
Synthesis and Evaluation of Trypanocidal Activity of Chromane-Type Compounds and Acetophenones


Abstract
:1. Introduction
2. Results
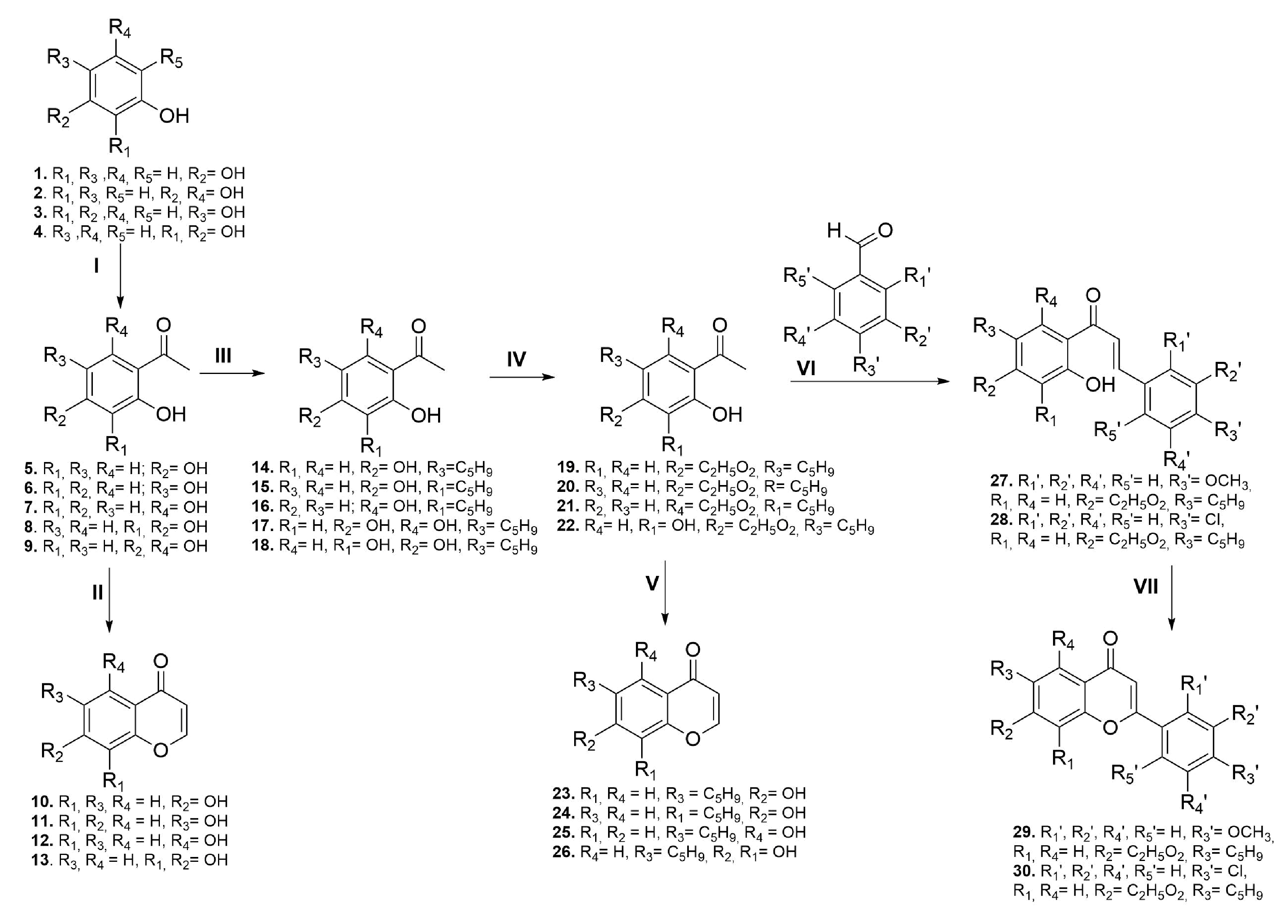
2.1. Compound Synthesis
2.2. In Vitro Cytotoxicity and Trypanocidal Activity
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.2. General Procedure for Preparation of 2-Hydroxyacetophenone Derivatives (I)
4.2.1. Compound (5)
4.2.2. Compound (6)
4.2.3. Compound (7)
4.2.4. Compound (8)
4.2.5. Compound (9)
4.3. General Procedure for Prenylation of 2-Hydroxyacetophenone Derivatives (III)
4.3.1. Compound (14)
4.3.2. Compound (15)
4.3.3. Compound (16)
4.3.4. Compound (17)
4.3.5. Compound (18)
4.4. General Procedure for Protection of Hydroxyl Groups (IV)
4.5. General Procedure for Preparation of Prenylated Chalcones (VI)
4.5.1. Compound (27)
4.5.2. Compound (28)
4.6. General Procedure for Preparation of Prenylated Flavones (VII)
4.6.1. Compound (29)
4.6.2. Compound (30)
4.7. General Procedure for Preparation of Chromones (II and V)
4.7.1. Compound (10)
4.7.2. Compound (11)
4.7.3. Compound (12)
4.7.4. Compound (13)
4.7.5. Compound (23)
4.7.6. Compound (24)
4.7.7. Compound (25)
4.7.8. Compound (26)
4.8. Cytotoxic Activity
4.9. Anti-Trypanosomal Activity
4.10. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pottie, K.; Girard, V. Common Infectious Diseases. Prim. Care Clin. Off. Pract. 2021, 48, 45–55. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization: Chagas Disease (American Trypanosomiasis). Available online: https://www.who.int/health-topics/chagas-disease#tab=tab_1 (accessed on 6 September 2021).
