
Energetics of Electron Pairs in Electrophilic Aromatic Substitutions


Abstract
:
1. Introduction
2. Results and Discussion
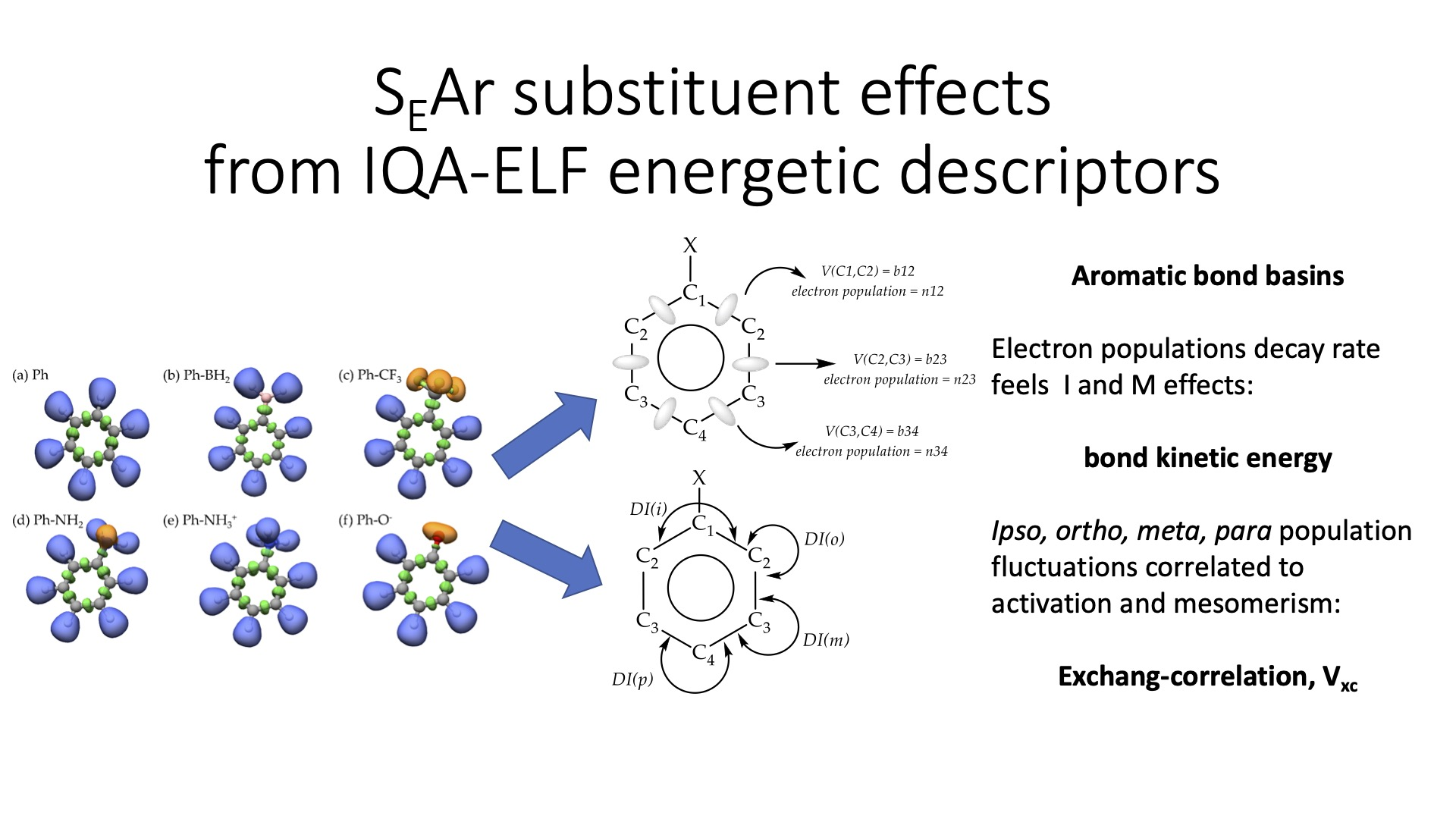
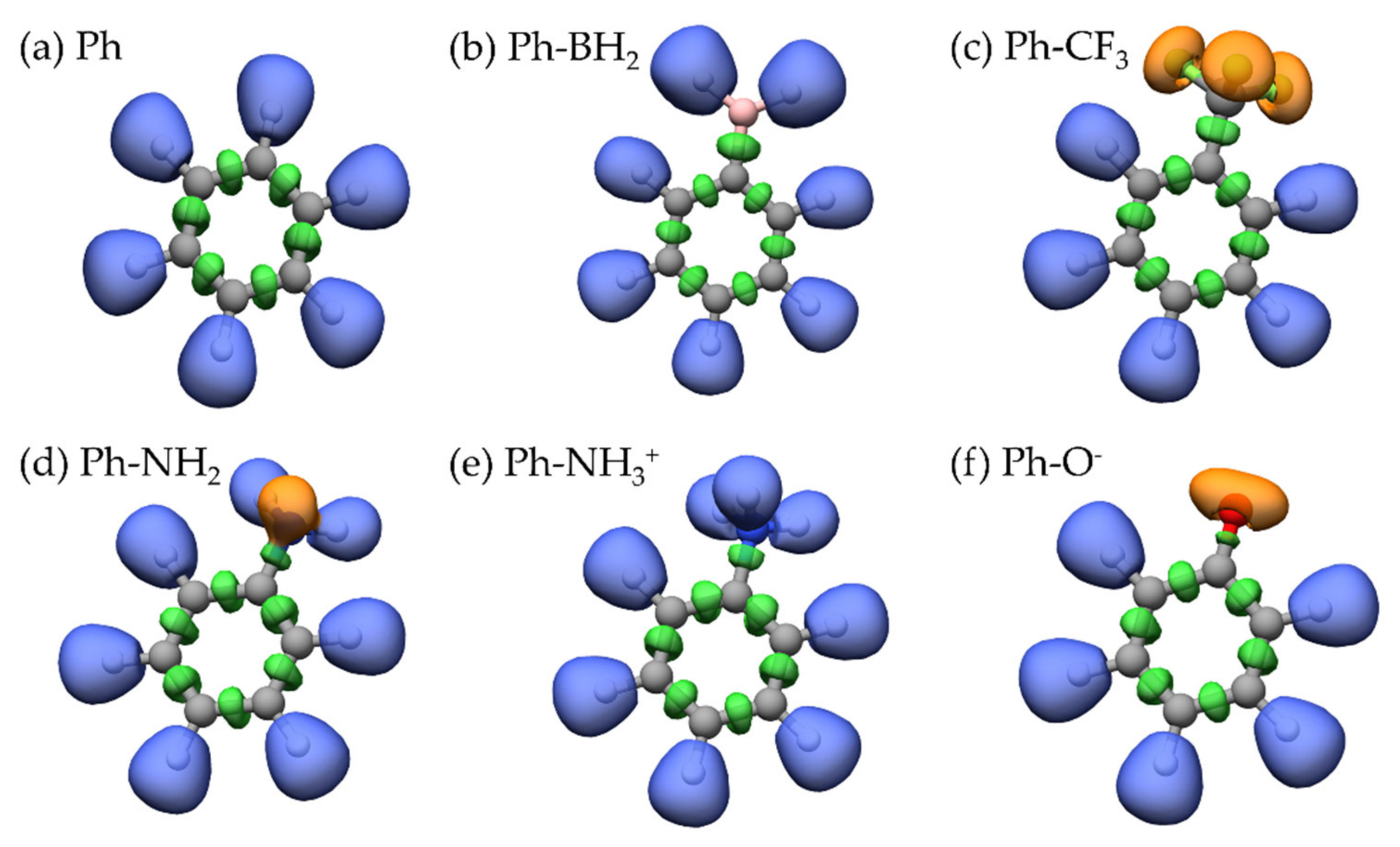
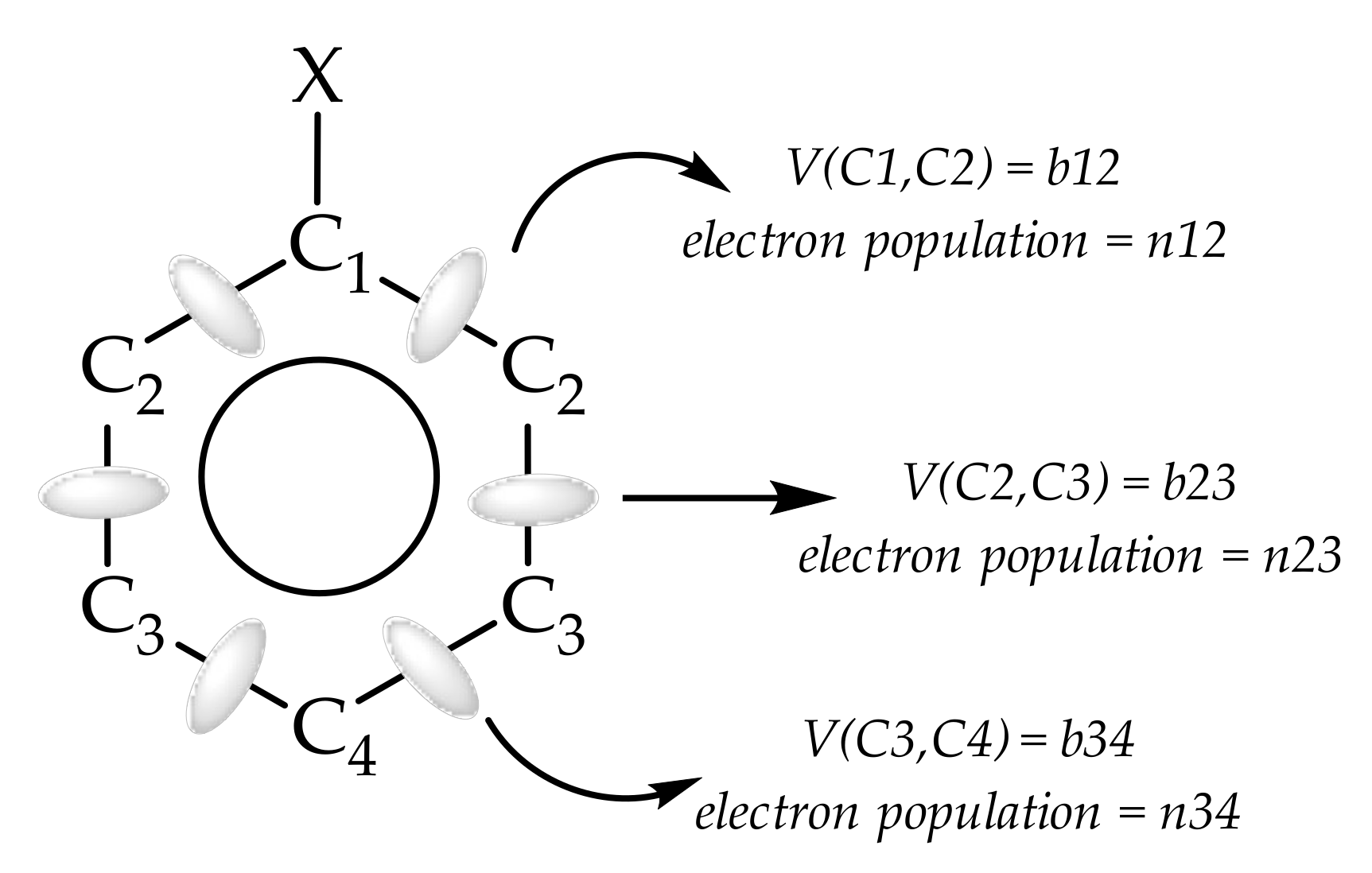
2.1. ELF Topology
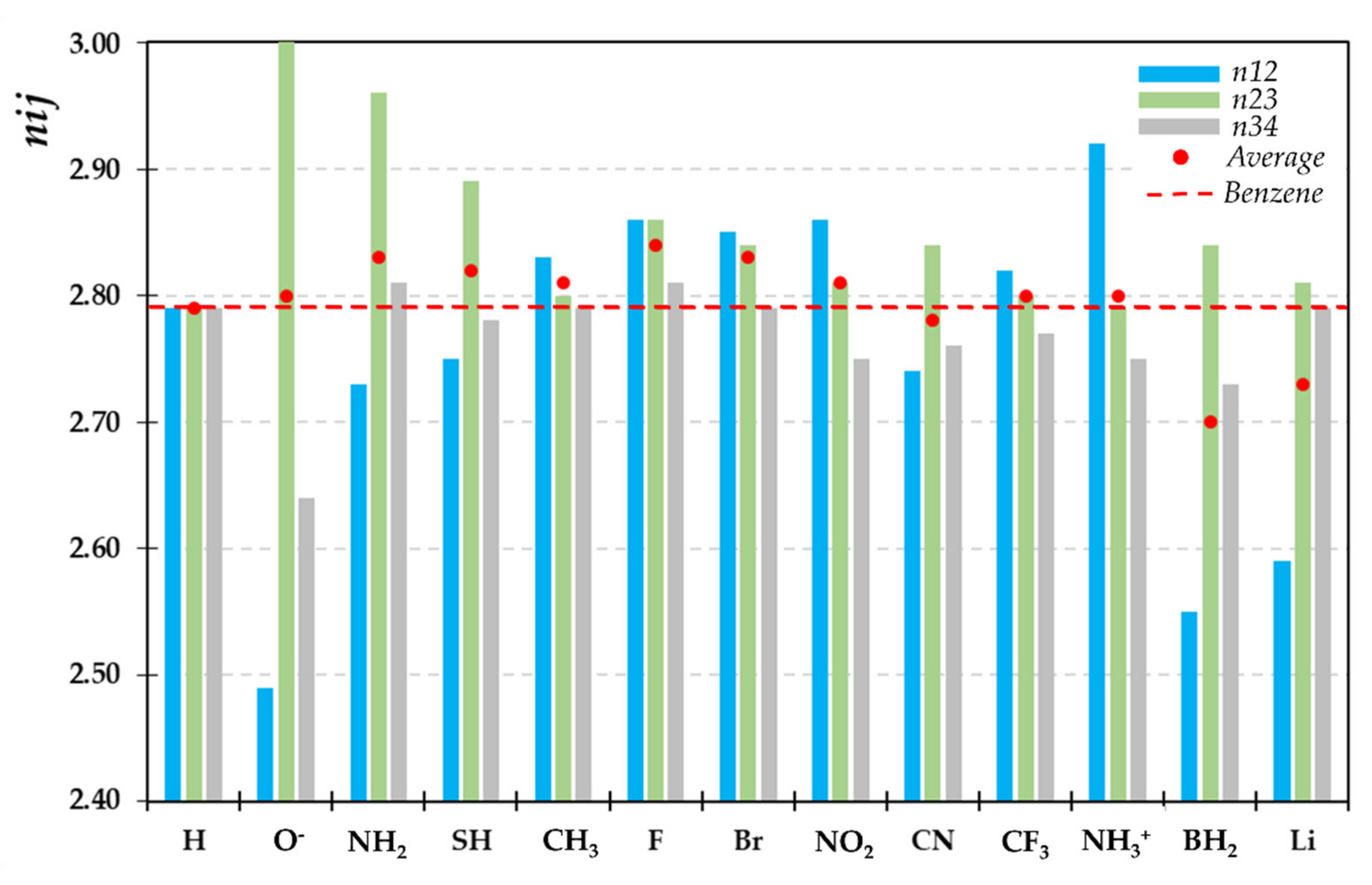
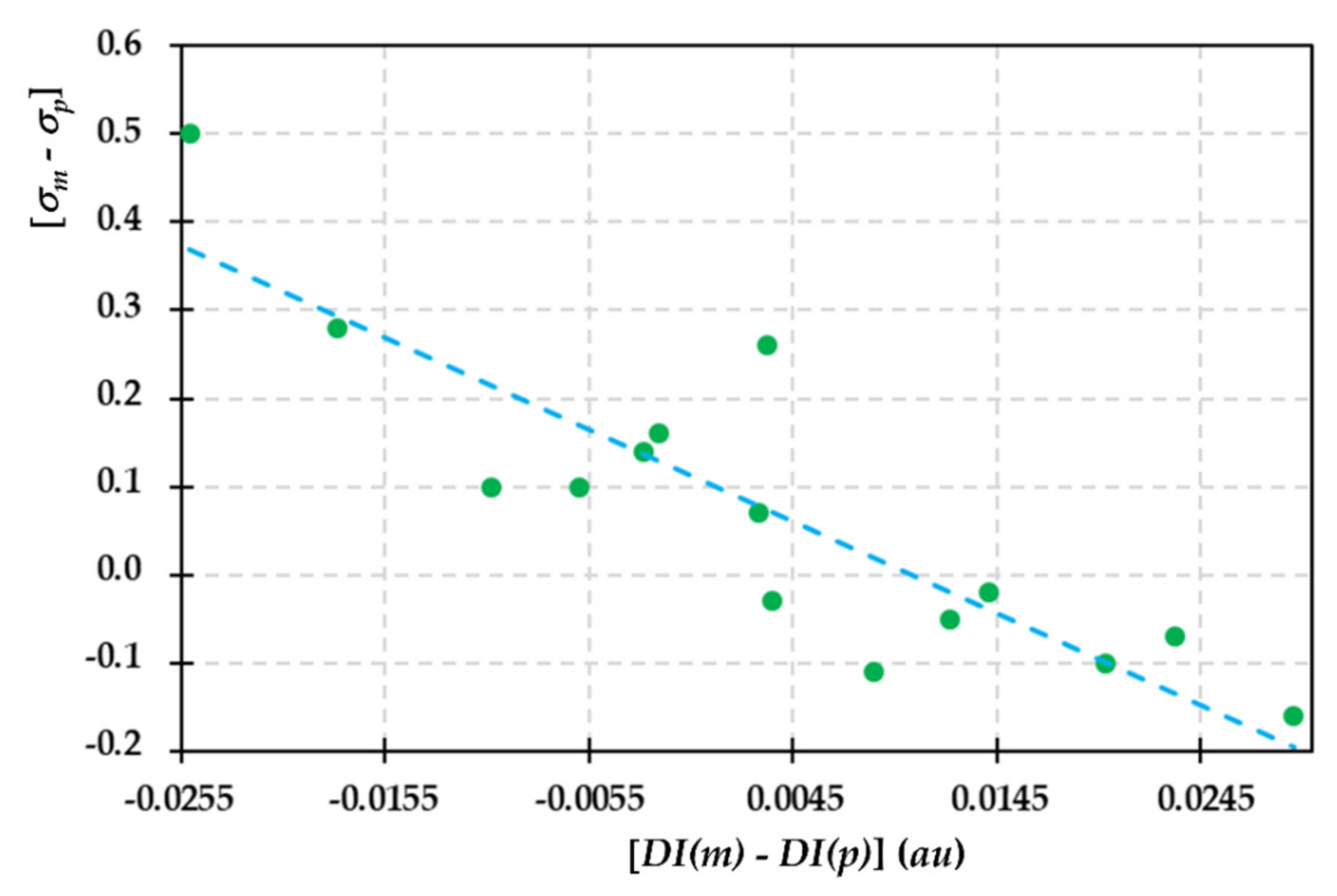
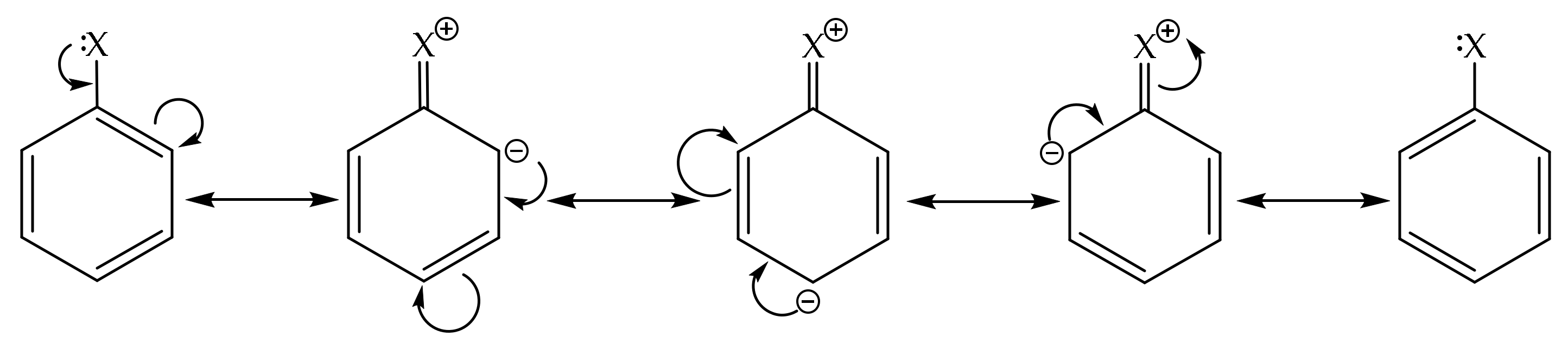
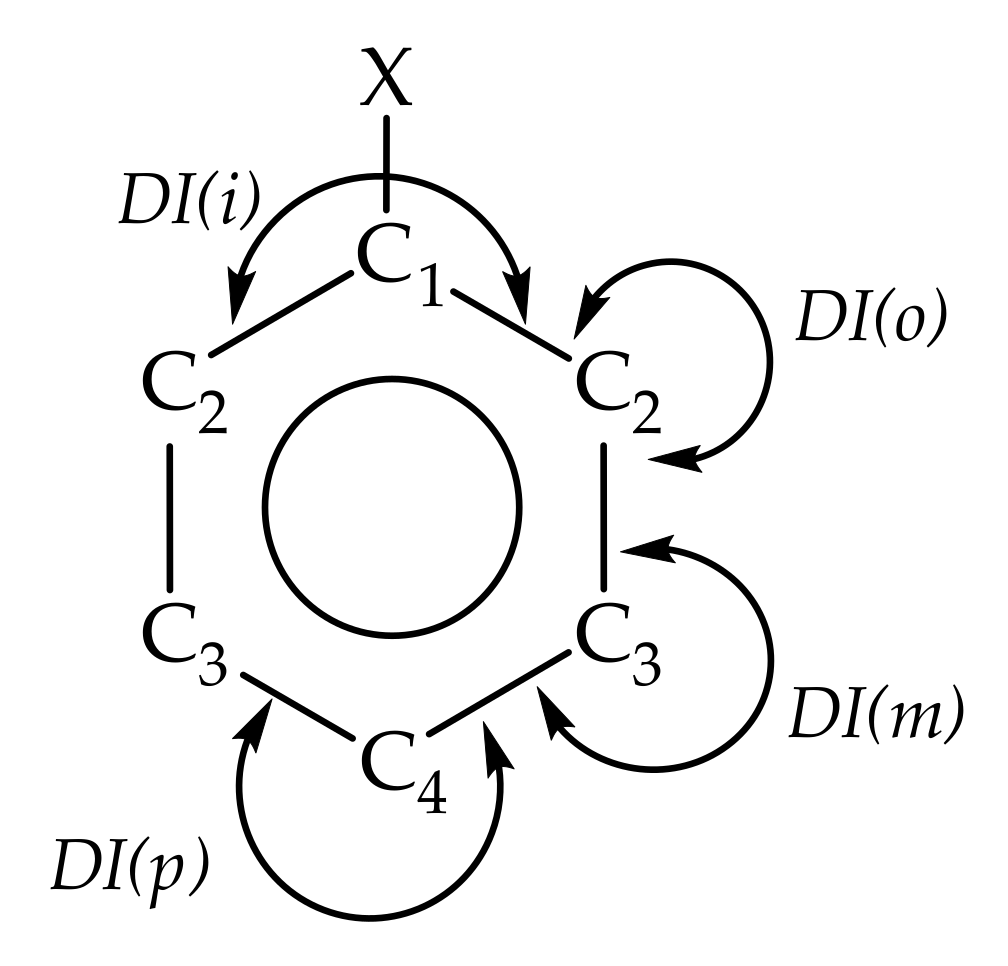
2.2. Recognizing Induction and Mesomerism
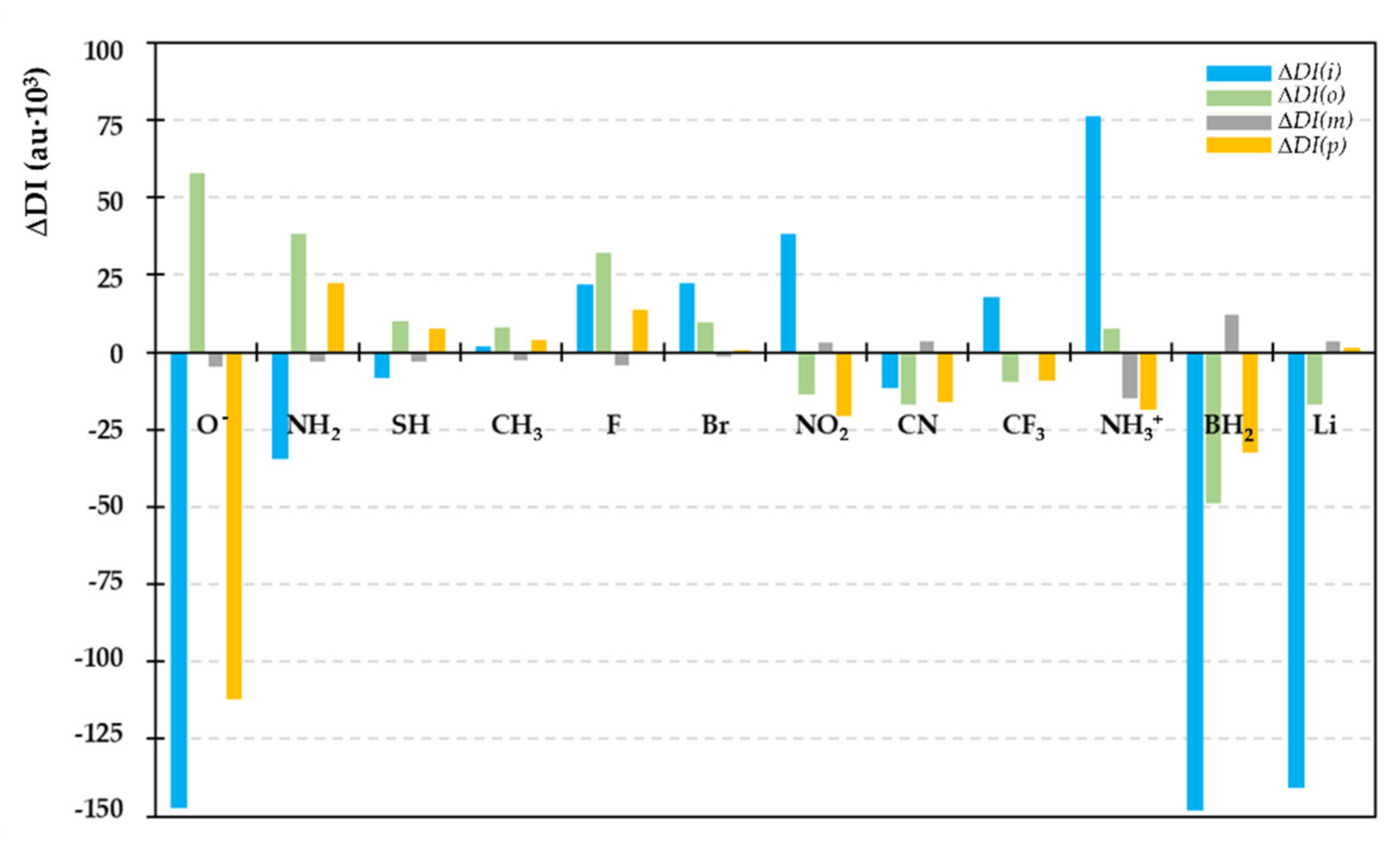
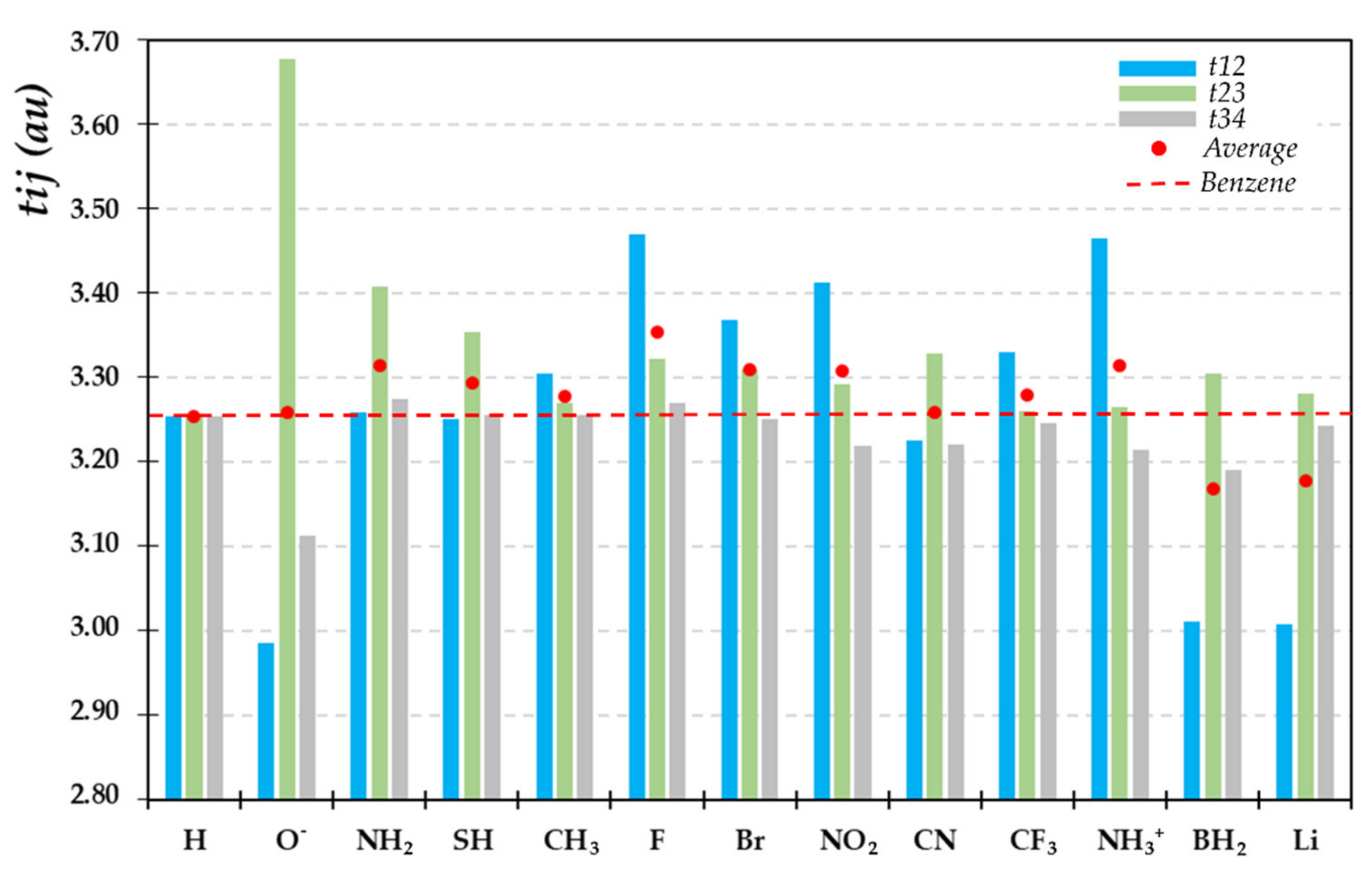
2.3. Bond Delocalization and Mesomerism
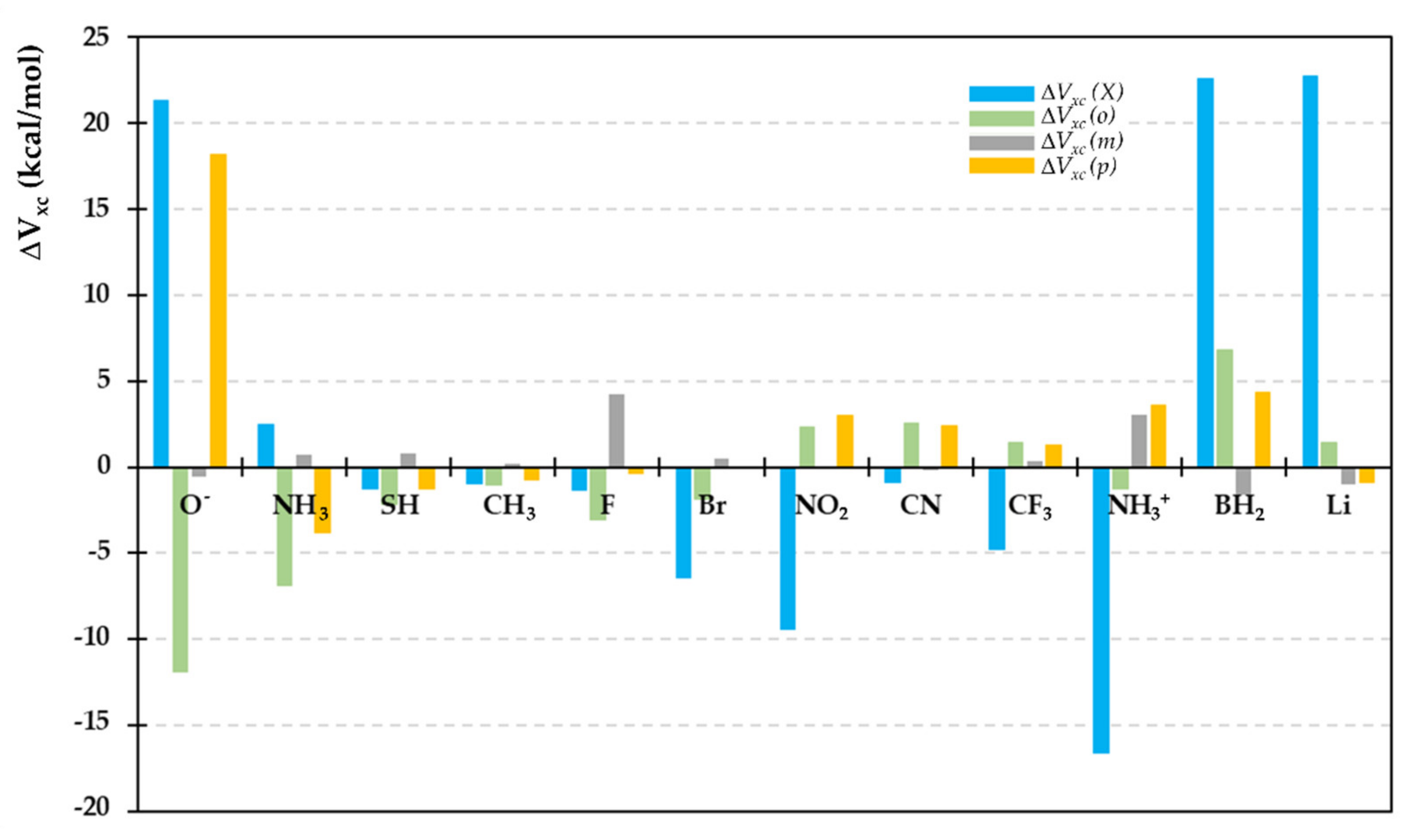
2.4. IQA-ELF Descriptors
3. Theoretical and Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Houk, K.N.; Liu, F. Holy Grails for Computational Organic Chemistry and Biochemistry. Acc. Chem. Res. 2017, 50, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Willems, H.M.G.; De Cesco, S.; Svensson, F. Computational Chemistry on a Budget: Supporting Drug Discovery with Limited Resources. J. Med. Chem. 2020, 63, 10158–10169. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-D.; Shang, C.; Zhang, X.-J.; Liu, Z.-P. Material discovery by combining stochastic surface walking global optimi-zation with a neural network. Chem. Sci. 2017, 8, 6327–6337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, K.T.; Frost, J.M.; Skelton, J.M.; Svane, K.L.; Walsh, A. Computational materials design of crystalline solids. Chem. Soc. Rev. 2016, 45, 6138–6146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtarolo, S.; Hart, G.L.; Nardelli, M.B.; Mingo, N.; Sanvito, S.; Levy, O. The high-throughput highway to computational materials design. Nat. Mater. 2013, 12, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Gooneie, A.; Schuschnigg, S.; Holzer, C. A Review of Multiscale Computational Methods in Polymeric Materials. Polymers 2017, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Mater, A.C.; Coote, M.L. Deep Learning in Chemistry. J. Chem. Inf. Model. 2019, 59, 2545–2559. [Google Scholar] [CrossRef]
