed on 10 November 2021).
- Zhao, B.; Smallwood, A.; Yang, J.; Koretke, K.; Nurse, K.; Calamari, A.; Kirkpatrick, R.B.; Lai, Z. Modulation of kinase-inhibitor interactions by auxiliary protein binding: Crystallography studies on Aurora A interactions with VX-680 and with TPX2. Protein Sci. 2008, 17, 1791–1797. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Ikezoe, T.; Nishioka, C.; Tasaka, T.; Taniguchi, A.; Kuwayama, Y.; Komatsu, N.; Bandobashi, K.; Togitani, K.; Koeffler, H.P.; et al. AZD1152, a novel and selective aurora B kinase inhibitor, induces growth arrest, apoptosis, and sensitization for tubulin depolymerizing agent or topoisomerase II inhibitor in human acute leukemia cells in vitro and In Vivo. Blood 2007, 110, 2034–2040. [Google Scholar] [CrossRef] [Green Version]
- Zekri, A.; Mesbahi, Y.; Boustanipour, E.; Sadr, Z.; Ghaffari, S.H. The Potential Contribution of microRNAs in Anti-cancer Effects of Aurora Kinase Inhibitor (AZD1152-HQPA). J. Mol. Neurosci. 2018, 65, 444–455. [Google Scholar] [CrossRef]
- Roshak, A.K.; Capper, E.A.; Imburgia, C.; Fornwald, J.; Scott, G.; Marshall, L.A. The human polo-like kinase, PLK, regulates cdc2/cyclin B through phosphorylation and activation of the cdc25C phosphatase. Cell. Signal. 2000, 12, 405–411. [Google Scholar] [CrossRef]
- Strebhardt, K. Multifaceted polo-like kinases: Drug targets and antitargets for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 643–660. [Google Scholar] [CrossRef]
- Ackermann, S.; Goeser, F.; Schulte, J.H.; Schramm, A.; Ehemann, V.; Hero, B.; Eggert, A.; Berthold, F.; Fischer, M. Polo-like kinase 1 is a therapeutic target in high-risk neuroblastoma. Clin. Cancer Res. 2011, 17, 731–741. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, D.; Steegmaier, M.; Hoffmann, M.; Grauert, M.; Baum, A.; Quant, J.; Haslinger, C.; Garin-Chesa, P.; Adolf, G.R. BI 6727, a polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin. Cancer Res. 2009, 15, 3094–3102. [Google Scholar] [CrossRef] [Green Version]
- Gorlick, R.; Kolb, E.A.; Keir, S.T.; Maris, J.M.; Reynolds, C.P.; Kang, M.H.; Carol, H.; Lock, R.; Billups, C.A.; Kurmasheva, R.T.; et al. Initial testing (stage 1) of the polo-like kinase inhibitor volasertib (BI 6727), by the pediatric preclinical testing program. Pediatr. Blood Cancer 2014, 61, 158–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, G. PLK1 in Complex with BI6727. Available online: https://www.wwpdb.org/pdb?id=pdb_00003fc2 (accessed on 10 November 2021).
- Wäsch, R.; Hasskarl, J.; Schnerch, D.; Lübbert, M. BI-2536—Targeting the mitotic kinase Polo-like kinase 1 (Plk1). Recent Results Cancer Res. 2010, 184, 215–218. [Google Scholar] [CrossRef]
- Chaturvedi, N.K.; McGuire, T.R.; Coulter, D.W.; Shukla, A.; McIntyre, E.M.; Sharp, J.G.; Joshi, S.S. Improved therapy for neuroblastoma using a combination approach: Superior efficacy with vismodegib and topotecan. Oncotarget 2016, 7, 15215–15229. [Google Scholar] [CrossRef]
- Li, Z.; Yang, C.; Li, X.; Du, X.; Tao, Y.; Ren, J.; Fang, F.; Xie, Y.; Li, M.; Qian, G.; et al. The dual role of BI 2536, a small-molecule inhibitor that targets PLK1, in induction of apoptosis and attenuation of autophagy in neuroblastoma cells. J. Cancer 2020, 11, 3274–3287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.-H.; Kothe, M.; Kohls, D.; Low, S. Structure of PLK1 in Complex with BI2536. Available online: https://www.rcsb.org/structure/2RKU (accessed on 10 November 2021).
- Kothe, M.; Kohls, D.; Low, S.; Coli, R.; Rennie, G.R.; Feru, F.; Kuhn, C.; Ding, Y.H. Research Article: Selectivity-determining Residues in Plk1. Chem. Biol. Drug Des. 2007, 70, 540–546. [Google Scholar] [CrossRef]
- Gilmartin, A.G.; Bleam, M.R.; Richter, M.C.; Erskine, S.G.; Kruger, R.G.; Madden, L.; Hassler, D.F.; Smith, G.K.; Gontarek, R.R.; Courtney, M.P.; et al. Distinct concentration-dependent effects of the polo-like kinase 1-specific inhibitor GSK461364A, including differential effect on apoptosis. Cancer Res. 2009, 69, 6969–6977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajtler, K.W.; Sadowski, N.; Ackermann, S.; Althoff, K.; Schönbeck, K.; Batzke, K.; Schäfers, S.; Odersky, A.; Heukamp, L.; Astrahantseff, K.; et al. The GSK461364 PLK1 inhibitor exhibits strong antitumoral activity in preclinical neuroblastoma models. Oncotarget 2017, 8, 6730–6741. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yosief, H.O.; Dai, L.; Huang, H.; Dhawan, G.; Zhang, X.; Muthengi, A.M.; Roberts, J.; Buckley, D.L.; Perry, J.A.; et al. Structure-Guided Design and Development of Potent and Selective Dual Bromodomain 4 (BRD4)/Polo-like Kinase 1 (PLK1) Inhibitors. J. Med. Chem. 2018, 61, 7785–7795. [Google Scholar] [CrossRef] [PubMed]
- Timme, N.; Han, Y.; Liu, S.; Yosief, H.O.; García, H.D.; Bei, Y.; Klironomos, F.; MacArthur, I.C.; Szymansky, A.; von Stebut, J.; et al. Small-Molecule Dual PLK1 and BRD4 Inhibitors are Active Against Preclinical Models of Pediatric Solid Tumors. Transl. Oncol. 2020, 13, 221–232. [Google Scholar] [CrossRef]
- Liu, X.; Klein, P.S. Glycogen synthase kinase-3 and alternative splicing. Wiley Interdiscip. Rev. RNA 2018, 9, e1501. [Google Scholar] [CrossRef]
- Pizarro, J.G.; Folch, J.; Esparza, J.L.; Jordan, J.; Pallàs, M.; Camins, A. A molecular study of pathways involved in the inhibition of cell proliferation in neuroblastoma B65 cells by the GSK-3 inhibitors lithium and SB-415286. J. Cell. Mol. Med. 2009, 13, 3906–3917. [Google Scholar] [CrossRef] [Green Version]
- Carter, Y.M.; Kunnimalaiyaan, S.; Chen, H.; Gamblin, T.C.; Kunnimalaiyaan, M. Specific glycogen synthase kinase-3 inhibition reduces neuroendocrine markers and suppresses neuroblastoma cell growth. Cancer Biol. Ther. 2014, 15, 510–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, R.; Xue, Y.; Berg, S.; Hellberg, S.; Ormo, M.; Nilsson, Y.; Radesater, A.C.; Jerning, E.; Markgren, P.O.; Borgegard, T.; et al. Crystal Structure of Glycogen Synthase Kinase 3 in Complexed with Inhibitor. Available online: https://www.rcsb.org/structure/1Q5K (accessed on 10 November 2021).
- Bhat, R.; Xue, Y.; Berg, S.; Hellberg, S.; Ormö, M.; Nilsson, Y.; Radesäter, A.C.; Jerning, E.; Markgren, P.O.; Borgegård, T.; et al. Structural Insights and Biological Effects of Glycogen Synthase Kinase 3-specific Inhibitor AR-A014418. J. Biol. Chem. 2003, 278, 45937–45945. [Google Scholar] [CrossRef] [Green Version]
- Duffy, D.J.; Krstic, A.; Schwarzl, T.; Higgins, D.G.; Kolch, W. GSK3 inhibitors regulate MYCN mRNA levels and reduce neuroblastoma cell viability through multiple mechanisms, including p53 and Wnt signaling. Mol. Cancer Ther. 2014, 13, 454–467. [Google Scholar] [CrossRef] [Green Version]
- Mathuram, T.L.; Ravikumar, V.; Reece, L.M.; Karthik, S.; Sasikumar, C.S.; Cherian, K.M. Tideglusib induces apoptosis in human neuroblastoma IMR32 cells, provoking sub-G0/G1 accumulation and ROS generation. Environ. Toxicol. Pharmacol. 2016, 46, 194–205. [Google Scholar] [CrossRef]
- Bahmad, H.F.; Chalhoub, R.M.; Harati, H.; Bou-Gharios, J.; Assi, S.; Ballout, F.; Monzer, A.; Msheik, H.; Araji, T.; Elajami, M.K.; et al. Tideglusib attenuates growth of neuroblastoma cancer stem/progenitor cells in vitro and in vivo by specifically targeting GSK-3β. Pharmacol. Rep. 2021, 73, 211–226. [Google Scholar] [CrossRef]
- Ugolkov, A.V.; Bondarenko, G.I.; Dubrovskyi, O.; Berbegall, A.P.; Navarro, S.; Noguera, R.; O’Halloran, T.V.; Hendrix, M.J.; Giles, F.J.; Mazar, A.P. 9-ING-41, a small-molecule glycogen synthase kinase-3 inhibitor, is active in neuroblastoma. Anticancer Drugs 2018, 29, 717–724. [Google Scholar] [CrossRef]
- Rizzieri, D.A.; Cooley, S.; Odenike, O.; Moonan, L.; Chow, K.H.; Jackson, K.; Wang, X.; Brail, L.; Borthakur, G. An open-label phase 2 study of glycogen synthase kinase-3 inhibitor LY2090314 in patients with acute leukemia. Leuk. Lymphoma 2016, 57, 1800–1806. [Google Scholar] [CrossRef]
- Kunnimalaiyaan, S.; Schwartz, V.K.; Jackson, I.A.; Clark Gamblin, T.; Kunnimalaiyaan, M. Antiproliferative and apoptotic effect of LY2090314, a GSK-3 inhibitor, in neuroblastoma in vitro. BMC Cancer 2018, 18, 560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrassa, L.; Damia, G. DNA damage response inhibitors: Mechanisms and potential applications in cancer therapy. Cancer Treat. Rev. 2017, 60, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Lowery, C.D.; VanWye, A.B.; Dowless, M.; Blosser, W.; Falcon, B.L.; Stewart, J.; Stephens, J.; Beckmann, R.P.; Lin, A.B.; Stancato, L.F. The checkpoint kinase 1 inhibitor prexasertib induces regression of preclinical models of human neuroblastoma. Clin. Cancer Res. 2017, 23, 4354–4363. [Google Scholar] [CrossRef] [Green Version]
- Lowery, C.D.; Dowless, M.; Renschler, M.; Blosser, W.; VanWye, A.B.; Stephens, J.R.; Iversen, P.W.; Lin, A.B.; Beckmann, R.P.; Krytska, K.; et al. Broad spectrum activity of the checkpoint kinase 1 inhibitor prexasertib as a single agent or chemopotentiator across a range of preclinical pediatric tumor models. Clin. Cancer Res. 2019, 25, 2278–2289. [Google Scholar] [CrossRef]
- Blasina, A.; Hallin, J.; Chen, E.; Arango, M.E.; Kraynov, E.; Register, J.; Grant, S.; Ninkovic, S.; Chen, P.; Nichols, T.; et al. Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol. Cancer Ther. 2008, 7, 2394–2404. [Google Scholar] [CrossRef] [Green Version]
- Ando, K.; Nakamura, Y.; Nagase, H.; Nakagawara, A.; Koshinaga, T.; Wada, S.; Makishima, M. Co-inhibition of the dna damage response and chk1 enhances apoptosis of neuroblastoma cells. Int. J. Mol. Sci. 2019, 20, 3700. [Google Scholar] [CrossRef] [Green Version]
- Asati, V.; Mahapatra, D.K.; Bharti, S.K. PIM kinase inhibitors: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2019, 172, 95–108. [Google Scholar] [CrossRef]
- Brunen, D.; de Vries, R.C.; Lieftink, C.; Beijersbergen, R.L.; Bernards, R. PIM kinases are a potential prognostic biomarker and therapeutic target in neuroblastoma. Mol. Cancer Ther. 2018, 17, 849–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trigg, R.M.; Lee, L.C.; Prokoph, N.; Jahangiri, L.; Reynolds, C.P.; Amos Burke, G.A.; Probst, N.A.; Han, M.; Matthews, J.D.; Kai Lim, H.; et al. The targetable kinase PIM1 drives ALK inhibitor resistance in high-risk neuroblastoma independent of MYCN status. Nat. Commun. 2019, 10, 5428. [Google Scholar] [CrossRef] [Green Version]
- Maddika, S.; Chen, J. Protein kinase DYRK2 is a scaffold that facilitates assembly of an E3 ligase. Nat. Cell Biol. 2009, 11, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Nonaka, Y.; Goto, T.; Ohnishi, E.; Hiramatsu, T.; Kii, I.; Yoshida, M.; Ikura, T.; Onogi, H.; Shibuya, H.; et al. Development of a novel selective inhibitor of the Down syndrome-related kinase Dyrk1A. Nat. Commun. 2010, 1, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhl, K.L.; Schultz, C.R.; Geerts, D.; Bachmann, A.S. Harmine, a dual-specificity tyrosine phosphorylation-regulated kinase (DYRK) inhibitor induces caspase-mediated apoptosis in neuroblastoma. Cancer Cell Int. 2018, 18, 82. [Google Scholar] [CrossRef]
- Nonaka, Y.; Hosoya, T.; Hagiwara, M.; Ito, N. Human DYRK1A/Harmine Complex. Available online: https://www.rcsb.org/structure/3anr (accessed on 10 November 2021).
