2.1. Finding the Limitations of [Ru(Phen)3]2+ Oxygen Sensitivity in CAM
The luminescence properties of [Ru(Phen)
3]
2+ were previously described [
3,
7,
8,
10,
32]. As a small molecule, [Ru(Phen)
3]
2+ leaks out of the vessels and remains in the interstitial space of blood vessel cells [
3]. Representative biodistribution of [Ru(Phen)
3]
2+ in the CAM at 1 and 20 min after its administration is presented in
Figure 1b. While bright luminescence of [Ru(Phen)
3]
2+ was detected inside the vessels at 1 min post-administration, the majority of [Ru(Phen)
3]
2+ luminescence was observed out of the vessels at 20 min.
The luminescence decays of [Ru(Phen)
3]
2+ presented a typical monoexponential character (
Figure 1c). Its lifetime rapidly changed with the absence of atmospheric oxygen. The [Ru(Phen)
3]
2+ luminescence lifetime sensitivity to oxygen in solutions is quite unambiguous [
32]. Since the CAM is a living organism, it is more complex and heterogeneous than cells monolayers. The ideal mono-exponential decay observed in homogeneous solutions is, therefore, no longer observed in the CAM. This can be explained by the complexity and multi-component character of this living system. While the CAM membrane plexus, composed of endodermal cells, represents a static component, the dynamic blood flow is present in the whole system.
Placement of the CAM into an atmosphere with low oxygen leads to significant changes in the oxygenation of the extracellular/interstitial space. However, the transport of oxygen at the cellular level and in the vessels is not a simple process. On the other hand, we had to keep the embryos alive during the measurements. For this reason, the chicken embryo kept residual oxygen supply as it can be recognized from
Table 1.
Due to the non-Gaussian character of the [Ru(Phen)
3]
2+ luminescence lifetime (values detected in different CAMs) distributions, the data treatment could not be carried out by obvious reduction to arithmetic central values (mean values). Therefore, several central trend indicators were provided, as part of the R-programming environment [
33]. The oxygenation values in
Table 1 were estimated from Equation (1) (see below) that represents a calibration curve obtained from our previous study of [Ru(Phen)
3]
2+ in an isotonic solution of 0.9% NaCl [
32]. The [Ru(Phen)
3]
2+ luminescence lifetimes measured at different regimes (0%, 10%, and 20% O
2 in N
2 for 5, 10, and 20 min) were converted to% oxygen by the Stern-Volmer relation as follows:
The ‘in’ values of oxygenation, which correspond to [Ru(Phen)3]2+ luminescence lifetimes inside the vessels, are higher than the ‘out’ values measured in the less vascularized areas. Moreover, the values detected during the first 5 min, which correspond to intravascular [Ru(Phen)3]2+, were also higher than at 10 and 20 min post-administration. We can see, that the % O2 in N2 (from 12 to 16) measured in the CAM at 20% and 10% O2 in N2 applied externally were not significantly different. At the contrary, 0% O2 in N2 regime (from 7 to 11) were significantly different from both 20 and 10% O2 in N2 regimes (*** p < 0.001).
A statistical overview of the measurements collected under defined conditions is shown in
Figure 2. This figure explicitly shows a structured six-dimensional summary(.) output of the program R [
33], as previously described in the methods section. We can see that the use of this representation also reveals some equivalence with [Ru(Phen)
3]
2+ luminescence lifetime distribution characteristics. Initial findings revealed remarkable dynamic differences in statistics of ‘out’ and ‘in’ cases, as well as significant variations between results obtained with early, intermediate, and late administration. Let us first look at the values of the arithmetical means. These values can be arranged into the chronological layout of
Table 1. Within this table, we specified the column where the generalized mean values of the [Ru(Phen)
3]
2+ luminescence lifetimes (τ) are transformed to the relative quantity.