- Dral, P.O. Quantum Chemistry in the Age of Machine Learning. J. Phys. Chem. Lett. 2020, 11, 2336–2347. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef]
- Leach, A. Molecular Modelling: Principles and Applications, 2nd ed.; Prentice Hall: Upper New Jersey River, NJ, USA, 2001. [Google Scholar]
- Brázdová, V.; Bowler, D.R. Atomistic Computer Simulations: A Practical Guide; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Martín Pendás, Á.; Francisco, E.; Blanco, M. Electron–electron interactions between ELF basins. Chem. Phys. Lett. 2008, 454, 396–403. [Google Scholar] [CrossRef]
- Munarriz, J.; Laplaza, R.; Martín Pendás, Á.; Contreras-García, J. A first step towards quantum energy potentials of electron pairs. Phys. Chem. Chem. Phys. 2019, 21, 4215–4223. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Borkman, R.F. Simple Bond-Charge Model for Potential-Energy Curves of Homonuclear Diatomic Molecules. J. Chem. Phys. 1968, 49, 1055–1058. [Google Scholar] [CrossRef]
- Borkman, R.F.; Parr, R.G. Toward an Understanding of Potential-Energy Functions for Diatomic Molecules. J. Chem. Phys. 1968, 48, 1116–1126. [Google Scholar] [CrossRef]
- Becke, A.D.; E Edgecombe, K. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nat. Cell Biol. 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Munarriz, J.; Calatayud, M.; Contreras-García, J. Valence-Shell Electron-Pair Repulsion Theory Revisited: An Explanation for Core Polarization. Chem. A Eur. J. 2019, 25, 10938–10945. [Google Scholar] [CrossRef]