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Chun, M.G.H.H.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.C.; Sheffler, D.J.; et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell 2015, 59, 285–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dower, C.M.; Bhat, N.; Gebru, M.T.; Chen, L.; Wills, C.A.; Miller, B.A.; Wang, H.G. Targeted inhibition of ULK1 promotes apoptosis and suppresses tumor growth and metastasis in neuroblastoma. Mol. Cancer Ther. 2018, 17, 2365–2376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Chen, N.; Cui, X.; Zheng, X.; Deng, L.; Price, S.; Karantza, V.; Minden, A. The protein kinase Pak4 disrupts mammary acinar architecture and promotes mammary tumorigenesis. Oncogene 2010, 29, 5883–5894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, B.W.; Guo, C.; Piraino, J.; Westwick, J.K.; Zhang, C.; Lamerdin, J.; Dagostino, E.; Knighton, D.; Loi, C.M.; Zager, M.; et al. Small-molecule p21-activated kinase inhibitor PF-3758309 is a potent inhibitor of oncogenic signaling and tumor growth. Proc. Natl. Acad. Sci. USA 2010, 107, 9446–9451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Li, X.; Xu, L.; Tao, Y.; Yang, C.; Chen, X.; Fang, F.; Wu, Y.; Ding, X.; Zhao, H.; et al. Inhibition of neuroblastoma proliferation by PF-3758309, a small-molecule inhibitor that targets p21-activated kinase 4. Oncol. Rep. 2017, 38, 2705–2716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knighton, D.R.; Deng, Y.; Murray, B.; Guo, C.; Piraino, J.; Westwick, J.; Zhang, C.; Lamerdin, J.; Dagostino, E.; Loi, C.-M.; et al. Crystal Structure of the Human p21-Activated Kinase 4 in Complex with PF-03758309. Available online: https://www.rcsb.org/structure/2X4Z (accessed on 10 November 2021).
- Schenone, S.; Brullo, C.; Musumeci, F.; Radi, M.; Botta, M. ATP-Competitive Inhibitors of mTOR: An Update. Curr. Med. Chem. 2011, 18, 2995–3014. [Google Scholar] [CrossRef]
- Spitzenberg, V.; König, C.; Ulm, S.; Marone, R.; Röpke, L.; Müller, J.P.; Grün, M.; Bauer, R.; Rubio, I.; Wymann, M.P.; et al. Targeting PI3K in neuroblastoma. J. Cancer Res. Clin. Oncol. 2010, 136, 1881–1890. [Google Scholar] [CrossRef]
- Seitz, C.; Hugle, M.; Cristofanon, S.; Tchoghandjian, A.; Fulda, S. The dual PI3K/mTOR inhibitor NVP-BEZ235 and chloroquine synergize to trigger apoptosis via mitochondrial-lysosomal cross-talk. Int. J. Cancer 2013, 132, 2682–2693. [Google Scholar] [CrossRef]
- Durbas, M.; Horwacik, I.; Boratyn, E.; Kamycka, E.; Rokita, H. GD2 ganglioside specific antibody treatment downregulates PI3K/Akt/mTOR signaling network in human neuroblastoma cell lines. Int. J. Oncol. 2015, 47, 1143–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.-D.; Liu, Z.; Liu, H.; Ran, D.-M.; Guo, J.-H.; Jiang, B.; Wu, Y.-L.; Gao, F.-H. Oridonin enhances the anticancer activity of NVP-BEZ235 against neuroblastoma cells in vitro and in vivo through autophagy. Int. J. Oncol. 2016, 49, 657–665. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, L.; Clarke, P.A.; Barker, K.; Chanthery, Y.; Gustafson, C.W.; Tucker, E.; Renshaw, J.; Raynaud, F.; Li, X.; Burke, R.; et al. Inhibition of mTOR-kinase destabilizes MYCN and is a potential therapy for MYCN-dependent tumors. Oncotarget 2016, 7, 57525–57544. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.-L.; Liu, Z.; Zhang, L.-D.; Zhu, H.-Q.; Guo, J.-H.; Zhao, M.; Wu, Y.-L.; Liu, F.; Gao, F.-H. GSK3β-dependent cyclin D1 and cyclin E1 degradation is indispensable for NVP-BEZ235 induced G0/G1 arrest in neuroblastoma cells. Cell Cycle 2017, 16, 2386–2395. [Google Scholar] [CrossRef]
- Kostopoulou, O.N.; Holzhauser, S.; Lange, B.K.A.; Ohmayer, A.; Andonova, T.; Bersani, C.; Dalianis, T.; Wickström, M. Analyses of FGFR3 and PIK3CA mutations in neuroblastomas and the effects of the corresponding inhibitors on neuroblastoma cell lines. Int. J. Oncol. 2019, 55, 1372–1384. [Google Scholar] [CrossRef]
- Mohlin, S.; Hansson, K.; Radke, K.; Martinez, S.; Blanco-Apiricio, C.; Garcia-Ruiz, C.; Welinder, C.; Esfandyari, J.; O’Neill, M.; Pastor, J.; et al. Anti-tumor effects of PIM/PI 3K/mTOR triple kinase inhibitor IBL-302 in neuroblastoma. EMBO Mol. Med. 2019, 11, e10058. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Alpelisib: First Global Approval. Drugs 2019, 79, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Holzhauser, S.; Lukoseviciute, M. Christina Papachristofi 1, Christina Vasilopoulou 1, Nikolas Herold 2, Malin Wickström 2, Ourania N Kostopoulou # 1, T.D.# 1 Effects of PI3K and FGFR inhibitors alone and in combination, and with/without cytostatics in childhood neuroblastoma cell lines. Int. J. Oncol. 2021, 58, 211–225. [Google Scholar] [CrossRef]
- Peirce, S.K.; Findley, H.W.; Prince, C.; Dasgupta, A.; Cooper, T.; Durden, D.L. The PI-3 kinase-Akt-MDM2-survivin signaling axis in high-risk neuroblastoma: A target for PI-3 kinase inhibitor intervention. Cancer Chemother. Pharmacol. 2011, 68, 325–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdreich-Epstein, A.; Singh, A.R.; Joshi, S.; Vega, F.M.; Guo, P.; Xu, J.; Groshen, S.; Ye, W.; Millard, M.; Campan, M.; et al. Association of high microvessel αvβ3 and low PTEN with poor outcome in stage 3 neuroblastoma: Rationale for using first in class dual PI3K/BRD4 inhibitor, SF1126. Oncotarget 2017, 8, 52193–52210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, E.H.; Pacold, M.E.; Perisic, O.; Stephens, L.; Hawkins, P.T.; Wymann, M.P.; Williams, R.L. Structure Determinants of Phosphoinositide 3-Kinase Inhibition by Wortmannin, LY294002, Quercetin, Myricetin and Staurosporine. Available online: https://www.rcsb.org/structure/1e7v (accessed on 10 November 2021).
