
Quantitative FRET (qFRET) Technology for the Determination of Protein–Protein Interaction Affinity in Solution


Abstract
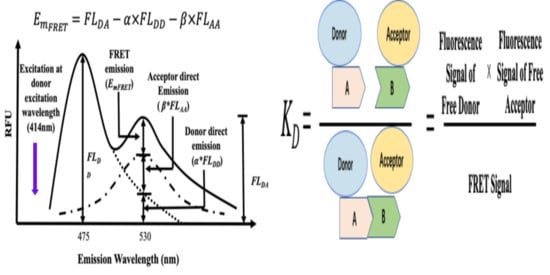
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
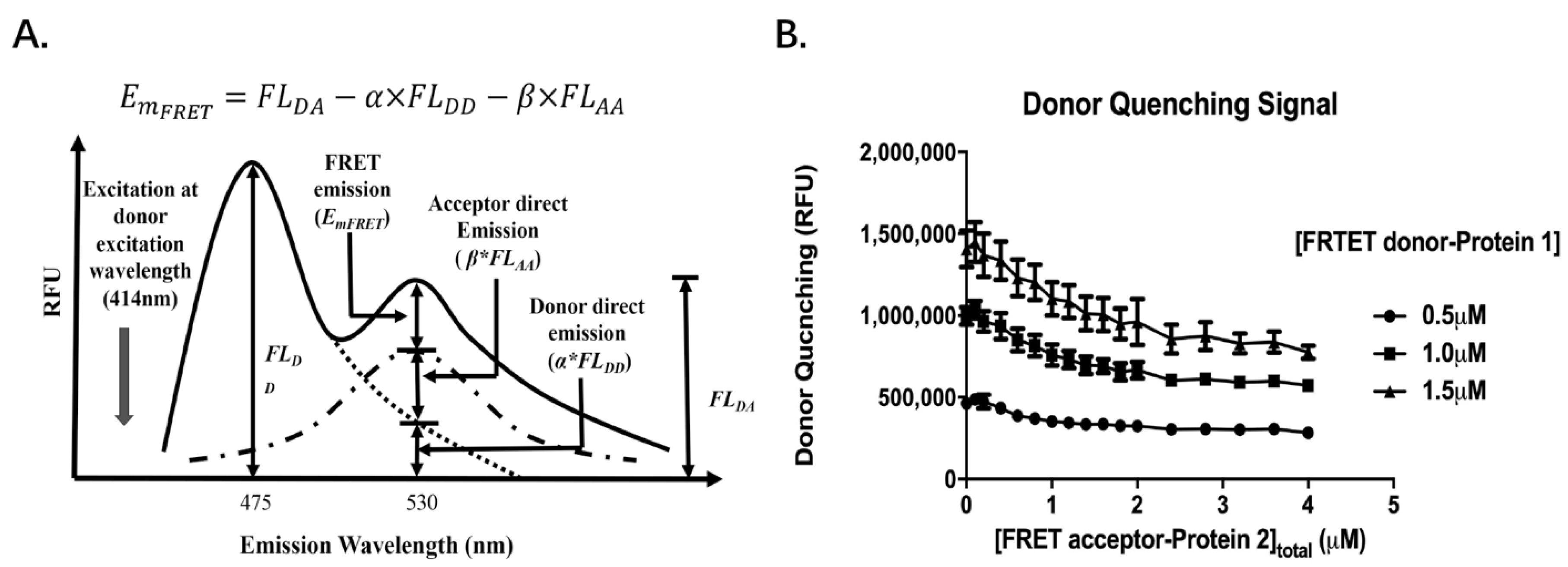{kind=link}
1. Approaches to Determine Protein–Protein Interaction Affinity (KD)
2. Theoretical Principles of Protein–Protein Interaction Affinity (KD) Determination from Acceptor Emission Using Quantitative FRET (qFRET) Analysis
3. Determination of Protein–Protein Interaction Affinity, KD, from FRET Donor Quenching
4. Comparison of Protein Interaction Affinity (KD) Determinations through FRET Acceptor Emission and FRET Donor Quenching Methods
5. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Braun, P.; Gingras, A.C. History of protein-protein interactions: From egg-white to complex networks. Proteomics 2012, 12, 1478–1498. [Google Scholar] [CrossRef]
- Nibbe, R.K.; Chowdhury, S.A.; Koyuturk, M.; Ewing, R.; Chance, M.R. Protein-protein interaction networks and subnetworks in the biology of disease. Wiley Interdiscip Rev. Syst. Biol. Med. 2011, 3, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Li, Q.; Wang, R. Current Experimental Methods for Characterizing Protein-Protein Interactions. ChemMedChem 2016, 11, 738–756. [Google Scholar] [CrossRef]
- Syafrizayanti; Betzen, C.; Hoheisel, J.D.; Kastelic, D. Methods for analyzing and quantifying protein-protein interaction. Expert Rev. Proteom. 2014, 11, 107–120. [Google Scholar] [CrossRef]
- Xing, S.; Wallmeroth, N.; Berendzen, K.W.; Grefen, C. Techniques for the Analysis of Protein-Protein Interactions in Vivo. Plant Physiol. 2016, 171, 727–758. [Google Scholar] [CrossRef] [Green Version]
- Rigaut, G.; Shevchenko, A.; Rutz, B.; Wilm, M.; Mann, M.; Seraphin, B. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 1999, 17, 1030–1032. [Google Scholar] [CrossRef]
- Krogan, N.J.; Cagney, G.; Yu, H.; Zhong, G.; Guo, X.; Ignatchenko, A.; Li, J.; Pu, S.; Datta, N.; Tikuisis, A.P.; et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 2006, 440, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545, 50–509. [Google Scholar] [CrossRef]
- Uetz, P.; Giot, L.; Cagney, G.; Mansfield, T.A.; Judson, R.S.; Knight, J.R.; Lockshon, D.; Narayan, V.; Srinivasan, M.; Pochart, P.; et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 2000, 403, 623–627. [Google Scholar] [CrossRef]
- Scott, D.E.; Bayly, A.R.; Abell, C.; Skidmore, J. Small molecules, big targets: Drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Discov. 2016, 15, 533–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent advances in the development of protein-protein interactions modulators: Mechanisms and clinical trials. Signal Transduct. Target. Ther. 2020, 5, 213. [Google Scholar] [CrossRef]
- Schuck, P. Use of surface plasmon resonance to probe the equilibrium and dynamic aspects of interactions between biological macromolecules. Annu. Rev. Biophys. Biomol. Struct. 1997, 26, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Douzi, B. Protein-Protein Interactions: Surface Plasmon Resonance. Methods Mol. Biol. 2017, 1615, 257–275. [Google Scholar]