Here Median τ is a symbol for the median calculated from the data sets of luminescence decay times. More precisely, these are the values obtained by exponential regression. As a basis for comparison, we use the arithmetic mean of luminescence lifetime values denoted as Mean τ. For such averages, the zero of relMd indicates Gaussian behavior. From a practical point of view, obviously, it is important to know how the relMd differs from zero. An application example arose in the process of identifying 10% of O2 in N2 as the anomalous condition. Further, relMd (consider 0.029, 0.024, 0.03 to be representatively high values) becomes even more pronounced at 0% of O2 in N2.
For a more detailed interpretation, let’s also consider the ‘out-in’ sensitivity indicator:
The indices ‘in’ and ‘out’ mean the selection of data under specific conditions. The relation is performed to conduct a statistical characterization of the corresponding ‘in’, ‘out’ pairs at given administration times under fixed O
2% in N
2 conditions.
Table 1 shows a variation of a few percent with the outlier value of rel
OI = 0.103.
Hypoxic conditions applied to certain cells of the CAM may induce the fatal effect. This is of importance since the luminescence lifetime of [Ru(Phen)
3]
2+ in the presence of DNA is longer than in aqueous solution, and comparable with the values obtained in the absence of oxygen [
10,
11]. CAM cells suffering from acute hypoxia are more likely to become necrotic [
34]. Cell membranes and nuclear envelopes of these cells can be more permeable for [Ru(Phen)
3]
2+ molecules. We hypothesize that this effect can explain the highest sensitivity to the oxygen level in the case of 10% O
2 in N
2 applied for 10 min to the CAM. Nevertheless, certain cells are not affected by these conditions. It is expected that more cells should suffer from hypoxia at the 0% O
2 in N
2 regimes. However, the [Ru(Phen)
3]
2+ luminescence lifetimes distribution is less dispersed. This identifiable anomaly is consistent with the results presented in
Figure 2 which are highlighting the differences between the ‘in’ and ‘out’ curves that relate to the [Ru(Phen)
3]
2+ luminescence lifetime statistics.
Analysis of data based on multidimensional statistical inputs requires visual comparison and additional interpretation. The implementation of hierarchical processing in this project is new, but more natural because the clustering provides advanced tools to achieve interpretable and even visual consequence. In the present work, we also emphasize that clustering does not takes place once, but rather as an iterative mechanism with feedback.
Figure 3 shows the results of the cluster analysis based on luminescence lifetimes obtained under different experimental conditions. The specific arrangement of the results and conditions is such that, while (a) focuses on the ‘in’ cases, panel (b) monitors the ‘out’ scenarios. The color and the corresponding contents reflect the differences between panels (a) and (b). In panel (a) the structure of the dendrogram is more fragmented, while (b) is more ordered. This can be explained by the experimental control (high tissue homogeneity) of the ‘out’ cases, whereas the ‘in’ case data also reflect more complex physiological internal processes.
The results obtained with 10% O2 in N2 are the most fragmented, as shown in the overall statistics in panel (c). This is in agreement with the aforementioned anomaly analysis expressed by means of the relOI indicator. The cophenetic distance in the case of the panel (c) also clearly shows that while 10% and 20% O2 in N2 are statistically close ([Ru(Phen)3]2+ luminescence lifetime height 150 ns, highest square arch), the case of 0% O2 in N2 dataset cases is a significant outlier ([Ru(Phen)3]2+ luminescence lifetime height = 270 ns).
However, one might ask where the roots of mismatch and fragmentation are within the ‘in’ cases. The answer is indicated by the three upper panels of
Figure 4, which were generated by the separate processing of (‘in’, 0%), (‘in’, 10%), and (‘in’, 20%) subsets grouped from the entire ‘in’ statistics. The administration time (read the dendrogram from left to right) of the legs (branches) changed significantly from the 0% sequence (5 min, 10 min, and 20 min) to (20 min, 5 min, and 10 min) in the cases of 10% and 20% O
2 in N
2. This means that conditions corresponding to the sensitive branch with a duration of 20 min behave as if it more closely mimics the distance conditions of ‘in’ case. Another perspective of data perception is established in the last three ‘in’ dendrograms where the selection of data for clustering is carried out according to the time after administration. Maintaining the canonical order (0%, 10%, and 20%) is a surprising result of these experiments. Here, too, an unusual dendrogram height shift occurs at 20 min.