- Walker, E.H.; Pacold, M.E.; Perisic, O.; Stephens, L.; Hawkins, P.T.; Wymann, M.P.; Williams, R.L. Structural Determinants of Phosphoinositide 3-Kinase Inhibition by Wortmannin, LY294002, Quercetin, Myricetin, and Staurosporine. Mol. Cell 2000, 6, 909–919. [Google Scholar] [CrossRef]
- Bender, A.; Opel, D.; Naumann, I.; Kappler, R.; Friedman, L.; Von Schweinitz, D.; Debatin, K.M.; Fulda, S. PI3K inhibitors prime neuroblastoma cells for chemotherapy by shifting the balance towards pro-apoptotic Bcl-2 proteins and enhanced mitochondrial apoptosis. Oncogene 2011, 30, 494–503. [Google Scholar] [CrossRef] [Green Version]
- Geng, X.; Xie, L.; Xing, H. PI3K Inhibitor Combined With Chemotherapy Can Enhance the Apoptosis of Neuroblastoma Cells In Vitro and In Vivo. Technol. Cancer Res. Treat. 2016, 15, 716–722. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Tan, F.; Liewehr, D.J.; Steinberg, S.M.; Thiele, C.J. In vitro and in vivo inhibition of neuroblastoma tumor cell growth by AKT inhibitor perifosine. J. Natl. Cancer Inst. 2010, 102, 758–770. [Google Scholar] [CrossRef]
- Li, Z.; Oh, D.Y.; Nakamura, K.; Thiele, C.J. Perifosine-induced inhibition of Akt attenuates brain-derived neurotrophic factor/TrkB-induced chemoresistance in neuroblastoma in vivo. Cancer 2011, 117, 5412–5422. [Google Scholar] [CrossRef] [Green Version]
- Becher, O.J.; Millard, N.E.; Modak, S.; Kushner, B.H.; Haque, S.; Spasojevic, I.; Trippett, T.M.; Gilheeney, S.W.; Khakoo, Y.; Lyden, D.C.; et al. A phase i study of single-agent perifosine for recurrent or refractory pediatric CNS and solid tumors. PLoS ONE 2017, 12, e0178593. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Shichino, H.; Kawamoto, H.; Kosaka, Y.; Chin, M.; Kato, K.; Mugishima, H. Phase I study of perifosine monotherapy in patients with recurrent or refractory neuroblastoma. Pediatr. Blood Cancer 2017, 64, e26623. [Google Scholar] [CrossRef]
- Kushner, B.H.; Cheung, N.K.V.; Modak, S.; Becher, O.J.; Basu, E.M.; Roberts, S.S.; Kramer, K.; Dunkel, I.J. A phase I/Ib trial targeting the Pi3k/Akt pathway using perifosine: Long-term progression-free survival of patients with resistant neuroblastoma. Int. J. Cancer 2017, 140, 480–484. [Google Scholar] [CrossRef] [Green Version]
- Becher, O.J.; Gilheeney, S.W.; Khakoo, Y.; Lyden, D.C.; Haque, S.; De Braganca, K.C.; Kolesar, J.M.; Huse, J.T.; Modak, S.; Wexler, L.H.; et al. A phase I study of perifosine with temsirolimus for recurrent pediatric solid tumors. Pediatr. Blood Cancer 2017, 64, e26409. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Hua, Z.; Dong, Y.; Zhan, Y.; Zhang, X.; Tian, W.; Liu, Z.; Thiele, C.J.; Li, Z. Proteome and Acetylome Analysis Identifies Novel Pathways and Targets Regulated by Perifosine in Neuroblastoma. Sci. Rep. 2017, 7, 42062. [Google Scholar] [CrossRef] [Green Version]
- Le Grand, M.; Kimpton, K.; Gana, C.C.; Valli, E.; Fletcher, J.I.; Kavallaris, M. Targeting Functional Activity of AKT Has Efficacy against Aggressive Neuroblastoma. ACS Pharmacol. Transl. Sci. 2020, 3, 148–160. [Google Scholar] [CrossRef]
- Misawa, A.; Hosoi, H.; Tsuchiya, K.; Sugimoto, T. Rapamycin inhibits proliferation of human neuroblastoma cells without suppression of MycN. Int. J. Cancer 2003, 104, 233–237. [Google Scholar] [CrossRef]
- Morgenstern, D.A.; Marzouki, M.; Bartels, U.; Irwin, M.S.; Sholler, G.L.S.; Gammon, J.; Yankanah, R.; Wu, B.; Samson, Y.; Baruchel, S. Phase i study of vinblastine and sirolimus in pediatric patients with recurrent or refractory solid tumors. Pediatr. Blood Cancer 2014, 61, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Han, L.; Weng, J.; Wang, K.; Chen, T. Rapamycin inhibits proliferation and induces autophagy in human neuroblastoma cells. Biosci. Rep. 2018, 38, BSR20181822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahmad, H.F.; Mouhieddine, T.H.; Chalhoub, R.M.; Assi, S.; Araji, T.; Chamaa, F.; Itani, M.M.; Nokkari, A.; Kobeissy, F.; Daoud, G.; et al. The Akt/mTOR pathway in cancer stem/progenitor cells is a potential therapeutic target for glioblastoma and neuroblastoma. Oncotarget 2018, 9, 33549–33561. [Google Scholar] [CrossRef] [Green Version]
- Hirai, H.; Sootome, H.; Nakatsuru, Y.; Miyama, K.; Taguchi, S.; Tsujioka, K.; Ueno, Y.; Hatch, H.; Majumder, P.K.; Pan, B.S.; et al. MK-2206, an allosteric akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 2010, 9, 1956–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, L.; Toyoda, H.; Xu, D.; Zhou, Y.; Sakurai, N.; Amano, K.; Kihira, K.; Hori, H.; Azuma, E.; Komada, Y.; et al. PDK1-mTOR signaling pathway inhibitors reduce cell proliferation in MK2206 resistant neuroblastoma cells. Cancer Cell Int. 2015, 15, 103. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Gong, W.; Hua, Z.; Chen, B.; Zhao, G.; Liu, Z.; Thiele, C.J.; Li, Z. Combination of Rapamycin and MK-2206 Induced Cell Death via Autophagy and Necroptosis in MYCN-Amplified Neuroblastoma Cell Lines. Front. Pharmacol. 2020, 11, 31. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Smith, M.; Lakoma, A.; Chen, Z.; Tao, L.; Scorsone, K.A.; Schild, L.; Aviles-Padilla, K.; Nikzad, R.; Zhang, Y.; Chakraborty, R.; et al. p53 nongenotoxic activation and mTORC1 inhibition lead to effective combination for neuroblastoma therapy. Clin. Cancer Res. 2017, 23, 6629–6639. [Google Scholar] [CrossRef] [Green Version]
- Mody, R.; Naranjo, A.; Van Ryn, C.; Yu, A.L.L.; London, W.B.B.; Shulkin, B.L.L.; Parisi, M.T.T.; Servaes, S.E.N.E.N.; Diccianni, M.B.B.; Sondel, P.M.M.; et al. Irinotecan–temozolomide with temsirolimus or dinutuximab in children with refractory or relapsed neuroblastoma (COG ANBL1221): An open-label, randomised, phase 2 trial. Lancet Oncol. 2017, 18, 946–957. [Google Scholar] [CrossRef] [Green Version]
- McGuire, T.R.; Coulter, D.W.; Bai, D.; Sughroue, J.A.; Li, J.; Yang, Z.; Qiao, Z.; Liu, Y.; Murry, D.J.; Chhonker, Y.S.; et al. Effects of novel pyrrolomycin MP1 in MYCN amplified chemoresistant neuroblastoma cell lines alone and combined with temsirolimus. BMC Cancer 2019, 19, 837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Tsai, H.W.; Tsai, Y.H.; Tseng, S.H. VS-5584, a PI3K/mTOR dual inhibitor, exerts antitumor effects on neuroblastomas in vitro and in vivo. J. Pediatr. Surg. 2021, 56, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.F.; Guan, K.L. Activation of MEK family kinases requires phosphorylation of two conserved Ser/Thr residues. EMBO J. 1994, 13, 1123–1131. [Google Scholar] [CrossRef]
- Eleveld, T.F.; Oldridge, D.A.; Bernard, V.; Koster, J.; Daage, L.C.; Diskin, S.J.; Schild, L.; Bentahar, N.B.; Bellini, A.; Chicard, M.; et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet. 2015, 47, 864–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Higashi, M.; Kimura, K.; Wakao, J.; Fumino, S.; Iehara, T.; Hosoi, H.; Sakai, T.; Tajiri, T. MEK inhibitors as a novel therapy for neuroblastoma: Their in vitro effects and predicting their efficacy. J. Pediatr. Surg. 2016, 51, 2074–2079. [Google Scholar] [CrossRef]
- Umapathy, G.; Guan, J.; Gustafsson, D.E.; Javanmardi, N.; Cervantes-Madrid, D.; Djos, A.; Martinsson, T.; Palmer, R.H.; Hallberg, B. MEK inhibitor trametinib does not prevent the growth of anaplastic lymphoma kinase (ALK)-addicted neuroblastomas. Sci. Signal. 2017, 10, 507. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, Y.; Tanaka, T.; Higashi, M.; Fumino, S.; Iehara, T.; Hosoi, H.; Sakai, T.; Tajiri, T. In vivo effects of short- and long-term MAPK pathway inhibition against neuroblastoma. J. Pediatr. Surg. 2018, 53, 2454–2459. [Google Scholar] [CrossRef]
- Puissant, A.; Frumm, S.M.; Alexe, G.; Bassil, C.F.; Qi, J.; Chanthery, Y.H.; Nekritz, E.A.; Zeid, R.; Gustafson, W.C.; Greninger, P.; et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013, 3, 309–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Healy, J.R.; Hart, L.S.; Shazad, A.L.; Gagliardi, M.E.; Tsang, M.; Elias, J.; Ruden, J.; Farrel, A.; Rokita, J.L.; Li, Y.; et al. Limited antitumor activity of combined BET and MEK inhibition in neuroblastoma. Pediatr. Blood Cancer 2020, 67, e28267. [Google Scholar] [CrossRef] [PubMed]
- Woodfield, S.E.; Zhang, L.; Scorsone, K.A.; Liu, Y.; Zage, P.E. Binimetinib inhibits MEK and is effective against neuroblastoma tumor cells with low NF1 expression. BMC Cancer 2016, 16, 172. [Google Scholar] [CrossRef] [Green Version]
- Andrlová, H.; Zeiser, R.; Meiss, F. Cobimetinib (GDC-0973, XL518). Recent Results Cancer Res. 2018, 211, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Ruan, Y.; Tippett, T.; Narendran, A. Targeted inhibition of MEK1 by cobimetinib leads to differentiation and apoptosis in neuroblastoma cells. J. Exp. Clin. Cancer Res. 2015, 34, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ultsch, M.H. Crystal Structure of MEK1 Kinase Bound to GDC0973. Available online: https://www.wwpdb.org/pdb?id=pdb_00004lmn (accessed on 10 November 2021).