- Pattnaik, P. Surface plasmon resonance: Applications in understanding receptor-ligand interaction. Appl. Biochem. Biotechnol. 2005, 126, 79–92. [Google Scholar] [CrossRef]
- Stahelin, R.V. Surface plasmon resonance: A useful technique for cell biologists to characterize biomolecular interactions. Mol. Biol. Cell 2013, 24, 883–886. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.H.; Park, J.; Kang, S.; Kim, M. Surface plasmon resonance: A versatile technique for biosensor applications. Sensors (Basel) 2015, 15, 10481–10510. [Google Scholar] [CrossRef] [Green Version]
- Helmerhorst, E.; Chandler, D.J.; Nussio, M.; Mamotte, C.D. Real-time and Label-free Bio-sensing of Molecular Interactions by Surface Plasmon Resonance: A Laboratory Medicine Perspective. Clin. Biochem. Rev. 2012, 33, 161–173. [Google Scholar]
- Schuck, P.; Zhao, H. The role of mass transport limitation and surface heterogeneity in the biophysical characterization of macromolecular binding processes by SPR biosensing. Methods Mol. Biol. 2010, 627, 15–54. [Google Scholar] [PubMed] [Green Version]
- Schuck, P.; Minton, A.P. Analysis of mass transport-limited binding kinetics in evanescent wave biosensors. Anal. Biochem. 1996, 240, 262–272. [Google Scholar] [CrossRef]
- Nieba, L.; Krebber, A.; Pluckthun, A. Competition BIAcore for measuring true affinities: Large differences from values determined from binding kinetics. Anal. Biochem. 1996, 234, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Wiseman, T.; Williston, S.; Brandts, J.F.; Lin, L.N. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal. Biochem. 1989, 179, 131–137. [Google Scholar] [CrossRef]
- Damian, L. Isothermal titration calorimetry for studying protein-ligand interactions. Methods Mol. Biol. 2013, 1008, 103–118. [Google Scholar]
- Tellinghuisen, J. Volume errors in isothermal titration calorimetry. Anal. Biochem. 2004, 333, 405–406. [Google Scholar] [CrossRef] [PubMed]
- Tellinghuisen, J.; Chodera, J.D. Systematic errors in isothermal titration calorimetry: Concentrations and baselines. Anal. Biochem. 2011, 414, 297–299. [Google Scholar] [CrossRef] [PubMed]
- Tellinghuisen, J. Calibration in isothermal titration calorimetry: Heat and cell volume from heat of dilution of NaCl(aq). Anal. Biochem. 2007, 360, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Raines, R.T. Fluorescence polarization assay to quantify protein-protein interactions: An update. Methods Mol. Biol. 2015, 1278, 323–327. [Google Scholar] [PubMed]
- Park, S.H.; Raines, R.T. Fluorescence polarization assay to quantify protein-protein interactions. Methods Mol. Biol. 2004, 261, 161–166. [Google Scholar]
- Gordon, G.W.; Berry, G.; Liang, X.H.; Levine, B.; Herman, B. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys. J. 1998, 74, 2702–2713. [Google Scholar] [CrossRef] [Green Version]
- Erickson, M.G.; Alseikhan, B.A.; Peterson, B.Z.; Yue, D.T. Preassociation of calmodulin with voltage-gated Ca(2+) channels revealed by FRET in single living cells. Neuron 2001, 31, 973–985. [Google Scholar] [CrossRef] [Green Version]