We recommend reading
Figure 3 panels in two directions. One can proceed from the details of the panels (a) and (b), and connect them in the direction of the composite panel (c) as the results. On the other hand, there is a top-down, reductionist way of interpreting panel (c) with less detailed visual representations of (a) and (b).
Figure 4 provides alternative and complementary methods for processing and understanding the results.
We know from previous examples as well as from practical experience that because of their flexibility, straightforwardness, and controllability, the dendrogram and cluster methods could still compete with many other forms of machine learning and data processing. They have become known and used not only in population genetics but also in many other bio-motivated fields, including brain sciences [
35].
We assumed that the change of oxygen atmosphere from normoxia to hypoxia in the gas chamber may have induced oxidative stress. The level of oxidative stress in CAM cells may then locally influence [Ru(Phen)3]2+ luminescence lifetime detection.
In the next step, we have evaluated the influence of the oxidative stress on the [Ru(Phen)3]2+ luminescence lifetime detection in glioma cells.
2.2. Localization of [Ru(Phen)3]2+ in Glioma Cells and Oxidative Stress
We have used a unique FLIM/PLIM system to see how [Ru(Phen)
3]
2+ localization influences the [Ru(Phen)
3]
2+ luminescence lifetime. Illustrative PLIM images of [Ru(Phen)
3]
2+ in the extracellular and intracellular spaces are presented in
Figure 5. U87 MG cells incubated during 1 h with 200 µM [Ru(Phen)
3]
2+ are recognized as the dark areas in the intensity and PLIM images. Extracellular [Ru(Phen)
3]
2+ luminescence lifetime was ≈650 ns (according to Equation (3) it corresponds to 13.7% of O
2 in N
2). [Ru(Phen)
3]
2+ was not recognized in the cytoplasm. However, the subcellular vesicles loaded with [Ru(Phen)
3]
2+ were identified at longer (24 h) incubations time. We have previously demonstrated that [Ru(Phen)
3]
2+ crossed the plasma membrane via endocytosis and maintained localized in the endocytotic vesicles, and peroxisomes [
7]. The intracellular lifetimes of [Ru(Phen)
3]
2+ were longer (≈800 ns and 7.3% of O
2 in N
2) than for extracellularly measured. Global lifetimes, i.e., the extracellular plus intracellular components, as represented by luminescence lifetime distribution histograms (
Figure 5), fall in the interval ranging between 600 and 900 ns (16.5% and 4.2% of O
2 in N
2). The increase of the intracellularly localized [Ru(Phen)
3]
2+ luminescence lifetime reflects lower oxygenation within the cells. However, the luminescence lifetime also may dependents on other environmental factors that could explain the longer luminescence lifetimes.
[Ru(Phen)
3]
2+ can be considered as a metalotoxin. However, it has a low toxicity and phototoxicity in comparison with porphyrins, as we have already demonstrated [
3,
7,
8]. The presence of metalotoxin in cells may induce oxidative stress. One of the most sensitive organelles to oxidative stress in cells are mitochondria. Representative confocal fluorescence images of CellROX green, a probe sensitive to oxidative stress induction in mitochondria, are presented in
Figure 6. We have selected this ROS unspecific probe due to its spectral properties and subcellular localization which are different from those of [Ru(Phen)
3]
2+ (
Figure 6b). Comparison of CellROX green localization with rhodamine 123 (a mitochondrial probe sensitive to mitochondria membrane potential), and [Ru(Phen)
3]
2+ is presented in
Figure S1 in Supplementary Materials. Intact mitochondria in U87 MG cells were detected in the presence of [Ru(Phen)
3]
2+. Application of light with the microscopy detection system-induced photodamages in cells, as revealed by the destabilization of the mitochondria membrane potential (tubular structures cannot be recognized, see
Figure S2 in Supplementary Materials) and an increase in oxidative stress (CellROX localization in the nucleus), as can be seen in
Figure 6c. The localization of [Ru(Phen)
3]
2+ before and after illumination did not change (
Figure 6b,c).