- Hatzivassiliou, G.; Haling, J.R.; Chen, H.; Song, K.; Price, S.; Heald, R.; Hewitt, J.F.M.; Zak, M.; Peck, A.; Orr, C.; et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature 2013, 501, 232–236. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Mossé, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Lee, J.S.; Guo, F.; Shin, J.; Perez-Atayde, A.R.; Kutok, J.L.; Rodig, S.J.; Neuberg, D.S.; Helman, D.; Feng, H.; et al. Activated ALK Collaborates with MYCN in Neuroblastoma Pathogenesis. Cancer Cell 2012, 21, 362–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallberg, B.; Palmer, R.H. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat. Rev. Cancer 2013, 13, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Umapathy, G.; Mendoza-Garcia, P.; Hallberg, B.; Palmer, R.H. Targeting anaplastic lymphoma kinase in neuroblastoma. APMIS 2019, 127, 288–302. [Google Scholar] [CrossRef] [Green Version]
- Javanmardi, N.; Fransson, S.; Djos, A.; Sjöberg, R.M.; Nilsson, S.; Truvé, K.; Kogner, P.; Martinsson, T. Low Frequency ALK Hotspots Mutations In Neuroblastoma Tumours Detected By Ultra-deep Sequencing: Implications For ALK Inhibitor Treatment. Sci. Rep. 2019, 9, 2199. [Google Scholar] [CrossRef]
- Zou, H.Y.; Li, Q.; Lee, J.H.; Arango, M.E.; McDonnell, S.R.; Yamazaki, S.; Koudriakova, T.B.; Alton, G.; Cui, J.J.; Kung, P.P.; et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007, 67, 4408–4417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, J.J.; Tran-Dubé, M.; Shen, H.; Nambu, M.; Kung, P.P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [Google Scholar] [CrossRef]
- Bresler, S.C.; Wood, A.C.; Haglund, E.A.; Courtright, J.; Belcastro, L.T.; Plegaria, J.S.; Cole, K.; Toporovskaya, Y.; Zhao, H.; Carpenter, E.L.; et al. Neuroblastoma: Differential inhibitor sensitivity of anaplastic lymphoma kinase variants found in neuroblastoma. Sci. Transl. Med. 2011, 3, 108ra114. [Google Scholar] [CrossRef] [Green Version]
- Krytska, K.; Ryles, H.T.; Sano, R.; Raman, P.; Infarinato, N.R.; Hansel, T.D.; Makena, M.R.; Song, M.M.; Reynolds, C.P.; Mossé, Y.P. Crizotinib synergizes with chemotherapy in preclinical models of neuroblastoma. Clin. Cancer Res. 2016, 22, 948–960. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wu, B.; Baruchel, S. Oral Metronomic Topotecan Sensitizes Crizotinib Antitumor Activity in ALKF1174L Drug-Resistant Neuroblastoma Preclinical Models. Transl. Oncol. 2017, 10, 604–611. [Google Scholar] [CrossRef]
- Chuang, Y.C.; Huang, B.Y.; Chang, H.W.; Yang, C.N. Molecular Modeling of ALK L1198F and/or G1202R Mutations to Determine Differential Crizotinib Sensitivity. Sci. Rep. 2019, 9, 11390. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Wang, M.; Zhang, A. Alectinib: A novel second generation anaplastic lymphoma kinase (ALK) inhibitor for overcoming clinically-acquired resistance. Acta Pharm. Sin. B 2015, 5, 34–37. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Guan, S.; Zhao, Y.; Yu, Y.; Woodfield, S.E.; Zhang, H.; Yang, K.L.; Bieerkehazhi, S.; Qi, L.; Li, X.; et al. The second-generation ALK inhibitor alectinib effectively induces apoptosis in human neuroblastoma cells and inhibits tumor growth in a TH-MYCN transgenic neuroblastoma mouse model. Cancer Lett. 2017, 400, 61–68. [Google Scholar] [CrossRef]
- Alam, M.W.; Borenäs, M.; Lind, D.E.; Cervantes-Madrid, D.; Umapathy, G.; Palmer, R.H.; Hallberg, B. Alectinib, an Anaplastic Lymphoma Kinase Inhibitor, Abolishes ALK Activity and Growth in ALK-Positive Neuroblastoma Cells. Front. Oncol. 2019, 9, 579. [Google Scholar] [CrossRef]
- Hagiwara, K.; Tokunaga, T.; Iida, H.; Nagai, H. Combined inhibition of ALK and HDAC induces synergistic cytotoxicity in neuroblastoma cell lines. Anticancer Res. 2019, 39, 3579–3584. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, B.; Jin, Z.; Yang, H.; Rao, Z. The Crystal Structure of COVID-19 Main Protease in Complex with an Inhibitor N3. Available online: https://www.rcsb.org/structure/6LU7 (accessed on 10 November 2021).
- Sakamoto, H.; Tsukaguchi, T.; Hiroshima, S.; Kodama, T.; Kobayashi, T.; Fukami, T.A.; Oikawa, N.; Tsukuda, T.; Ishii, N.; Aoki, Y. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011, 19, 679–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsilje, T.H.; Pei, W.; Chen, B.; Lu, W.; Uno, T.; Jin, Y.; Jiang, T.; Kim, S.; Li, N.; Warmuth, M.; et al. Synthesis, structure-activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro- N 2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)- N 4-(2-(isopropylsulfonyl) phenyl)pyrimidine-2,4. J. Med. Chem. 2013, 56, 5675–5690. [Google Scholar] [CrossRef]
- Lee, C.C.; Spraggon, G. Crystal Structure of Anaplastic Lymphoma Kinase Complexed with LDK378. Available online: https://www.rcsb.org/structure/4MKC (accessed on 10 November 2021).
- Friboulet, L.; Li, N.; Katayama, R.; Lee, C.C.; Gainor, J.F.; Crystal, A.S.; Michellys, P.Y.; Awad, M.M.; Yanagitani, N.; Kim, S.; et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014, 4, 662–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, R.E.; Sanda, T.; Hanna, M.; Fröhling, S.; Luther, W.; Zhang, J.; Ahn, Y.; Zhou, W.; London, W.B.; McGrady, P.; et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008, 455, 975–978. [Google Scholar] [CrossRef]
- Debruyne, D.N.; Bhatnagar, N.; Sharma, B.; Luther, W.; Moore, N.F.; Cheung, N.K.; Gray, N.S.; George, R.E. ALK inhibitor resistance in ALKF1174L-driven neuroblastoma is associated with AXL activation and induction of EMT. Oncogene 2016, 35, 3681–3691. [Google Scholar] [CrossRef] [Green Version]
- Guan, J.; Fransson, S.; Siaw, J.T.; Treis, D.; Van Den Eynden, J.; Chand, D.; Umapathy, G.; Ruuth, K.; Svenberg, P.; Wessman, S.; et al. Clinical response of the novel activating ALK-I1171T mutation in neuroblastoma to the ALK inhibitor ceritinib. Cold Spring Harb. Mol. Case Stud. 2018, 4, a002550. [Google Scholar] [CrossRef]
- Siaw, J.T.; Wan, H.; Pfeifer, K.; Rivera, V.M.; Guan, J.; Palmer, R.H.; Hallberg, B. Brigatinib, an anaplastic lymphoma kinase inhibitor, abrogates activity and growth in ALK-positive neuroblastoma cells, Drosophila and mice. Oncotarget 2016, 7, 29011–29022. [Google Scholar] [CrossRef]
- Dougan, D.R.; Zhou, T. Crystal Structure of Anaplastic Lymphoma Kinase (ALK) Bound by Brigatinib. Available online: https://www.wwpdb.org/pdb?id=pdb_00006mx8 (accessed on 10 November 2021).
- Huang, W.S.; Liu, S.; Zou, D.; Thomas, M.; Wang, Y.; Zhou, T.; Romero, J.; Kohlmann, A.; Li, F.; Qi, J.; et al. Discovery of Brigatinib (AP26113), a Phosphine Oxide-Containing, Potent, Orally Active Inhibitor of Anaplastic Lymphoma Kinase. J. Med. Chem. 2016, 59, 4948–4964. [Google Scholar] [CrossRef]
- Johnson, T.W.; Richardson, P.F.; Bailey, S.; Brooun, A.; Burke, B.J.; Collins, M.R.; Cui, J.J.; Deal, J.G.; Deng, Y.L.; Dinh, D.; et al. Discovery of (10 R)-7-Amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h ][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and broad-spectrum potency against ALK-resistant mutations. J. Med. Chem. 2014, 57, 4720–4744. [Google Scholar] [CrossRef]
- Infarinato, N.R.; Park, J.H.; Krytska, K.; Ryles, H.T.; Sano, R.; Szigety, K.M.; Li, Y.; Zou, H.Y.; Lee, N.V.; Smeal, T.; et al. The ALK/ROS1 inhibitor PF-06463922 overcomes primary resistance to crizotinib in ALK-driven neuroblastoma. Cancer Discov. 2016, 6, 96–107. [Google Scholar] [CrossRef] [Green Version]
- Guan, J.; Tucker, E.R.; Wan, H.; Chand, D.; Danielson, L.S.; Ruuth, K.; El Wakil, A.; Wite, B.; Jamin, Y.; Umapathy, G.; et al. The ALK inhibitor PF-06463922 is effective as a single agent in neuroblastoma driven by expression of ALK and MYCN. DMM Dis. Model. Mech. 2016, 9, 941–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redaelli, S.; Ceccon, M.; Zappa, M.; Sharma, G.G.; Mastini, C.; Mauri, M.; Nigoghossian, M.; Massimino, L.; Cordani, N.; Farina, F.; et al. Lorlatinib treatment elicits multiple on- and off-target mechanisms of resistance in ALK-driven cancer. Cancer Res. 2018, 78, 6866–6880. [Google Scholar] [CrossRef] [Green Version]
- McTigue, M.A.; Deng, Y.L.; Liu, W.; Brooun, A.; Stewart, A.E. Structure of the Human Anaplastic Lymphoma Kinase in Complex with PF- 06463922 ((10R)-7-Amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16, 17-tetrahydro-2H-8,4-(metheno)pyrazolo(4,3-h)(2,5,11) benzoxadiazacyclotetradecine-3-carbonitrile). Available online: https://www.rcsb.org/structure/4CLI (accessed on 10 November 2021).
- Ardini, E.; Menichincheri, M.; Banfi, P.; Bosotti, R.; De Ponti, C.; Pulci, R.; Ballinari, D.; Ciomei, M.; Texido, G.; Degrassi, A.; et al. Entrectinib, a Pan-TRK, ROS1, and ALK inhibitor with activity in multiple molecularly defined cancer indications. Mol. Cancer Ther. 2016, 15, 628–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignyta Receives Orphan Drug Designation in the European Union for Entrectinib for the Treatment of Neuroblastoma. Business Wire. Available online: https://www.businesswire.com/news/home/20151208005075/en/Ignyta-Receives-Orphan-Drug-Designation-in-the-European-Union-for-Entrectinib-for-the-Treatment-of-Neuroblastoma (accessed on 10 November 2021).
- Iyer, R.; Wehrmann, L.; Golden, R.L.; Naraparaju, K.; Croucher, J.L.; MacFarland, S.P.; Guan, P.; Kolla, V.; Wei, G.; Cam, N.; et al. Entrectinib is a potent inhibitor of Trk-driven neuroblastomas in a xenograft mouse model. Cancer Lett. 2016, 372, 179–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aveic, S.; Pantile, M.; Seydel, A.; Esposito, M.R.; Zanon, C.; Li, G.; Tonini, G.P. Combating autophagy is a strategy to increase cytotoxic effects of novel ALK inhibitor entrectinib in neuroblastoma cells. Oncotarget 2016, 7, 5646–5663. [Google Scholar] [CrossRef] [Green Version]
- MacFarland, S.P.; Naraparaju, K.; Iyer, R.; Guan, P.; Kolla, V.; Hu, Y.; Tan, K.; Brodeur, G.M. Mechanisms of entrectinib resistance in a neuroblastoma xenograft model. Mol. Cancer Ther. 2020, 19, 920–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galkin, A.V.; Melnick, J.S.; Kim, S.; Hood, T.L.; Li, N.; Li, L.; Xia, G.; Steensma, R.; Chopiuk, G.; Jiang, J.; et al. Identification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM-ALK. Proc. Natl. Acad. Sci. USA 2007, 104, 270–275. [Google Scholar] [CrossRef] [Green Version]
- Regairaz, M.; Munier, F.; Sartelet, H.; Castaing, M.; Marty, V.; Renauleaud, C.; Doux, C.; Delbé, J.; Courty, J.; Fabre, M.; et al. Mutation-independent activation of the anaplastic lymphoma kinase in neuroblastoma. Am. J. Pathol. 2016, 186, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Najem, S.; Langemann, D.; Appl, B.; Trochimiuk, M.; Hundsdoerfer, P.; Reinshagen, K.; Eschenburg, G. Smac mimetic LCL161 supports neuroblastoma chemotherapy in a drug class-dependent manner and synergistically interacts with ALK inhibitor TAE684 in cells with ALK mutation F1174L. Oncotarget 2016, 7, 72634–72653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, C.E.; Gao, Y.; Tan, L.; Donovan, K.A.; Nowak, R.P.; Loehr, A.; Bahcall, M.; Fischer, E.S.; Jänne, P.A.; George, R.E.; et al. Chemically Induced Degradation of Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2018, 61, 4249–4255. [Google Scholar] [CrossRef]
- Bossi, R.T.; Saccardo, M.B.; Ardini, E.; Menichincheri, M.; Rusconi, L.; Magnaghi, P.; Orsini, P.; Fogliatto, G.; Bertrand, J.A. Structure of Human Anaplastic Lymphoma Kinase in Complex with NVP- TAE684. Available online: https://www.rcsb.org/structure/2XB7 (accessed on 10 November 2021).