- Contreras-García, J.; Mori-Sánchez, P.; Silvi, B.; Recio, J.M. A Quantum Chemical Interpretation of Compressibility in Solids. J. Chem. Theory Comput. 2009, 5, 2108–2114. [Google Scholar] [CrossRef]
- Andrés, J.; González-Navarrete, P.; Safont, V.S.; Silvi, B. Curly arrows, electron flow, and reaction mechanisms from the perspective of the bonding evolution theory. Phys. Chem. Chem. Phys. 2017, 19, 29031–29046. [Google Scholar] [CrossRef]
- Munárriz, J.; Laplaza, R.; Polo, V. A bonding evolution theory study on the catalytic Noyori hydrogenation reaction. Mol. Phys. 2019, 117, 1315–1324. [Google Scholar] [CrossRef]
- Blanco, M.A.; Martín Pendás, A.; Francisco, E. Interacting Quantum Atoms: A Correlated Energy Decomposition Scheme Based on the Quantum Theory of Atoms in Molecules. J. Chem. Theory Comput. 2005, 1, 1096–1109. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.; Martín Pendás, Á.; Popelier, P.L.A. Extension of the interacting quantum atoms (IQA) approach to B3LYP level density functional theory (DFT). Phys. Chem. Chem. Phys. 2016, 18, 20986–21000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guevara-Vela, J.M.; Francisco, E.; Rocha-Rinza, E.; Martín Pendás, A. Interacting Quantum Atoms—A Review. Molecules 2020, 25, 4028. [Google Scholar] [CrossRef] [PubMed]
- Munarriz, J.; Velez, E.; Casado, M.A.; Polo, V. Understanding the reaction mechanism of the oxidative addition of ammonia by (PXP)Ir(i) complexes: The role of the X group. Phys. Chem. Chem. Phys. 2018, 20, 1105–1113. [Google Scholar] [CrossRef]
- Fernández-Alarcón, A.; Guevara-Vela, J.M.; Casals-Sainz, J.L.; Costales, A.; Francisco, E.; Martín Pendás, Á.; Rinza, T.R. Photochemistry in Real Space: Batho- and Hypsochromism in the Water Dimer. Chem. A Eur. J. 2020, 26, 17035–17045. [Google Scholar] [CrossRef]