To see the stability of [Ru(Phen)
3]
2+ distribution and luminescence at external induction of oxidative stress, we have applied H
2O
2 into the cell culture. The presence of 200 µM H
2O
2 in U87 MG resulted in significant reduction of intracellular catalase and superoxide dismutase 1 (see
Figure S3 in the Supplementary Materials). Zhang et al. observed that apoptosis triggered by H
2O
2 mediated oxidative stress in hepatoma cells involved decreasing of catalase and superoxide dismutase activity [
36]. It is in agreement with our findings. In our study, we have applied H
2O
2 for short time (<1 h) to induce acute stress, and prevent its complete metabolism. We have previously demonstrated by MitoSOX
TM Red [
15] that superoxide production increased in U87 MG cells treated with H
2O
2. Besides, application of H
2O
2 resulted in significant ROS production extracellularly and intracellularly detected by DCFDA/H2DCFDA assay (see
Figure S4 in Supplementary Materials), and further caused significant lipid peroxidation inside the cells (see
Figure S5 in Supplementary Materials).
The extracellular application of H
2O
2 (without molecular probes) induced dissipation of mitochondrial membrane potential, and fission of mitochondria in U87 MG cells (
Figure 6d and
Figure S2). The same effect as in the absence of [Ru(Phen)
3]
2+ (fission of mitochondria) was observed on mitochondria in the presence of [Ru(Phen)
3]
2+ and H
2O
2 keeping in the cell culture medium (
Figure 6e). [Ru(Phen)
3]
2+ distribution and luminescence were not changed.
A similar experiment was performed in cells that were incubated for 24 h with [Ru(Phen)
3]
2+, at that time it crossed plasma membrane via endocytosis [
7]. In this case, [Ru(Phen)
3]
2+ was removed from the cell culture medium prior observation. The expected fission of mitochondria induced by H
2O
2 was less effective (
Figure 6f). Indeed, oxidative stress induced by H
2O
2 caused partial relocalization of CellROX into the nucleus. The number of [Ru(Phen)
3]
2+ loaded vesicular structures previously identified as the peroxisomes [
7] were localized nearby tubular structured mitochondria (see
Figures S1 and S6 in Supplementary Materials), where it may attenuate the oxidative stress. This finding strongly suggests the antioxidants-like effects of [Ru(Phen)
3]
2+. The partial localization of CellROX in the nuclei, on the other hand, suggests that oxidative stress level increased in these cells due to H
2O
2.