- Chao, M.N.; Storey, M.; Li, C.; Rodriguez, M.G.; Di Salvo, F.; Szajnman, S.H.; Moreno, S.N.J.; Docampo, R.; Rodriguez, J.B. Selenium-containing analogues of WC-9 are extremely potent inhibitors of Trypanosoma cruzi proliferation. Bioorg. Med. Chem. 2017, 25, 6435–6449. [Google Scholar] [CrossRef] [PubMed]
- Espinoza-Hicks, J.C.; Chacón-Vargas, K.F.; Hernández-Rivera, J.L.; Nogueda-Torres, B.; Tamariz, J.; Sánchez-Torres, L.E.; Camacho-Dávila, A. Novel prenyloxy chalcones as potential leishmanicidal and trypanocidal agents: Design, synthesis, and evaluation. Eur. J. Med. Chem. 2019, 167, 402–413. [Google Scholar] [CrossRef]
- Passalacqua, T.G.; Dutra, L.A.; De Almeida, L.; Velásquez, A.M.A.; Torres Esteves, F.A.; Yamasaki, P.R.; Dos Santos Bastos, M.; Regasini, L.O.; Michels, P.A.M.; Da Silva Bolzani, V.; et al. Synthesis and evaluation of novel prenylated chalcone derivatives as anti-leishmanial and anti-trypanosomal compounds. Bioorg. Med. Chem. Lett. 2015, 25, 3342–3345. [Google Scholar] [CrossRef] [PubMed]
- Garcia, E.; Coa, J.C.; Otero, E.; Carda, M.; Velez, I.D.; Robledo, S.M.; Cardona, W.I. Synthesis, and antiprotozoal activity of furanchalcone-quinoline, furanchalcone-chromone and furanchalcone-imidazole hybrids. Med. Chem. Res. 2017, 27, 497–511. [Google Scholar] [CrossRef]
- Gomes, K.S.; da Costa-Silva, T.A.; Oliveira, I.H.; Aguilar, A.M.; Oliveira-Silva, D.; Uemi, M.; Silva, W.A.; Melo, L.R.; Andrade, C.K.Z.; Tempone, A.G.; et al. Structure-activity relationship study of antitrypanosomal chalcone derivatives using multivariate analysis. Bioorg. Med. Chem. Lett. 2019, 29, 1459–1462. [Google Scholar] [CrossRef] [PubMed]
- Arioka, S.; Sakagami, M.; Uematsu, R.; Yamaguchi, H.; Togame, H.; Takemoto, H.; Hinou, H.; Nishimura, S.I. Potent inhibitor scaffold against Trypanosoma cruzi trans-sialidase. Bioorg. Med. Chem. 2010, 18, 1633–1640. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhou, R.-G.; Wu, T.; Yang, T.; Qin, Q.-X.; Li, L.; Yang, B.; Yang, J. Total synthesis of apigenin. J. Chem. Res. 2012, 36, 121–122. [Google Scholar] [CrossRef]
- Hoarau, C.; Pettus, T.R.R. Strategies for the preparation of differentially protected ortho-prenylated phenols. Synlett 2003, 1, 127–137. [Google Scholar] [CrossRef]
- Gomes, M.; Muratov, E.; Pereira, M.; Peixoto, J.; Rosseto, L.; Cravo, P.; Andrade, C.; Neves, B. Chalcone Derivatives: Promising Starting Points for Drug Design. Molecules 2017, 22, 1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev 2017, 117, 7762–7810. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, J.A.; Farrell, D.F. The chemistry of 2′-amino analogs of 2′- hydroxychalcone and its derivatives. J. Org. Chem. 1990, 55, 1757–1761. [Google Scholar] [CrossRef]
- Lahyani, A.; Trabelsi, M. Ultrasonic-assisted synthesis of flavones by oxidative cyclization of 2′-hydroxychalcones using iodine monochloride. Ultrason. Sonochem. 2016, 31, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Wang, Y.; Zhu, Y.Y.; Han, J.; Zhou, Y.F.; Koirala, D.; Li, D.W.; Hu, C. Synthesis, characterization, crystal structure and cytotoxicities of 2- aroyl-3-aryl-5H-furo[3,2-g]chromene derivatives. Arkivoc 2010, 11, 204–214. [Google Scholar]