- Contreras-García, J.; Marqués, M.; Menéndez, J.M.; Recio, J.M. From ELF to Compressibility in Solids. Int. J. Mol. Sci. 2015, 16, 8151–8167. [Google Scholar] [CrossRef] [Green Version]
- Laplaza, R.; Polo, V.; Contreras-García, J. A Bond Charge Model Ansatz for Intrinsic Bond Energies: Application to C–C Bonds. J. Phys. Chem. A 2020, 124, 176–184. [Google Scholar] [CrossRef]
- Galabov, B.; Nalbantova, D.; Schleyer, P.V.R.; Schaefer, I.H.F. Electrophilic Aromatic Substitution: New Insights into an Old Class of Reactions. Accounts Chem. Res. 2016, 49, 1191–1199. [Google Scholar] [CrossRef]
- Vollhardt, K.P.C.; Schore, N.E. Organic Chemistry: Structure and Function; W.H. Freeman, Macmillan Learning: New York, NY, USA, 2018. [Google Scholar]
- Taylor, R. Electrophilic Aromatic Substitution; Wiley: New York, NY, USA, 1990. [Google Scholar]
- Brown, J.J.; Cockroft, S.L. Aromatic reactivity revealed: Beyond resonance theory and frontier orbitals. Chem. Sci. 2013, 4, 1772–1780. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Parr, R.G. Activation hardness: New index for describing the orientation of electrophilic aromatic substitution. J. Am. Chem. Soc. 1990, 112, 5720–5724. [Google Scholar] [CrossRef]
- Koleva, G.; Galabov, B.; Wu, J.I.; Schaefer, H.F., III; Schleyer, P.v.R. Electrophile Affinity: A Reactivity Measure for Aromatic Substitution. J. Am. Chem. Soc. 2009, 131, 14722–14727. [Google Scholar] [CrossRef] [PubMed]
- Schnatter, W.F.K.; Rogers, D.W.; Zavitsas, A.A. Electrophilic Aromatic Substitution: Enthalpies of Hydrogenation of the Ring Determine Reactivities of C6H5X. The Direction of the C6H5–X Bond Dipole Determines Orientation of the Substitution. J. Phys. Chem. A 2013, 117, 13079–13088. [Google Scholar] [CrossRef] [PubMed]