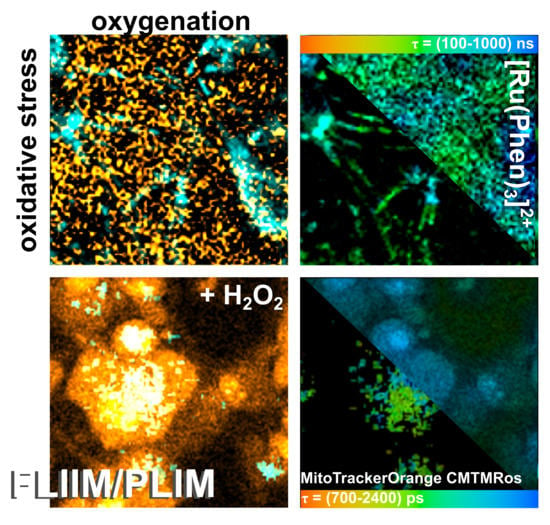
2.4. MTO Fluorescence and [Ru(Phen)3]2+ Luminescence Lifetimes Sensitivity during Oxidative Stress Induced in Glioma Cells
In our previous study, we have demonstrated that oxidative stress induced in cancer cells fragmented MTO such a way that one fraction was bound to DNA in the nucleus resulting in an increase of MTO fluorescence lifetime [
15]. Interestingly, MTO fluorescence lifetime increased in mitochondria in which oxidative stress level increased. For this reason, we have selected MTO to detect oxidative stress levels and [Ru(Phen)
3]
2+ to detect oxygenation in cells with a unique time-resolved microscopy system. With this setup, excitation and emission of MTO and [Ru(Phen)
3]
2+ were clearly discriminated, i.e., no signals of MTO was detected in PLIM, whereas no [Ru(Phen)
3]
2+ luminescence was detected in the MTO FLIM channel.
Oxidative stress was induced externally by H
2O
2.
Figure 8 shows FLIM images of MTO fluorescence lifetimes and PLIM images of [Ru(Phen)
3]
2+ luminescence lifetimes in U87MG cells in the presence and absence of H
2O
2. We can recognize the increase of MTO fluorescence lifetime (blue-colored) in the presence of H
2O
2 (
Figure 8a). Two main regions of interest (ROI) in cells were recognized. One ROI is in the perinuclear area (in, ≈1500 ps) with long MTO fluorescence lifetimes, whereas the second ROI is in the periphery, nearby the plasma membrane (out, ≈1200 ps) with shorter MTO fluorescence lifetimes (
Figure 8b,c). As the intracellular H
2O
2 concentration increased, the MTO delocalizes into the nucleus and the MTO fluorescence lifetime dramatically increased in this region (≈1900 ps).
PLIM measurements of [Ru(Phen)
3]
2+ were performed within 1 h after its administration, i.e., when [Ru(Phen)
3]
2+ localized extracellularly. Dark areas in images showing the cell localization of [Ru(Phen)
3]
2+ in PLIM can be recognized in
Figure 8a. Application of H
2O
2 resulted in a decrease of the extracellular [Ru(Phen)
3]
2+ luminescence lifetimes from ≈650 ns (13.7% of O
2 in N
2) to ≈400 ns (35% of O
2 in N
2), as presented in
Figure 8b. As observed in solutions, this effect could be caused by the H
2O
2 decomposition. The PLIM detection at longer incubation time (1 h) after the H
2O
2 application developed a bimodal character of [Ru(Phen)
3]
2+ luminescence lifetimes (see the distribution histograms in
Figure 8c). The shorter lifetimes < 600 ns (>16.5% of O
2 in N
2) were detected in the extracellular area, whereas the longer lifetime > 600 ns (<16.5% of O
2 in N
2) were detected in cells. It should be noted that the nuclei of the cells were brightly labeled with [Ru(Phen)
3]
2+ and with the longest luminescence lifetimes. This can be explained by the interaction of [Ru(Phen)
3]
2+ with cellular DNA. Indeed, Komor et al. described that the luminescence of [Ru(Phen)
3]
2+ increased in the presence of DNA [
10,
11]. We assume that the H
2O
2 applied destabilized the plasma membrane, which became permeable to [Ru(Phen)
3]
2+ molecules. These results suggest that the evaluation of [Ru(Phen)
3]
2+ lifetimes and subsequent determination of oxygenation level strongly depend on cell fitness.