- Bossi, R.T.; Saccardo, M.B.; Ardini, E.; Menichincheri, M.; Rusconi, L.; Magnaghi, P.; Orsini, P.; Avanzi, N.; Borgia, A.L.; Nesi, M.; et al. Crystal structures of anaplastic lymphoma kinase in complex with ATP competitive inhibitors. Biochemistry 2010, 49, 6813–6825. [Google Scholar] [CrossRef] [PubMed]
- Emdal, K.B.; Pedersen, A.K.; Bekker-Jensen, D.B.; Lundby, A.; Claeys, S.; De Preter, K.; Speleman, F.; Francavilla, C.; Olsen, J.V. Integrated proximal proteomics reveals IRS2 as a determinant of cell survival in ALK-driven neuroblastoma. Sci. Signal. 2018, 11, 557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, E.; Heidenreich, D.; Tucker, E.; Raab, M.; Strebhardt, K.; Chesler, L.; Knapp, S.; Bellenie, B.; Hoelder, S. Designing Dual Inhibitors of Anaplastic Lymphoma Kinase (ALK) and Bromodomain-4 (BRD4) by Tuning Kinase Selectivity. J. Med. Chem. 2019, 62, 2618–2637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munira, S.; Yuki, R.; Saito, Y.; Nakayama, Y. ALK inhibitors-induced M phase delay contributes to the suppression of cell proliferation. Cancers 2020, 12, 1054. [Google Scholar] [CrossRef]
- Nakagawara, A. Trk receptor tyrosine kinases: A bridge between cancer and neural development. Cancer Lett. 2001, 169, 107–114. [Google Scholar] [CrossRef]
- Eggert, A.; Grotzer, M.A.; Ikegaki, N.; Liu, X.G.; Evans, A.E.; Brodeur, G.M. Expression of the neurotrophin receptor TrkA down-regulates expression and function of angiogenic stimulators in SH-SY5Y neuroblastoma cells (Cancer Research (2002) 62, (1802–1808)). Cancer Res. 2012, 72, 1588. [Google Scholar] [CrossRef] [Green Version]
- Jaboin, J.; Kim, C.J.; Kaplan, D.R.; Thiele, C.J. Brain-derived neurotrophic factor activation of TrkB protects neuroblastoma cells from chemotherapy-induced apoptosis via phosphatidylinositol 3′-kinase pathway. Cancer Res. 2002, 62, 6756–6763. [Google Scholar]
- Khotskaya, Y.B.; Holla, V.R.; Farago, A.F.; Mills Shaw, K.R.; Meric-Bernstam, F.; Hong, D.S. Targeting TRK family proteins in cancer. Pharmacol. Ther. 2017, 173, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Thress, K.; MacIntyre, T.; Wang, H.; Whitston, D.; Liu, Z.Y.; Hoffmann, E.; Wang, T.; Brown, J.L.; Webster, K.; Omer, C.; et al. Identification and preclinical characterization of AZ-23, a novel, selective, and orally bioavailable inhibitor of the Trk kinase pathway. Mol. Cancer Ther. 2009, 8, 1818–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Lamb, M.L.; Block, M.H.; Davies, A.M.; Han, Y.; Hoffmann, E.; Ioannidis, S.; Josey, J.A.; Liu, Z.; Lyne, P.D.; et al. Human TrkA in Complex with the Inhibitor AZ-23. Available online: https://www.wwpdb.org/pdb?id=pdb_00004aoj (accessed on 10 November 2021).
- Wang, T.; Lamb, M.L.; Block, M.H.; Davies, A.M.; Han, Y.; Hoffmann, E.; Ioannidis, S.; Josey, J.A.; Liu, Z.Y.; Lyne, P.D.; et al. Discovery of disubstituted imidazo[4,5-b ]pyridines and purines as potent TrkA inhibitors. ACS Med. Chem. Lett. 2012, 3, 705–709. [Google Scholar] [CrossRef] [Green Version]
- Croucher, J.L.; Iyer, R.; Li, N.; Molteni, V.; Loren, J.; Gordon, W.P.; Tuntland, T.; Liu, B.; Brodeur, G.M. TrkB inhibition by GNF-4256 slows growth and enhances chemotherapeutic efficacy in neuroblastoma xenografts. Cancer Chemother. Pharmacol. 2015, 75, 131–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, J.; Zhang, Z.; Xu, F.; Cui, S.; Qi, C.; Luo, J.; Wang, Z.; Lu, X.; Tu, Z.; Ren, X.; et al. GZD2202, a novel TrkB inhibitor, suppresses BDNF-mediated proliferation and metastasis in neuroblastoma models. J. Drug Target. 2019, 27, 442–450. [Google Scholar] [CrossRef]
- Cui, S.; Wang, Y.; Wang, Y.; Tang, X.; Ren, X.; Zhang, L.; Xu, Y.; Zhang, Z.; Zhang, Z.M.; Lu, X.; et al. Design, synthesis and biological evaluation of 3-(imidazo[1,2-a]pyrazin-3-ylethynyl)-2-methylbenzamides as potent and selective pan-tropomyosin receptor kinase (TRK) inhibitors. Eur. J. Med. Chem. 2019, 179, 470–482. [Google Scholar] [CrossRef]
- Miura, A.; Sootome, H.; Fujita, N.; Suzuki, T.; Fukushima, H.; Mizuarai, S.; Masuko, N.; Ito, K.; Hashimoto, A.; Uto, Y.; et al. TAS-119, a novel selective Aurora A and TRK inhibitor, exhibits antitumor efficacy in preclinical models with deregulated activation of the Myc, β-Catenin, and TRK pathways. Investig. New Drugs 2021, 39, 724–735. [Google Scholar] [CrossRef]
- Ghedini, G.C.; Ronca, R.; Presta, M.; Giacomini, A. Future applications of FGF/FGFR inhibitors in cancer. Expert Rev. Anticancer Ther. 2018, 18, 861–872. [Google Scholar] [CrossRef]
- Markham, A. Erdafitinib: First Global Approval. Drugs 2019, 79, 1017–1021. [Google Scholar] [CrossRef]
- Ogg, D.; Breed, J. Fibroblast Growth Factor Receptor 1 in Complex with JNJ-4275693. Available online: https://www.rcsb.org/structure/5EW8 (accessed on 10 November 2021).
- Patani, H.; Bunney, T.D.; Thiyagarajan, N.; Norman, R.A.; Ogg, D.; Breed, J.; Ashford, P.; Potterton, A.; Edwards, M.; Williams, S.V.; et al. Landscape of activating cancer mutations in FGFR kinases and their differential responses to inhibitors in clinical use. Oncotarget 2016, 7, 24252–24268. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.; Wang, M.; Tian, X.; Zhang, X. An overview of the binding models of FGFR tyrosine kinases in complex with small molecule inhibitors. Eur. J. Med. Chem. 2017, 126, 476–490. [Google Scholar] [CrossRef] [PubMed]
- Phanhthilath, N.; Hakim, S.; Su, C.; Liu, A.; Subramonian, D.; Lesperance, J.; Zage, P.E. Mechanisms of efficacy of the FGFR1–3 inhibitor AZD4547 in pediatric solid tumor models. Investig. New Drugs 2020, 38, 1677–1686. [Google Scholar] [CrossRef]
- Tucker, J.; Klein, T.; Breed, J.; Breeze, A.; Overman, R.; Phillips, C.; Norman, R.A. FGFR1 in Complex with AZD4547. Available online: https://www.rcsb.org/structure/4V05 (accessed on 10 November 2021).
- Tucker, J.A.; Klein, T.; Breed, J.; Breeze, A.L.; Overman, R.; Phillips, C.; Norman, R.A. Structural insights into FGFR kinase isoform selectivity: Diverse binding modes of AZD4547 and ponatinib in complex with FGFR1 and FGFR4. Structure 2014, 22, 1764–1774. [Google Scholar] [CrossRef] [Green Version]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef]
- Wecker, H.; Waller, C.F. Afatinib. Recent Results Cancer Res. 2018, 211, 199–215. [Google Scholar] [CrossRef]
- Mao, X.; Chen, Z.; Zhao, Y.; Yu, Y.; Guan, S.; Woodfield, S.E.; Vasudevan, S.A.; Tao, L.; Pang, J.C.; Lu, J.; et al. Novel multi-targeted ErbB family inhibitor afatinib blocks EGF-induced signaling and induces apoptosis in neuroblastoma. Oncotarget 2017, 8, 1555–1568. [Google Scholar] [CrossRef] [Green Version]
- Solca, F.; Dahl, G.; Zoephel, A.; Bader, G.; Sanderson, M.; Klein, C.; Kraemer, O.; Himmelsbach, F.; Haaksma, E.; Adolf, G.R. Crystal Structure of EGFR Kinase in Complex with BIBW2992. Available online: https://www.rcsb.org/structure/4G5J (accessed on 10 November 2021).