- Franco-Pérez, M.; Polanco-Ramírez, C.A.; Gázquez, J.L.; Ayers, P.W.; Vela, A. Study of organic reactions using chemical re-activity descriptors derived through a temperature-dependent approach. Theor. Chem. Acc. 2020, 139, 44. [Google Scholar] [CrossRef]
- Hadzic, M.; Braïda, B.; Volatron, F. Wheland Intermediates: An ab Initio Valence Bond Study. Org. Lett. 2011, 13, 1960–1963. [Google Scholar] [CrossRef] [PubMed]
- Kromann, J.C.; Jensen, J.H.; Kruszyk, M.; Jessing, M.; Jørgensen, M. Fast and accurate prediction of the regioselectivity of electrophilic aromatic substitution reactions. Chem. Sci. 2017, 9, 660–665. [Google Scholar] [CrossRef] [Green Version]
- Fievez, T.; Pinter, B.; Geerlings, P.; Bickelhaupt, F.M.; De Proft, F. Regioselectivity in Electrophilic Aromatic Substitution: Insights from Interaction Energy Decomposition Potentials. Eur. J. Org. Chem. 2011, 2011, 2958–2968. [Google Scholar] [CrossRef]
- Cheshmedzhieva, D.; Ilieva, S.; Galabov, B. Computational evaluation of σI and σR substituent constants. J. Mol. Struct. 2010, 976, 427–430. [Google Scholar] [CrossRef]
- Stuyver, T.; Danovich, D.; De Proft, F.; Shaik, S. Electrophilic Aromatic Substitution Reactions: Mechanistic Landscape, Electrostatic and Electric-Field Control of Reaction Rates, and Mechanistic Crossovers. J. Am. Chem. Soc. 2019, 141, 9719–9730. [Google Scholar] [CrossRef]
- Hubig, S.M.; Kochi, J.K. Structure and Dynamics of Reactive Intermediates in Reaction Mechanisms. σ- and π-Complexes in Electrophilic Aromatic Substitutions. J. Org. Chem. 2000, 65, 6807–6818. [Google Scholar] [CrossRef]
- Koleva, G.; Galabov, B.; Kong, J.; Schaefer, H.F.; Schleyer, P.V.R. Electrophilic Aromatic Sulfonation with SO3: Concerted or Classic SEAr Mechanism? J. Am. Chem. Soc. 2011, 133, 19094–19101. [Google Scholar] [CrossRef]