- González, L.A.; Upegui, Y.A.; Rivas, L.; Echeverri, F.; Escobar, G.; Robledo, S.M.; Quiñones, W. Effect of substituents in the A and B rings of chalcones on antiparasite activity. Arch. Pharm. 2020, 353, 2000157. [Google Scholar] [CrossRef]
- Baell, J.; Walters, M. Chemistry: Chemical con artists foil drug discovery. Nature 2014, 513, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Torres, F.; Robledo, S.M.; Quiñones, W.; Escobar, G.; Archbold, R.; Correa, E.; Gil, J.F.; Arbeláez, N.; Murillo, J.; Echeverri, F. Exploring Antiparasitic Molecule Sources from Timber by-Product Industries-Leishmanicidal and Trypanocidal Compounds from Clathrotropis brunnea Amshoff. Front Pharmacol. 2020, 11, 584668. [Google Scholar] [CrossRef] [PubMed]
- Buckner, F.S.; Verlinde, C.L.; La Flamme, A.C.; Van Voorhis, W.C. Efficient technique for screening drugs for activity against Trypanosoma cruzi using parasites expressing beta-galactosidase. Antimicrob. Agents Chemother. 1996, 40, 2592–2597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuno, K.; Burrows, J.N.; Duncan, K.; van Huijsduijnen, R.H.; Kaneko, T.; Kita, K.; Mowbray, C.E.; Schmatz, D.; Warner, P.; Slingsby, B.T. Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nat. Rev. Drug Discov. 2015, 14, 751–758. [Google Scholar] [CrossRef]


{kind=link}
| Series | N | Cytotoxicity (LC50 a (µM)) | Trypanocidal Activity (EC50 b (µM)) | SI c |
|---|---|---|---|---|
A | 5 | 54.0 ± 0.5 | 17.8 ± 0.8 | 3.0 |
| 6 | 6.3 ± 0 | 3.3 ± 0.1 | 1.9 | |
| 7 | 15.3 ± 1.9 | 10.5 ± 0.6 | 1.5 | |
| 8 | >200 | 18.3 ± 1.1 | >10.9 | |
| 9 | >200 | 49.3 ± 9.0 | >4.1 | |
B | 10 | 23.4 ± 6.4 | >11.7 * | <2 |
| 11 | 28.0 ± 1.9 | 11.8 ± 0.3 | 2.4 | |
| 12 | >100 | 19.9 ± 1.3 | >5.0 | |
| 13 | 43.0 ± 0.9 | 154.4 ± 26.8 | 0.3 | |
C | 14 | 33.98 ± 4.9 | >17 * | <2 |
| 15 | 25.21 ± 4.6 | 20.7 ± 1.1 | 1.2 | |
| 16 | 7.8 ± 3.1 | >4 * | <2 | |
| 17 | 21.2 ± 6.9 | 7.5 ± 0 | 2.8 | |
| 18 | 5.0 ± 1.7 | 2.6 ± 0.1 | 1.9 | |
D | 23 | >200 | >20 * | >10 |
| 24 | >100 | 111.1 ± 18.5 | >0.9 | |
| 25 | 47.1 ± 5.5 | 17.3 ± 0.4 | >5.8 | |
| 26 | >200 | 10.3 ± 0 | 4.59 | |
E | 27 | 43.4 ± 7.3 | 35.3 ± 1.1 | 1.2 |
| 28 | 0.5 ± 0.12 | >0.3 | <2 | |
F | 29 | 17.0 ± 0.2 | >9.8 * | <2 |
| 30 | 27.2 ± 3.3 | >13.6 * | <2 | |
| BNZ d | >768.5 | 56.5 ± 1.5 | >16.8 | |
| DOX e | 0.5 ± 0 | N/A f | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González, L.A.; Robledo, S.; Upegui, Y.; Escobar, G.; Quiñones, W. Synthesis and Evaluation of Trypanocidal Activity of Chromane-Type Compounds and Acetophenones. Molecules 2021, 26, 7067. https://doi.org/10.3390/molecules26237067
González LA, Robledo S, Upegui Y, Escobar G, Quiñones W. Synthesis and Evaluation of Trypanocidal Activity of Chromane-Type Compounds and Acetophenones. Molecules. 2021; 26(23):7067. https://doi.org/10.3390/molecules26237067
Chicago/Turabian StyleGonzález, Luis A., Sara Robledo, Yulieth Upegui, Gustavo Escobar, and Wiston Quiñones. 2021. "Synthesis and Evaluation of Trypanocidal Activity of Chromane-Type Compounds and Acetophenones" Molecules 26, no. 23: 7067. https://doi.org/10.3390/molecules26237067