- Solca, F.; Dahl, G.; Zoephel, A.; Bader, G.; Sanderson, M.; Klein, C.; Kraemer, O.; Himmelsbach, F.; Haaksma, E.; Adolf, G.R. Target Binding Properties and Cellular Activity of Afatinib (BIBW 2992), an Irreversible ErbB Family Blocker. J. Pharmacol. Exp. Ther. 2012, 343, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Peifer, M.; Hertwig, F.; Roels, F.; Dreidax, D.; Gartlgruber, M.; Menon, R.; Krämer, A.; Roncaioli, J.L.; Sand, F.; Heuckmann, J.M.; et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 2015, 526, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Bellini, A.; Bernard, V.; Leroy, Q.; Frio, T.R.; Pierron, G.; Combaret, V.; Lapouble, E.; Clement, N.; Rubie, H.; Thebaud, E.; et al. Deep sequencing reveals occurrence of subclonal ALK mutations in neuroblastoma at diagnosis. Clin. Cancer Res. 2015, 21, 4913–4921. [Google Scholar] [CrossRef] [Green Version]
- Graham, D.K.; Deryckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine-sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, X.; Bi, S.; Zhao, K.; Yu, C. Inhibition of Mer and Axl receptor tyrosine kinases leads to increased apoptosis and improved chemosensitivity in human neuroblastoma. Biochem. Biophys. Res. Commun. 2015, 457, 461–466. [Google Scholar] [CrossRef]
- Myers, S.H.; Brunton, V.G.; Unciti-Broceta, A. AXL Inhibitors in Cancer: A Medicinal Chemistry Perspective. J. Med. Chem. 2016, 59, 3593–3608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aveic, S.; Corallo, D.; Porcù, E.; Pantile, M.; Boso, D.; Zanon, C.; Viola, G.; Sidarovich, V.; Mariotto, E.; Quattrone, A.; et al. TP-0903 inhibits neuroblastoma cell growth and enhances the sensitivity to conventional chemotherapy. Eur. J. Pharmacol. 2018, 818, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Owens, L.V.; Xu, L.H.; Craven, R.J.; Dent, G.A.; Weiner, T.M.; Kornberg, L.; Liu, E.T.; Cance, W.G. Overexpression of the Focal Adhesion Kinase (p125FAK) in Invasive Human Tumors. Cancer Res. 1995, 55, 2752–2755. [Google Scholar] [PubMed]
- Waters, A.; Beierle, E. The Interaction Between FAK, MYCN, p53 and Mdm2 in Neuroblastoma. Anticancer Agents Med. Chem. 2014, 14, 46–51. [Google Scholar] [CrossRef]
- Cooper, J.; Giancotti, F.G. Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell 2019, 35, 347–367. [Google Scholar] [CrossRef]
- Beierle, E.A.; Massoll, N.A.; Hartwich, J.; Kurenova, E.V.; Golubovskaya, V.M.; Cance, W.G.; McGrady, P.; London, W.B. Focal adhesion kinase expression in human neuroblastoma: Immunohistochemical and real-time PCR analyses. Clin. Cancer Res. 2008, 14, 3299–3305. [Google Scholar] [CrossRef] [Green Version]
- Beierle, E.A.; Ma, X.; Stewart, J.; Nyberg, C.; Trujillo, A.; Cance, W.G.; Golubovskaya, V.M. Inhibition of focal adhesion kinase decreases tumor growth in human neuroblastoma. Cell Cycle 2010, 9, 1005–1015. [Google Scholar] [CrossRef] [Green Version]
- Stafman, L.L.; Williams, A.P.; Marayati, R.; Aye, J.M.; Markert, H.R.; Garner, E.F.; Quinn, C.H.; Lallani, S.B.; Stewart, J.E.; Yoon, K.J.; et al. Focal Adhesion Kinase Inhibition Contributes to Tumor Cell Survival and Motility in Neuroblastoma Patient-Derived Xenografts. Sci. Rep. 2019, 9, 13259. [Google Scholar] [CrossRef]
- Slack-Davis, J.K.; Martin, K.H.; Tilghman, R.W.; Iwanicki, M.; Ung, E.J.; Autry, C.; Luzzio, M.J.; Cooper, B.; Kath, J.C.; Roberts, W.G.; et al. Cellular characterization of a novel focal adhesion kinase inhibitor. J. Biol. Chem. 2007, 282, 14845–14852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golubovskaya, V.M.; Nyberg, C.; Zheng, M.; Kweh, F.; Magis, A.; Ostrov, D.; Cance, W.G. A small molecule inhibitor, 1,2,4,5-benzenetetraamine tetrahydrochloride, targeting the Y397 site of focal adhesion kinase decreases tumor growth. J. Med. Chem. 2008, 51, 7405–7416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, V.; Macchia, M.; Tuccinardi, T.; Poli, G. Three-dimensional interactions analysis of the anticancer target c-src kinase with its inhibitors. Cancers 2020, 12, 2327. [Google Scholar] [CrossRef] [PubMed]
- Rossari, F.; Minutolo, F.; Orciuolo, E. Past, present, and future of Bcr-Abl inhibitors: From chemical development to clinical efficacy. J. Hematol. Oncol. 2018, 11, 84. [Google Scholar] [CrossRef] [Green Version]
- Vitali, R.; Mancini, C.; Cesi, V.; Tanno, B.; Piscitelli, M.; Mancuso, M.; Sesti, F.; Pasquali, E.; Calabretta, B.; Dominici, C.; et al. Activity of tyrosine kinase inhibitor Dasatinib in neuroblastoma cells in vitro and in orthotopic mouse model. Int. J. Cancer 2009, 125, 2547–2555. [Google Scholar] [CrossRef]
- Chen, J. Inhibition of immune responses by Dasatinib may account for its different effects on neuroblastoma between in vitro and in vivo. Int. J. Cancer 2010, 127, 1249–1250. [Google Scholar] [CrossRef]
- Klei, H.E. X-ray Crystal Structure of Dasatinib (BMS-354825) Bound to Activated ABL Kinase Domain. Available online: https://www.wwpdb.org/pdb?id=pdb_00002gqg (accessed on 10 November 2021).
- Tokarski, J.S.; Newitt, J.A.; Chang, C.Y.J.; Cheng, J.D.; Wittekind, M.; Kiefer, S.E.; Kish, K.; Lee, F.Y.F.; Borzillerri, R.; Lombardo, L.J.; et al. The Structure of Dasatinib (BMS-354825) Bound to Activated ABL Kinase Domain Elucidates Its Inhibitory Activity against Imatinib-Resistant ABL Mutants. Cancer Res. 2006, 66, 5790–5797. [Google Scholar] [CrossRef] [Green Version]
- Bieerkehazhi, S.; Chen, Z.; Zhao, Y.; Yu, Y.; Zhang, H.H.; Vasudevan, S.A.; Woodfield, S.E.; Tao, L.; Yi, J.S.; Muscal, J.A.; et al. Novel Src/Abl tyrosine kinase inhibitor bosutinib suppresses neuroblastoma growth via inhibiting Src/Abl signaling. Oncotarget 2017, 8, 1469–1480. [Google Scholar] [CrossRef] [Green Version]
- Boxer, S.G.; Levinson, N.M. Structural and Spectroscopic Analysis of the Kinase Inhibitor Bosutinib Binding to the Abl Tyrosine Kinase Domain. Available online: https://www.wwpdb.org/pdb?id=pdb_00003ue4 (accessed on 10 November 2021).
- Levinson, N.M.; Boxer, S.G. Structural and spectroscopic analysis of the kinase inhibitor bosutinib and an isomer of bosutinib binding to the Abl tyrosine kinase domain. PLoS ONE 2012, 7, e29828. [Google Scholar] [CrossRef]
- Whittle, S.B.; Patel, K.; Zhang, L.; Woodfield, S.E.; Du, M.; Smith, V.; Zage, P.E. The novel kinase inhibitor ponatinib is an effective anti-angiogenic agent against neuroblastoma. Investig. New Drugs 2016, 34, 685–692. [Google Scholar] [CrossRef]
- Singh, A.; Meier-Stephenson, V.; Jayanthan, A.; Narendran, A. In Vitro Sensitivity Profiling of Neuroblastoma Cells Against A Comprehensive Small Molecule Kinase Inhibitor Library to Identify Agents for Future Therapeutic Studies. Curr. Cancer Drug Targets 2017, 17, 569–584. [Google Scholar] [CrossRef]
- Li, H.; Wang, Y.; Chen, Z.; Lu, J.; Pan, J.; Yu, Y.; Zhao, Y.; Zhang, H.; Hu, T.; Liu, Q.; et al. Novel multiple tyrosine kinase inhibitor ponatinib inhibits bFGF activated signaling in neuroblastoma cells and suppresses neuroblastoma growth in vivo. Oncotarget 2017, 8, 5874–5884. [Google Scholar] [CrossRef] [PubMed]
- Sidarovich, V.; De Mariano, M.; Aveic, S.; Pancher, M.; Adami, V.; Gatto, P.; Pizzini, S.; Pasini, L.; Croce, M.; Parodi, F.; et al. A high-content screening of anticancer compounds suggests the multiple tyrosine kinase inhibitor ponatinib for repurposing in neuroblastoma therapy. Mol. Cancer Ther. 2018, 17, 1405–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corallo, D.; Pastorino, F.; Pantile, M.; Mariotto, E.; Caicci, F.; Viola, G.; Ponzoni, M.; Tonini, G.P.; Aveic, S. Autophagic flux inhibition enhances cytotoxicity of the receptor tyrosine kinase inhibitor ponatinib. J. Exp. Clin. Cancer Res. 2020, 39, 195. [Google Scholar] [CrossRef]
- Zhou, T. AP24534, a Pan-BCR-ABL Inhibitor for Chronic Myeloid Leukemia, Potently Inhibits the T315I Mutant and Overcomes Mutation-Based Resistance. Available online: https://www.rcsb.org/structure/3IK3 (accessed on 10 November 2021).
- O’Hare, T.; Shakespeare, W.C.; Zhu, X.; Eide, C.A.; Rivera, V.M.; Wang, F.; Adrian, L.T.; Zhou, T.; Huang, W.S.; Xu, Q.; et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 2009, 16, 401–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vignaroli, G.; Mencarelli, M.; Sementa, D.; Crespan, E.; Kissova, M.; Maga, G.; Schenone, S.; Radi, M.; Botta, M. Exploring the chemical space around the privileged pyrazolo[3,4-d] pyrimidine scaffold: Toward novel allosteric inhibitors of T315I-mutated Abl. ACS Comb. Sci. 2014, 16, 168–175. [Google Scholar] [CrossRef]
- Navarra, M.; Celano, M.; Maiuolo, J.; Schenone, S.; Botta, M.; Angelucci, A.; Bramanti, P.; Russo, D. Antiproliferative and pro-apoptotic effects afforded by novel Src-kinase inhibitors in human neuroblastoma cells. BMC Cancer 2010, 10, 602. [Google Scholar] [CrossRef] [Green Version]
- Radi, M.; Brullo, C.; Crespan, E.; Tintori, C.; Musumeci, F.; Biava, M.; Schenone, S.; Dreassi, E.; Zamperini, C.; Maga, G.; et al. Identification of potent c-Src inhibitors strongly affecting the proliferation of human neuroblastoma cells. Bioorg. Med. Chem. Lett. 2011, 21, 5928–5933. [Google Scholar] [CrossRef] [PubMed]
- Tintori, C.; Fallacara, A.L.; Radi, M.; Zamperini, C.; Dreassi, E.; Crespan, E.; Maga, G.; Schenone, S.; Musumeci, F.; Brullo, C.; et al. Combining X-ray crystallography and molecular modeling toward the optimization of pyrazolo[3,4-d ]pyrimidines as potent c-Src inhibitors active in vivo against neuroblastoma. J. Med. Chem. 2015, 58, 347–361. [Google Scholar] [CrossRef]
- Fallacara, A.L.; Mancini, A.; Zamperini, C.; Dreassi, E.; Marianelli, S.; Chiariello, M.; Pozzi, G.; Santoro, F.; Botta, M.; Schenone, S. Pyrazolo[3,4-d]pyrimidines-loaded human serum albumin (HSA) nanoparticles: Preparation, characterization and cytotoxicity evaluation against neuroblastoma cell line. Bioorg. Med. Chem. Lett. 2017, 27, 3196–3200. [Google Scholar] [CrossRef]
- Molinari, A.; Fallacara, A.L.; Di Maria, S.; Zamperini, C.; Poggialini, F.; Musumeci, F.; Schenone, S.; Angelucci, A.; Colapietro, A.; Crespan, E.; et al. Efficient optimization of pyrazolo[3,4-d]pyrimidines derivatives as c-Src kinase inhibitors in neuroblastoma treatment. Bioorg. Med. Chem. Lett. 2018, 28, 3454–3457. [Google Scholar] [CrossRef]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef]
- Okada, K.; Nakano, Y.; Yamasaki, K.; Nitani, C.; Fujisaki, H.; Hara, J. Sorafenib treatment in children with relapsed and refractory neuroblastoma: An experience of four cases. Cancer Med. 2016, 5, 1947–1949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barford, D.; Roe, S.M.; Wan, P.T.C. Cancer Genome Project The Complex of Wild Type B-RAF and BAY439006. Available online: https://www.wwpdb.org/pdb?id=pdb_00001uwh (accessed on 10 November 2021).