- Galabov, B.; Koleva, G.; Simova, S.; Hadjieva, B.; Schaefer, H.F., III; Schleyer, P.v.R. Arenium Ions are not Obligatory. In-termediates in Electrophilic Aromatic Substitution. Proc. Natl. Acad. Sci. USA 2014, 111, 10067–10072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwaltney, S.R.; Rosokha, S.V.; Head-Gordon, M.; Kochi, J.K. Charge-Transfer Mechanism for Electrophilic Aromatic Nitration and Nitrosation via the Convergence of (ab Initio) Molecular-Orbital and Marcus–Hush Theories with Experiments. J. Am. Chem. Soc. 2003, 125, 3273–3283. [Google Scholar] [CrossRef] [PubMed]
- Shernyukov, A.; Genaev, A.M.; Salnikov, G.E.; Rzepa, H.S.; Shubin, V.G. Noncatalytic bromination of benzene: A combined computational and experimental study. J. Comput. Chem. 2016, 37, 210–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuster, F.; Sevin, A.A.; Silvi, B. Topological Analysis of the Electron Localization Function (ELF) Applied to the Electrophilic Aromatic Substitution. J. Phys. Chem. A 2000, 104, 852–858. [Google Scholar] [CrossRef]
- Silvi, B.; Kryachko, E.S.; Tishchenko, O.; Fuster, F.; Nguyen, M.T. Key properties of monohalogen substituted phenols: In-terpretation in terms of the electron localization function. Mol. Phys. 2002, 100, 1659–1675. [Google Scholar] [CrossRef]
- Fuster, F.; Sevin, A.; Silvi, B. Determination of substitutional sites in heterocycles from the topological analysis of the electron localization function (ELF). J. Comput. Chem. 2000, 21, 509–514. [Google Scholar] [CrossRef]
- Poater, J.; Duran, M.; Solà, M.; Silvi, B. Theoretical Evaluation of Electron Delocalization in Aromatic Molecules by Means of Atoms in Molecules (AIM) and Electron Localization Function (ELF) Topological Approaches. Chem. Rev. 2005, 105, 3911–3947. [Google Scholar] [CrossRef]