One original aspect of our work was to apply simultaneously MTO and [Ru(Phen)
3]
2+ in cell culture media and to record FLIM and PLIM images from the same cells to monitor oxidative stress level and oxygenation changes. MTO FLIM and [Ru(Phen)
3]
2+ PLIM images were detected in the absence and presence of H
2O
2. No detectable FLIM and PLIM signals were observed in U87 MG cells in the absence of MTO and [Ru(Phen)
3]
2+ (
Figure 9a). As can be seen in
Figure 9, we have obtained results comparable to those observed when MTO and [Ru(Phen)
3]
2+ were applied independently. The MTO fluorescence lifetimes increased and the nucleus localized MTO appeared after H
2O
2 application. The [Ru(Phen)
3]
2+ luminescence lifetimes decreased shortly after H
2O
2 application and increased with time and nuclear distribution (
Figure 9b,c). It should be noted that the oxidative stress level evidenced by the MTO fluorescence lifetime increased in the presence of [Ru(Phen)
3]
2+ in the perinuclear area. Besides, the [Ru(Phen)
3]
2+ luminescence lifetime slightly increased in the presence of MTO. Therefore, the mutual influence of [Ru(Phen)
3]
2+ and MTO should be taken into consideration when oxidative stress level and oxygenation are estimated simultaneously.
2.5. MTO Fluorescence and [Ru(Phen)3]2+ Luminescence Lifetimes Changes Observed during PDT Induced by Curcumin and Blue Light in Glioma Cells
In the following, we aimed to apply the simultaneous FLIM/PLIM detection of MTO and [Ru(Phen)3]2+ after intracellular induction of oxidative stress by PDT. We have tested the hypothesis to detect oxygen consumption in the extracellular medium (with [Ru(Phen)3]2+), and oxidative stress (with MTO) induction within the cells by PDT.
The absorption curves of [Ru(Phen)
3]
2+, MTO, and curcumin are partially overlapping. Therefore, intensity-based techniques (e.g., fluorescence microscopy) present obstacles to discriminate their emissions. In our laboratory, we utilized a time-resolved microscopy system that enabled us to detect fluorescence (FLIM) and phosphorescence (PLIM) lifetimes from sample (cell) after its excitation with a white femtosecond laser. Thanks to this system we could differentiate by time-resolved spectroscopy the [Ru(Phen)
3]
2+, MTO, and curcumin localization in the same cell. Moreover, the lifetimes of MTO-FLIM and [Ru(Phen)
3]
2+ provided information regarding the oxidative stress level and oxygenation of the sample [
3,
15].
As mentioned in the introduction, curcumin can be a scavenger of singlet oxygen but, under irradiation with blue light, it can produce superoxide radicals, singlet oxygen and other reactive oxygen species [
29,
30,
31]. A combination of dual-detection (MTO-[Ru(Phen)
3]
2+) could reveal both, oxidative stress and antioxidant effect of curcumin during PDT in the same cells.
In the present study, we have incubated U87 MG cells with 10 µM curcumin during 1 h. Representative fluorescence intensity images of curcumin, MTO, and [Ru(Phen)
3]
2+ are presented in
Figure 10. We have observed an intracellular localization of curcumin, whereas MTO localizes in mitochondria, and [Ru(Phen)
3]
2+ in the extracellular space. Irradiation (detection time less than 3 min, laser was set at 2% of the total power of the laser: 405, 488, and 555 nm) coming from the microscopy system during fluorescence imaging triggered photoreactions generated oxidative stress production, photobleaching of curcumin fluorescence, and nuclear localization of MTO (
Figure 10c). The intracellular localization of curcumin was not specifically in mitochondria. This is in agreement with the observations performed by Sala de Oyanguren et al. who demonstrated specific localization of curcumin in the endoplasmic reticulum and described its role in the process of autophagy and apoptosis [
37].