- Wan, P.T.C.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Project, C.G.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Grüllich, C. Cabozantinib: Multi-kinase Inhibitor of MET, AXL, RET, and VEGFR2. Recent Results Cancer Res. 2018, 211, 67–75. [Google Scholar] [CrossRef]
- Zhang, L.; Scorsone, K.; Woodfield, S.E.; Zage, P.E. Sensitivity of neuroblastoma to the novel kinase inhibitor cabozantinib is mediated by ERK inhibition. Cancer Chemother. Pharmacol. 2015, 76, 977–987. [Google Scholar] [CrossRef]
- Daudigeos-Dubus, E.; Le Dret, L.; Bawa, O.; Opolon, P.; Vievard, A.; Villa, I.; Bosq, J.; Vassal, G.; Geoerger, B. Dual inhibition using cabozantinib overcomes HGF/MET signaling mediated resistance to pan-VEGFR inhibition in orthotopic and metastatic neuroblastoma tumors. Int. J. Oncol. 2017, 50, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Sennino, B.; Ishiguro-Oonuma, T.; Wei, Y.; Naylor, R.M.; Williamson, C.W.; Bhagwandin, V.; Tabruyn, S.P.; You, W.K.; Chapman, H.A.; Christensen, J.G.; et al. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov. 2012, 2, 270–287. [Google Scholar] [CrossRef] [Green Version]
- Musumeci, F.; Schenone, S.; Desogus, A.; Nieddu, E.; Deodato, D.; Botta, L. Click Chemistry, A Potent Tool in Medicinal Sciences. Curr. Med. Chem. 2015, 22, 2022–2050. [Google Scholar] [CrossRef] [PubMed]
- Perisa, M.P.; Storey, M.; Streby, K.A.; Ranalli, M.A.; Skeens, M.; Shah, N. Cabozantinib for relapsed neuroblastoma: Single institution case series. Pediatr. Blood Cancer 2020, 67, e28317. [Google Scholar] [CrossRef] [PubMed]
- James, J.; Ruggeri, B.; Armstrong, R.C.; Rowbottom, M.W.; Jones-Bolin, S.; Gunawardane, R.N.; Dobrzanski, P.; Gardner, M.F.; Zhao, H.; Cramer, M.D.; et al. CEP-32496: A novel orally active BRAF V600E inhibitor with selective cellular and in vivo antitumor activity. Mol. Cancer Ther. 2012, 11, 930–941. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yu, Y.; Zhao, Y.; Wu, D.; Yu, X.; Lu, J.; Chen, Z.; Zhang, H.; Hu, Y.; Zhai, Y.; et al. Small molecule inhibitor agerafenib effectively suppresses neuroblastoma tumor growth in mouse models via inhibiting ERK MAPK signaling. Cancer Lett. 2019, 457, 129–141. [Google Scholar] [CrossRef]
- Flynn, S.M.; Lesperance, J.; Macias, A.; Phanhthilath, N.; Paul, M.R.; Kim, J.W.; Tamayo, P.; Zage, P.E. The multikinase inhibitor RXDX-105 is effective against neuroblastoma In Vitro and In Vivo. Oncotarget 2019, 10, 6323–6333. [Google Scholar] [CrossRef]
- Ettrich, T.J.; Seufferlein, T. Regorafenib. Recent Results Cancer Res. 2018, 211, 45–56. [Google Scholar] [CrossRef]
- Chen, Z.; Zhao, Y.; Yu, Y.; Pang, J.C.; Woodfield, S.E.; Tao, L.; Guan, S.; Zhang, H.; Bieerkehazhi, S.; Shi, Y.; et al. Small molecule inhibitor regorafenib inhibits RET signaling in neuroblastoma cells and effectively suppresses tumor growth in vivo. Oncotarget 2017, 8, 104090–104103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramonian, D.; Phanhthilath, N.; Rinehardt, H.; Flynn, S.; Huo, Y.; Zhang, J.; Messer, K.; Mo, Q.; Huang, S.; Lesperance, J.; et al. Regorafenib is effective against neuroblastoma in vitro and in vivo and inhibits the RAS/MAPK, PI3K/Akt/mTOR and Fos/Jun pathways. Br. J. Cancer 2020, 123, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Zafar, A.; Wang, W.; Liu, G.; Wang, X.; Xian, W.; McKeon, F.; Foster, J.; Zhou, J.; Zhang, R. Molecular targeting therapies for neuroblastoma: Progress and challenges. Med. Res. Rev. 2021, 41, 961–1021. [Google Scholar] [CrossRef] [PubMed]
- Aveic, S.; Pantile, M.; Polo, P.; Sidarovich, V.; Mariano, M.; Quattrone, A.; Longo, L.; Tonini, G.P. Autophagy inhibition improves the cytotoxic effects of receptor tyrosine kinase inhibitors. Cancer Cell Int. 2018, 18, 63. [Google Scholar] [CrossRef] [PubMed]
- Moreno, L.; Barone, G.; DuBois, S.G.; Molenaar, J.; Fischer, M.; Schulte, J.; Eggert, A.; Schleiermacher, G.; Speleman, F.; Chesler, L.; et al. Accelerating drug development for neuroblastoma: Summary of the Second Neuroblastoma Drug Development Strategy forum from Innovative Therapies for Children with Cancer and International Society of Paediatric Oncology Europe Neuroblastoma. Eur. J. Cancer 2020, 136, 52–68. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