- Hammett, L.P. Some Relations between Reaction Rates and Equilibrium Constants. Chem. Rev. 1935, 17, 125–136. [Google Scholar] [CrossRef]
- Gallo-Bueno, A.; Francisco, E.; Martín Pendás, Á. Decay rate of real space delocalization measures: A comparison between analytical and test systems. Phys. Chem. Chem. Phys. 2016, 18, 11772–11780. [Google Scholar] [CrossRef] [Green Version]
- Gallo-Bueno, A.; Kohout, M.; Martín Pendás, A. Decay Rate of Correlated Real-Space Delocalization Measures: Insights into Chemical Bonding and Mott Transitions from Hydrogen Chains. J. Chem. Theory Comput. 2016, 12, 3053–3062. [Google Scholar] [CrossRef]
- Kohn, W. Theory of the Insulating State. Phys. Rev. 1964, 133, A171–A181. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, R.W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Francisco, E.; Crespo, D.M.; Costales, A.; Martín Pendás, Á. A multipolar approach to the interatomic covalent interaction energy. J. Comput. Chem. 2017, 38, 816–829. [Google Scholar] [CrossRef] [Green Version]
- Martín Pendás, A.; Blanco, M.A.; Francisco, E. Two-electron integrations in the quantum theory of atoms in molecules. J. Chem. Phys. 2004, 120, 4581–4592. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Dunning, T.H. Gaussian Basis Functions for Use in Molecular Calculations. III. Contraction of (10s6p) Atomic Basis Sets for the First-Row Atoms. J. Chem. Phys. 1971, 55, 716–723. [Google Scholar] [CrossRef]
- Dunning, T.H.; Peterson, K.A.; Wilson, A.K. Gaussian basis sets for use in correlated molecular calculations. X. The atoms aluminum through argon revisited. J. Chem. Phys. 2001, 114, 9244–9253. [Google Scholar] [CrossRef] [Green Version]