The fluorescence lifetime of curcumin is short (≈hundreds of ps), as reported by Khopde et al. in different solvent [
38]. FLIM and PLIM images of curcumin were detected in the same spectral window as [Ru(Phen)
3]
2+. Fluorescence lifetime histograms of curcumin have a bimodal character (
Figure 10e). Short components (≈350 ps) were localized in the perinuclear area, whereas longer fluorescence lifetimes of curcumin (≈580 ps) were observed in the periphery near the plasma membrane. In comparison to fluorescence, very little is known about curcumin phosphorescence. In the nineties, Chignell et al. described the photobleaching of curcumin and reported that its phosphorescence depends on the excitation wavelength [
29]. A broad phosphorescence spectrum > 650 nm was observed at 77 K and 282 nm excitation by this group. In the present study, the phosphorescence lifetime of curcumin was observed in the same PLIM detection window as it was for [Ru(Phen)
3]
2+ (
Figure 10d). Phosphorescence lifetimes of ≈590 ns were detected within U87 MG cells. To the best of our knowledge, this is the first time that curcumin phosphorescence imaging is demonstrated. We should note, that possible other product of curcumin degradation in the cytoplasm could be detected in PLIM mode. This effect should be verified in the future study. Very often the PS is degraded during PDT due to oxidative stress [
39]. For this reason, the curcumin PLIM, in the present study, may be considered as a marker for PDT efficacy in cells. With this regard, curcumin degradation should result in decrease of its phosphorescence.
In the following, curcumin was irradiated in cells with blue light emitted by a diode (463 ± 10 nm and 100 µW/cm
2 irradiance) for 30 and 90 min. Our time-resolved microscopy system detected the FLIM image of MTO and PLIM images of curcumin and [Ru(Phen)
3]
2+ in the same cell. Irradiation of cells in the presence of [Ru(Phen)
3]
2+ only and in combination with MTO in the absence of curcumin did not result in significantly different changes in FLIM/PLIM images (see
Figures S7 and S8 in Supplementary Materials). Indeed, irradiation of cells in the presence of [Ru(Phen)
3]
2+ resulted in significant ROS formation, and detected with H2DCFDA sensor in the extracellular area (see
Figure S4 in Supplementary Materials). It should be noted that higher light dose may induce certain damages as it was observed by confocal fluorescence microscopy. However, the presence of MTO during irradiation resulted to photodamage of the cells that affected FLIM/PLIM detection. The FLIM and PLIM images of MTO, curcumin, and [Ru(Phen)
3]
2+ are presented in
Figure 11. We can see that the presence of curcumin in U87 MG cells without irradiation increases the MTO fluorescence lifetimes (>2000 ps). This suggests that curcumin increases the oxidative stress in cells. The luminescence lifetimes of [Ru(Phen)
3]
2+ were not modified by the presence of curcumin (≈700 ns; 11.28% of O
2 in N
2). The histograms of curcumin phosphorescence lifetimes revealed values as short as ≈400 ns.
Irradiation dramatically changed the MTO and curcumin luminescence lifetime images. Indeed, the MTO fluorescence lifetimes decreased below 1400 ps after 30 min irradiation with blue light. Longer irradiation induced an increase of MTO fluorescence lifetimes. This suggests that the oxidative stress firstly decreased and later increased in cells along the irradiation time. We can recognize that the phosphorescence of curcumin was quenched by the 30 min irradiation with blue light, which led to a slight increase of the [Ru(Phen)
3]
2+ luminescence lifetime from 631 to 658 ns (from 14.7% to 13.3% of O
2 in N
2). These changes can be seen in the histograms of [Ru(Phen)
3]
2+ and curcumin phosphorescence lifetimes (
Figure 11). It could be that these changes are due to a slight oxygen consumption. On the contrary, the longest 90 min irradiation resulted in decreasing [Ru(Phen)
3]
2+ luminescence lifetimes down to 613 ns (15.7% of O
2 in N
2). It should be noted that the irradiation of cells during FLIM/PLIM detection did not induce any photodamage as it was the case for the confocal fluorescence microscopy.
