
Microwave-Assisted Synthesis, Characterization and Modeling of CPO-27-Mg Metal-Organic Framework for Drug Delivery

 ,
,
Abstract
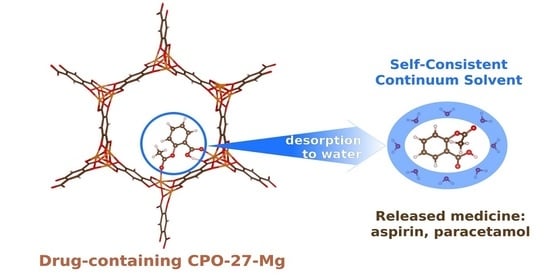
:
1. Introduction
2. Results and Discussion
- (1)
- we chose CPO-27(Mg) as the most suitable MOF structure from the point of view of its potential toxicity (it is the least toxic among similar structures) and pore volume/pore size characteristics that need to be suitable to accommodate as much drug as possible,
- (2)
- then we performed DFT calculations to predict the behavior of the two drugs in the chosen CPO-27(Mg) structure,
- (3)
- then we synthesized the selected CPO-27(Mg) structure and chose the microwave (MW) method, based on our previous experience on the microwave synthesis of MIL-100, MIL-53, and ZIF-8 structures and some literature data [12,13,14] that demonstrates that the MW synthesis provides certain advantages over the conventional solvothermal protocol, such as reduced duration of synthesis, reduced temperature of the synthesis, reduced pressure (instead of the autogenous pressure we used 1 atm), and controllable and uniform particle size,
- (4)
- and finally, we performed experiments on adsorption of aspirin and paracetamol in the predicted and synthesized CPO-27(Mg) structure.
2.1. DFT Simulations
2.2. Synthesis and Adsorption
3. Materials and Methods
3.1. Computational Details
3.2. Material Synthesis
3.3. Material Characterization
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Gref, R.; Minamitake, Y.; Peracchia, M.T.; Trubetskoy, V.; Torchilin, V.; Langer, R. Biodegradable long-circulating polymeric nanospheres. Science 1994, 263, 1600–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabizon, A.A. Stealth liposomes and tumor targeting: One step further in the quest for the magic bullet. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2001, 7, 223–225. [Google Scholar]
- Hasan, A.S.; Socha, M.; Lamprecht, A.; El Ghazouani, F.; Sapin, A.; Hoffman, M.; Maincent, P.; Ubrich, N. Effect of the microencapsulation of nanoparticles on the reduction of burst release. Int. J. Pharm. 2007, 344, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Horcajada, P.; Serre, C.; Maurin, G.; Ramsahye, N.A.; Balas, F.; Vallet-Regi, M.; Sebban, M.; Taulelle, F.; Férey, G. Flexible porous metal-organic frameworks for a controlled drug delivery. J. Am. Chem. Soc. 2008, 130, 6774–6780. [Google Scholar] [CrossRef]
- Horcajada, P.; Chalati, T.; Serre, C.; Gillet, B.; Sebrie, C.; Baati, T.; Eubank, J.F.; Heurtaux, D.; Clayette, P.; Kreuz, C.; et al. Porous metal-organic-framework nanoscale carriers as a potential platform for drug delivery and imaging. Nat. Mater. 2010, 9, 172–178. [Google Scholar] [CrossRef]
- Horcajada, P.; Gref, R.; Baati, T.; Allan, P.K.; Maurin, G.; Couvreur, P.; Ferey, G.; Morris, R.E.; Serre, C. Metal-organic frameworks in biomedicine. Chem. Rev. 2012, 112, 1232–1268. [Google Scholar] [CrossRef]
- Dietzel, P.D.C.; Blom, R.; Fjellvåg, H. Base-Induced Formation of Two Magnesium Metal-Organic Framework Compounds with a Bifunctional Tetratopic Ligand. Eur. J. Inorg. Chem. 2008, 2008, 3624–3632. [Google Scholar] [CrossRef]
- Bae, T.-H.; Long, J.R. CO2/N2 separations with mixed-matrix membranes containing Mg2(dobdc) nanocrystals. Energy Environ. Sci. 2013, 6, 3565–3569. [Google Scholar] [CrossRef]
- Rojas, S.; Wheatley, P.S.; Quartapelle-Procopio, E.; Gil, B.; Marszalek, B.; Morris, R.E.; Barea, E. Metal–organic frameworks as potential multi-carriers of drugs. CrystEngComm 2013, 15, 9364–9367. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Yu, J.; Liu, M.; Liu, A.; Dou, Z.; Yang, Y. A low cytotoxic cationic metal-organic framework carrier for controllable drug release. J. Med. Chem. 2014, 57, 5679–5685. [Google Scholar] [CrossRef]
- Allan, P.K.; Wheatley, P.S.; Aldous, D.; Mohideen, M.I.; Tang, C.; Hriljac, J.A.; Megson, I.L.; Chapman, K.W.; De Weireld, G.; Vaesen, S.; et al. Metal-organic frameworks for the storage and delivery of biologically active hydrogen sulfide. Dalton Trans. 2012, 41, 4060–4066. [Google Scholar] [PubMed]