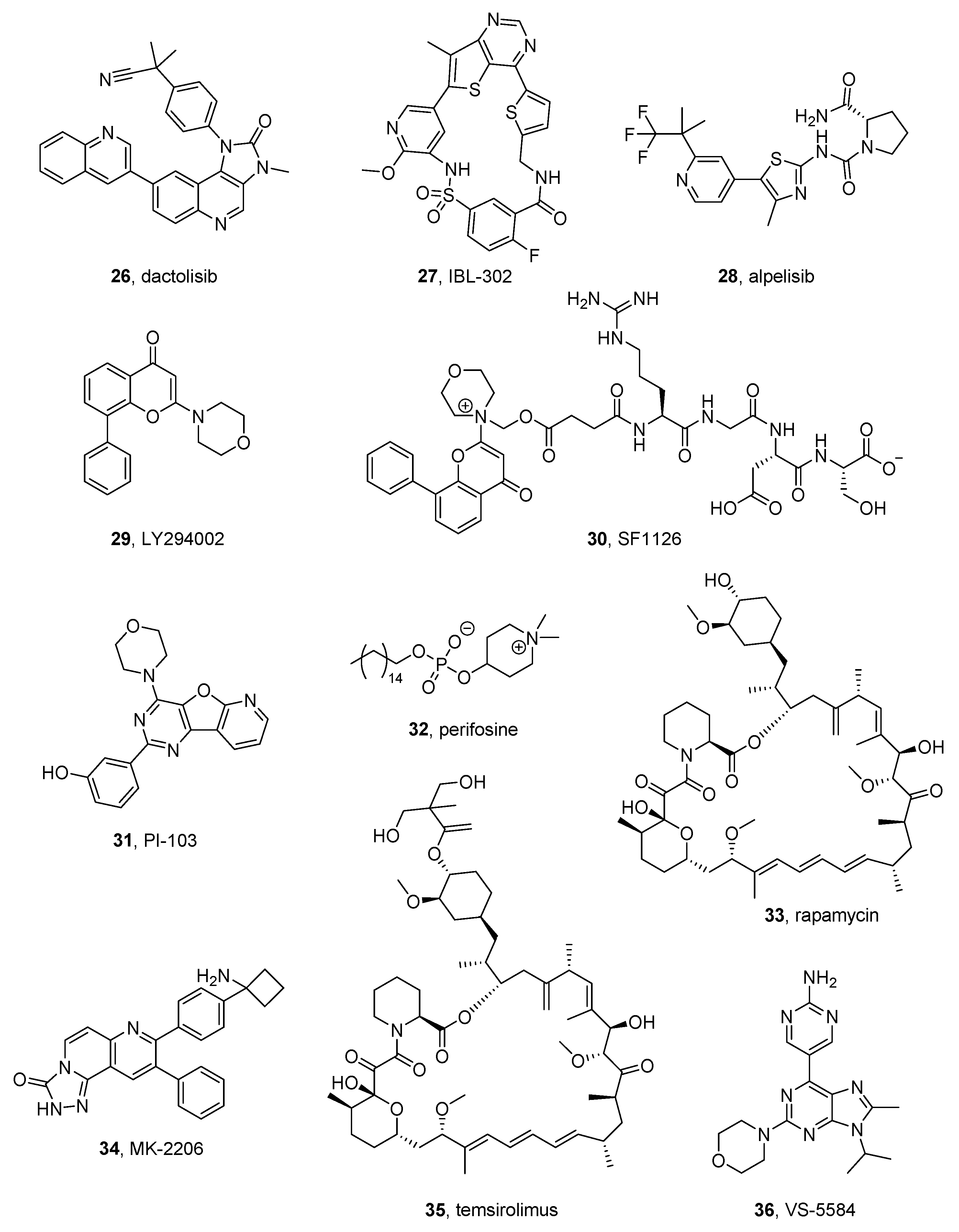{kind=link}
{kind=link}
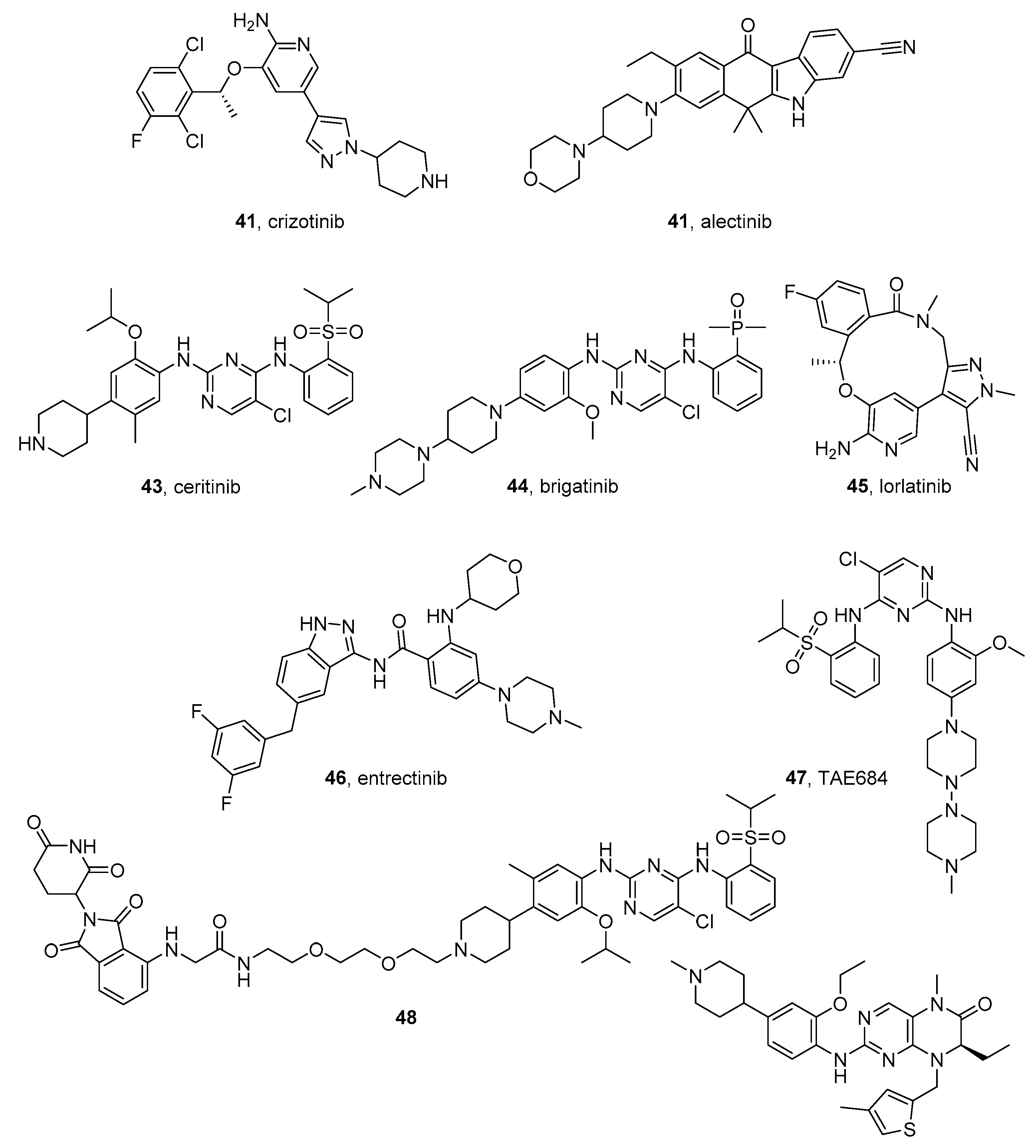{kind=link}
{kind=link}
{kind=link}
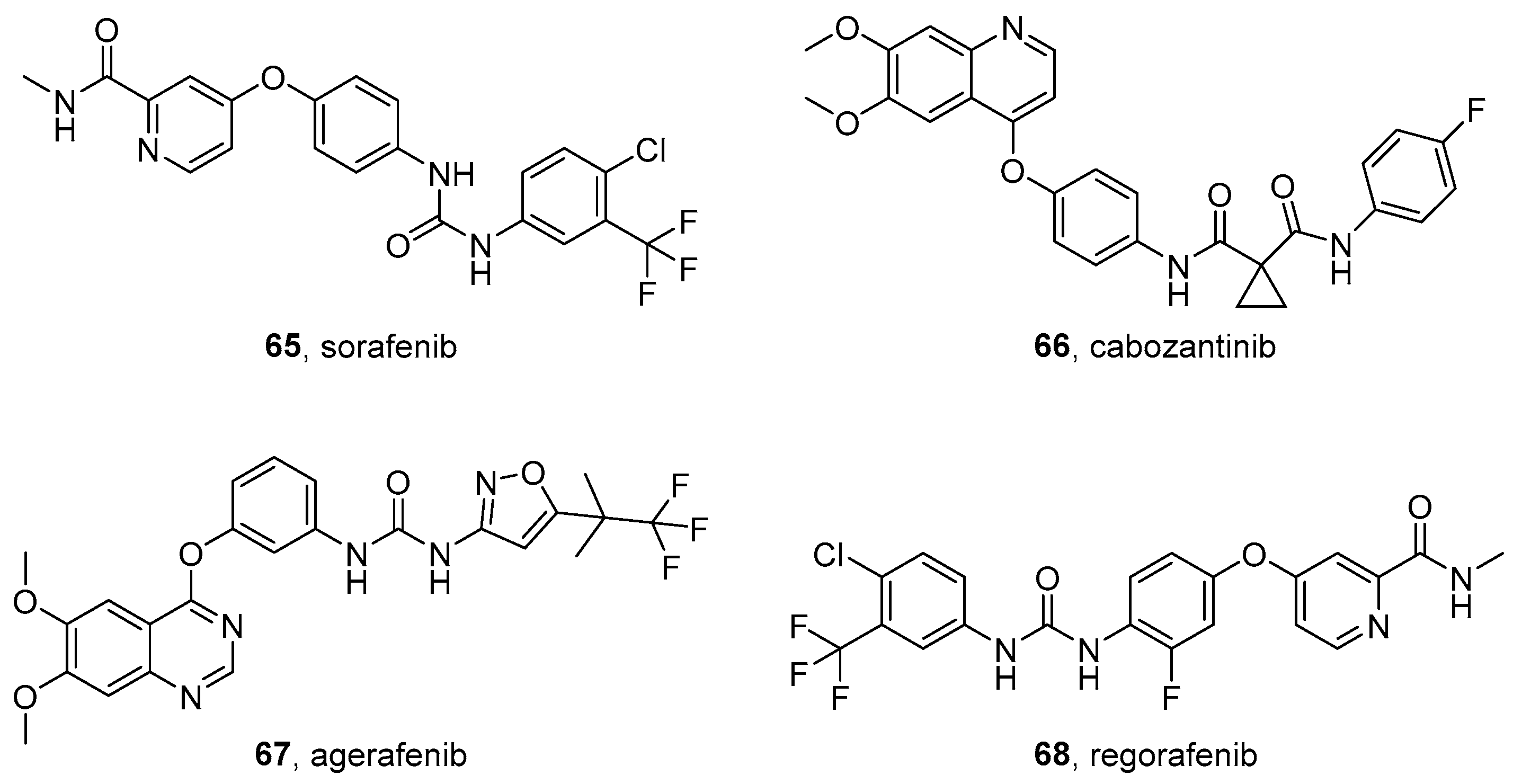{kind=link}
| Compound | Pharmaceutical Company | Targeted Kinase | NCT Number | Drug Combined with | Phase | Status |
|---|---|---|---|---|---|---|
| Ribociclib, 2 | Novartis | CDK4/6 | NCT01747876 | - | 1 | terminated, has results |
| NCT02780128 | - | 1 | recruiting | |||
| NCT03434262 | trametinib | 1 | recruiting | |||
| Palbociclib, 3 | Pfizer | CDK4/6 | NCT03526250 | 2 | recruiting | |
| NCT03709680 | temozolomide | 1 | recruiting | |||
| Alisertib, 8 | Takeda | AURKA | NCT01601535 | iritonotecan temozolomide | 1–2 | completed |
| NCT02444884 | - | 1 | completed | |||
| NCT01154816 | - | 2 | completed | |||
| 9-ING-41, 18 | Actuate Therapeutics | GSK-3β | NCT04239092 | alone or with irinotecan | 1 | recruiting |
| SF1126, 30 | SignalRx Pharmaceuticals | PI3K | NCT02337309 | - | 1 | terminated |
| Rapamycin, 33 | mTOR | NCT01467986 | dasatinib irinotecan temozolomide | 2 | completed | |
| NCT01331135 | - | 1 | completed | |||
| NCT02574728 | celecoxib etoposide cyclophosphamide | 2 | recruiting | |||
| Temsirolimus, 35 | mTOR | NCT01767194 | dinutuximab irinotecan sargramostim temozolomide | 2 | active, not recruiting, has results | |
| NCT00808899 | irinotecan cyclophosphamide doxorubicin etoposide cisplatin topotecan 13-cis-retinoic acid radiation | 2 | terminated, has results | |||
| NCT01204450 | valproic acid | 1 | terminated | |||
| Trametinib, 37 | Novartis | MEK | NCT03434262 | gemcitabine ribociclib sonidegib | 1 | recruiting |
| NCT02124772 | dabrafenib | 1–2 | completed | |||
| Crizotinib, 41 | Pfizer | ALK | NCT03126916 | different anticancer agents | 3 | recruiting |
| NCT01606878 | different anticancer agents | 1 | completed | |||
| NCT00939770 | - | 1–2 | completed, has results | |||
| NCT03107988 | lorlatinib cyclophosphamide topotecan | 1 | recruiting | |||
| NCT01121588 | - | 1 | active not recruiting | |||
| Ceritinib, 43 | Novartis | NCT02780128 | - | 1 | recruiting | |
| NCT02559778 | dasatinib sorafenib vorinostat difluoromethylornithine | 2 | recruiting | |||
| NCT01742286 | - | 1 | completed | |||
| Lorlatinib, 45 | Pfizer | ALK | NCT04753658 | - | 1? | recruiting |
| NCT03107988 | cyclophosphamide topotecan | 1 | recruiting | |||
| Entrectinib, 46 | Ignyta | ALK | NCT02650401 | - | 1–2 | recruiting |
| Erdafitinib, 54 | Janssen | FGFR | NCT03210714 | - | 2 | recruiting |
| NCT03155620 | different anticancer agents | 2 | recruiting | |||
| Dasatinib, 60 | Bristol-Myers Squibb | Src, multitarget | NCT01467986 | rapamycin irinotecan temozolomide | 2 | completed |
| NCT02559778 | ceritinib sorafenib | 2 | recruiting | |||
| NCT00788125 | carboplatin etoposide ifosfamide | 1–2 | active, not recruiting | |||
| Sorafenib, 65 | Bayer-Onyx | multitarget | NCT02298348 | cyclophospamide topotecan | 1 | recruiting |
| NCT02559778 | ceritinib dasatinib vorinostat | 2 | recruiting | |||
| NCT01518413 | rinotecan | 1 | completed | |||
| Regorafenib, 68 | Bayer | multitarget | NCT02085148 | irinotecan vincristine | 1 | active, not recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musumeci, F.; Cianciusi, A.; D’Agostino, I.; Grossi, G.; Carbone, A.; Schenone, S. Synthetic Heterocyclic Derivatives as Kinase Inhibitors Tested for the Treatment of Neuroblastoma. Molecules 2021, 26, 7069. https://doi.org/10.3390/molecules26237069
Musumeci F, Cianciusi A, D’Agostino I, Grossi G, Carbone A, Schenone S. Synthetic Heterocyclic Derivatives as Kinase Inhibitors Tested for the Treatment of Neuroblastoma. Molecules. 2021; 26(23):7069. https://doi.org/10.3390/molecules26237069
Chicago/Turabian StyleMusumeci, Francesca, Annarita Cianciusi, Ilaria D’Agostino, Giancarlo Grossi, Anna Carbone, and Silvia Schenone. 2021. "Synthetic Heterocyclic Derivatives as Kinase Inhibitors Tested for the Treatment of Neuroblastoma" Molecules 26, no. 23: 7069. https://doi.org/10.3390/molecules26237069