- Erickson, M.G.; Liang, H.; Mori, M.X.; Yue, D.T. FRET two-hybrid mapping reveals function and location of L-type Ca2+ channel CaM preassociation. Neuron 2003, 39, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.F.; Tatham, M.H.; Hay, R.T.; Samuel, I.D. Quantitative analysis of multi-protein interactions using FRET: Application to the SUMO pathway. Protein. Sci. 2008, 17, 777–784. [Google Scholar] [CrossRef] [Green Version]
- Mehta, K.; Hoppe, A.D.; Kainkaryam, R.; Woolf, P.J.; Linderman, J.J. A computational approach to inferring cellular protein-binding affinities from quantitative fluorescence resonance energy transfer imaging. Proteomics 2009, 9, 5371–5383. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Madahar, V.; Liao, J. Development of FRET Assay into Quantitative and High-throughput Screening Technology Platforms for Protein-Protein Interactions. Ann. Biomed. Eng. 2011, 39, 1224–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, J.Y.; Song, Y.; Liu, Y. A new trend to determine biochemical parameters by quantitative FRET assays. Acta Pharm. Sin. 2015, 36, 1408–1415. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Rodgers, V.G.; Schultz, J.S.; Liao, J. Protein interaction affinity determination by quantitative FRET technology. Biotechnol. Bioeng. 2012, 109, 2875–2883. [Google Scholar] [CrossRef]
- Wallrabe, H.; Elangovan, M.; Burchard, A.; Periasamy, A.; Barroso, M. Confocal FRET microscopy to measure clustering of ligand-receptor complexes in endocytic membranes. Biophys. J. 2003, 85, 559–571. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Huang, C.; Emery, B.P.; Sedgwick, A.C.; Bull, S.D.; He, X.P.; Tian, H.; Yoon, J.; Sessler, J.L.; James, T.D. Forster resonance energy transfer (FRET)-based small-molecule sensors and imaging agents. Chem. Soc. Rev. 2020, 49, 5110–5139. [Google Scholar] [CrossRef]
- Velick, S.F.; Parker, C.W.; Eisen, H.N. Excitation energy transfer and the quantitative study of the antibody hapten reaction. Proc Natl Acad Sci USA 1960, 46, 1470–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.L.; Schultz, J.S. Equilibrium binding in immunosensors. IEEE Trans Biomed Eng 1986, 33, 133–138. [Google Scholar] [CrossRef]
- Jiang, L.; Xiong, Z.; Song, Y.; Lu, Y.; Chen, Y.; Schultz, J.S.; Li, J.; Liao, J. Protein-Protein Affinity Determination by Quantitative FRET Quenching. Sci Rep 2019, 9, 2050. [Google Scholar] [CrossRef] [PubMed]
- Tatham, M.H.; Chen, Y.; Hay, R.T. Role of two residues proximal to the active site of Ubc9 in substrate recognition by the Ubc9.SUMO-1 thiolester complex. Biochemistry 2003, 42, 3168–3179. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, J.; Madahar, V.; Dang, R.; Jiang, L. Quantitative FRET (qFRET) Technology for the Determination of Protein–Protein Interaction Affinity in Solution. Molecules 2021, 26, 6339. https://doi.org/10.3390/molecules26216339
Liao J, Madahar V, Dang R, Jiang L. Quantitative FRET (qFRET) Technology for the Determination of Protein–Protein Interaction Affinity in Solution. Molecules. 2021; 26(21):6339. https://doi.org/10.3390/molecules26216339
Chicago/Turabian StyleLiao, Jiayu, Vipul Madahar, Runrui Dang, and Ling Jiang. 2021. "Quantitative FRET (qFRET) Technology for the Determination of Protein–Protein Interaction Affinity in Solution" Molecules 26, no. 21: 6339. https://doi.org/10.3390/molecules26216339