- Haque, E.; Jhung, S.H. Synthesis of isostructural metal-organic frameworks, CPO-27s, with ultrasound, microwave, and conventional heating: Effect of synthesis methods and metal ions. Chem. Eng. J. 2011, 173, 866–872. [Google Scholar] [CrossRef]
- Isaeva, V.I.; Tarasov, A.L.; Chernyshev, V.V.; Kustov, L.M. Control of morphology and size of microporous framework MIL-53 (Al) crystals by synthesis procedure. Mendeleev Commun. 2015, 25, 466–467. [Google Scholar]
- Isaeva, V.I.; Kustov, L.M. Microwave activation as an alternative production of metal-organic frameworks. Russ. Chem. Bull. 2016, 65, 2103–2114. [Google Scholar]
- Goedecker, S.; Teter, M.; Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 1996, 54, 1703–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. QUICKSTEP: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef] [Green Version]
- Byrd, R.H.; Lu, P.; Nocedal, J.; Zhu, C. A limited memory algorithm for bound constrained optimization. Siam J. Sci. Comput. 1995, 16, 1190–1208. [Google Scholar] [CrossRef]
- Yin, W.J.; Krack, M.; Li, X.; Chen, L.Z.; Liu, L.M. Periodic continuum solvation model integrated with first-principles calculations for solid surfaces. Prog. Nat. Sci. Mater. Int. 2017, 27, 283–288. [Google Scholar] [CrossRef]
- Chung, Y.G.; Camp, J.; Haranczyk, M.; Sikora, B.J.; Bury, W.; Krungleviciute, V.; Yildirim, T.; Farha, O.K.; Sholl, D.S.; Snurr, R.Q. Computation-ready, experimental metal-organic frameworks. A tool to enable high-throughput screening of nanoporous crystals. Chem. Mater. 2014, 26, 6185–6192. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef]
- Marques, M.A.; Oliveira, M.J.; Burnus, T. Libxc: A library of exchange and correlation functionals for density functional theory. Computer Phys. Commun. 2012, 183, 2272–2281. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Murray, É.D.; Kong, L.; Lundqvist, B.I.; Langreth, D.C. Higher-accuracy van der Waals density functional. Phys. Rev. B 2010, 82, 081101. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Cadot, S.; Veyre, L.; Luneau, D.; Farrusseng, D.; Quadrelli, E.A. A water-based and high space-time yield synthetic route to MOF Ni2(dhtp) and its linker 2,5-dihydroxyterephthalic acid. J. Mater. Chem. A 2014, 2, 17757–17763. [Google Scholar] [CrossRef]
- Kljachko-Gurvich, A.L. An improved method of determining surface area by the adsorption of air. Bull. Acad. Sci. USSR, Div. Chem. Sci. 1961, 10, 1884–1886. [Google Scholar] [CrossRef]
- Wojdyr, M.; Fityk, A. general-purpose peak fitting program. J. Appl. Crystallogr. 2010, 43, 1126–1128. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functional | Δa/a0, % | Δb/b0, % | Δc/c0, % | Δα/α0, % | Δβ/β0, % | Δγ/γ0, % | ΔV/V0, % |
|---|---|---|---|---|---|---|---|
| HSE06 | −11.62 | −6.25 | −4.44 | −1.64 | 0.93 | −0.35 | −24.43 |
| PBEsol | −1.25 | 0.09 | −0.01 | 0.04 | 0.07 | −0.15 | 1.02 |
| revPBE | −2.77 | −0.87 | −0.87 | −0.17 | 0.16 | −0.18 | −4.51 |
| PBEsol-D3 | −0.77 | −0.16 | −0.06 | −0.04 | 0.00 | −0.14 | −0.45 |
| revPBE-D3 | −2.35 | −0.74 | −0.80 | −0.01 | 0.08 | −0.17 | −3.80 |
| Complex | Edes (Water) | Edes (Ethanol) |
|---|---|---|
| Asp@CPO-27-Mg | −5.6 | 0.7 |
| Par@CPO-27-Mg | 51.8 | 77.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kudelin, A.I.; Papathanasiou, K.; Isaeva, V.; Caro, J.; Salmi, T.; Kustov, L.M. Microwave-Assisted Synthesis, Characterization and Modeling of CPO-27-Mg Metal-Organic Framework for Drug Delivery. Molecules 2021, 26, 426. https://doi.org/10.3390/molecules26020426
Kudelin AI, Papathanasiou K, Isaeva V, Caro J, Salmi T, Kustov LM. Microwave-Assisted Synthesis, Characterization and Modeling of CPO-27-Mg Metal-Organic Framework for Drug Delivery. Molecules. 2021; 26(2):426. https://doi.org/10.3390/molecules26020426
Chicago/Turabian StyleKudelin, Anton I., Konstantinos Papathanasiou, Vera Isaeva, Juergen Caro, Tapio Salmi, and Leonid M. Kustov. 2021. "Microwave-Assisted Synthesis, Characterization and Modeling of CPO-27-Mg Metal-Organic Framework for Drug Delivery" Molecules 26, no. 2: 426. https://doi.org/10.3390/molecules26020426