One can reasonably assume that the antioxidant effects of irradiated curcumin prevail for short irradiation times. The photobleaching of curcumin probably induced oxygen consumption ([Ru(Phen)3]2+ luminescence lifetimes increasing). Moreover, degraded curcumin fractions, active as antioxidants, reduced the oxidative stress level in cells (MTO fluorescence lifetime decreasing).
Long-time irradiation of U87 MG cells probably reversed the curcumin antioxidant effect to photodamage induction. The oxidative stress level (MTO fluorescence lifetimes) increased in those cells. The cells became leakier for [Ru(Phen)3]2+ molecules, which were internalized in the cells. However, we could not distinguish between the signal from [Ru(Phen)3]2+ and possible photoproducts of curcumin that may remain in cells after irradiation and create bias in the [Ru(Phen)3]2+ luminescence lifetime determination.
As we have demonstrated above (
Section 2.2), extracellular H
2O
2 induced significant oxidative stress in cells. In one of our previous study, we have demonstrated that this effect can be reduced by catalase (antioxidant) application [
15]. One can assume that curcumin-PDT may induce H
2O
2 production in cells and subsequently increase intracellular catalase concentration. For this reason, the oxidative stress was induced extracellularly by H
2O
2 in U87 MG cells treated with curcumin (
Figure 11). Indeed, we have first observed a decrease of oxidative stress (decreased MTO fluorescence lifetimes), and a decrease of [Ru(Phen)
3]
2+ luminescence lifetimes. This is consistent with the results obtained in the absence of curcumin (
Figure 8 and
Figure 9). Interestingly, short phosphorescence lifetimes of curcumin were detected in cells. As expected, an increase of MTO fluorescence and [Ru(Phen)
3]
2+ luminescence lifetimes were observed 1 h after H
2O
2 administration. These results suggest that the H
2O
2 concentration was too high to be reduced by curcumin.
Although we have demonstrated that curcumin possesses antioxidant properties this compound is not as antioxidating as catalase. On the other hand, curcumin can be degraded with H2O2. The resulting products of this reaction could result in their interaction with curcumin, and decrease its phosphorescence lifetime. Low concentrations of curcumin at 1 h after H2O2 administration disable the antioxidant effect of curcumin and could be another reason for higher for the oxidative stress.
Photodamages induced in cells by curcumin differ from massive injury caused by H2O2. For this reason, we have conducted an assessment of the metabolic activity and phototoxicity in U87 MG cells in the presence of all studied molecules before and after irradiation with the blue light. Those results also define the limits of simultaneous MTO and [Ru(Phen)3]2+ luminescence lifetimes detection with minimal phototoxicity.
Two irradiation times were applied: 5 min and 30 min. We have chosen to irradiate cells for 5 min to evaluate if the irradiation during PLIM/FLIM measurement may induce photodamages in the cells. The evaluation was performed in living cells 3 h after irradiation with the WST-8 kit (
Figure 12a,b). This kit is biocompatible for living cells and does not requires DMSO administration. Standard MTT-assays were performed 24 h after cell irradiations (
Figure 12c,d). It should be noted, that the absorption of WST-8 overlaps with that of [Ru(Phen)
3]
2+, MTO, and curcumin. For this reason, a level of significance was estimated within the studied sets. A significant decrease of metabolic activity was found 30 min and 3 h after the irradiations in U87 MG cells treated with 10 µM curcumin, and in the presence of MTO (
Figure 12a). The metabolic activity of those cells was remarkably suppressed 24 h after the irradiation (
Figure 12c). Presence of [Ru(Phen)
3]
2+ in cells amplified the photoeffect (
Figure 12b,d). However, the viability of these cells did not drop below 60%. These results suggest that the antioxidizing activity of curcumin is more important than the apoptotic one. Our results suggest that a simultaneous detection of [Ru(Phen)
3]
2+ and MTO can be performed without photodamages if the irradiation from excitation laser maintain less than 30 min when the light dose is equal to 100 µW/cm
2.


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}