- Dobson, J.F. Interpretation of the Fermi hole curvature. J. Chem. Phys. 1991, 94, 4328–4333. [Google Scholar] [CrossRef]
- Savin, A.; Jepsen, O.; Flad, J.; Andersen, O.K.; Preuss, H.; Von Schnering, H.G. Electron Localization in Solid-State Structures of the Elements: The Diamond Structure. Angew. Chem. Int. Ed. 1992, 31, 187–188. [Google Scholar] [CrossRef]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Computational tools for the electron localization function topological analysis. Comput. Chem. 1999, 23, 597–604. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visual-ization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goddard, T.D.; Huang, C.C.; Ferrin, T.E. Visualizing density maps with UCSF Chimera. J. Struct. Biol. 2007, 157, 281–287. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| X. | Activation | Electronic Effect | Directing Effect |
|---|---|---|---|
| O− | +e | +I/+M | o/p |
| NH2 | +s | −I/+M | o/p |
| SH | +m | −I/+M | o/p |
| CH3,R | +w | +I | o/p |
| F [a] | +w | −I/+M | p |
| F [a] | −w | −I/+M | o |
| Cl,Br,I | −w | −I/+M | o/p |
| NO2,CN | −s | −I/−M | m |
| CF3,NH3+ | −s | −I | m |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munárriz, J.; Gallegos, M.; Contreras-García, J.; Martín Pendás, Á. Energetics of Electron Pairs in Electrophilic Aromatic Substitutions. Molecules 2021, 26, 513. https://doi.org/10.3390/molecules26020513
Munárriz J, Gallegos M, Contreras-García J, Martín Pendás Á. Energetics of Electron Pairs in Electrophilic Aromatic Substitutions. Molecules. 2021; 26(2):513. https://doi.org/10.3390/molecules26020513
Chicago/Turabian StyleMunárriz, Julen, Miguel Gallegos, Julia Contreras-García, and Ángel Martín Pendás. 2021. "Energetics of Electron Pairs in Electrophilic Aromatic Substitutions" Molecules 26, no. 2: 513. https://doi.org/10.3390/molecules26020513