
Glucose 6-Phosphate Dehydrogenase from Trypanosomes: Selectivity for Steroids and Chemical Validation in Bloodstream Trypanosoma brucei

 ,
,
Abstract
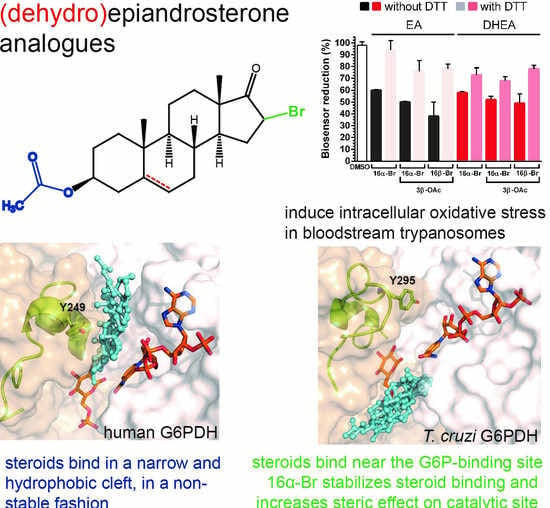
:
1. Introduction
2. Results and Discussion
2.1. G6PDH Is a Low Abundant and, Predominantly, Cytosolic Protein in Non-Infective T. cruzi
2.2. Epiandrosterone (EA) Targets G6PDH in T. cruzi
2.3. Inhibition of G6PDH by New (Dehydro)Epiandrosterone Derivatives
2.4. Differential Binding Mode of Steroids to Human and T. cruzi G6PDH
2.5. Biological Activity of Steroid Derivatives
2.6. Steroid Derivatives Affect the Intracellular Redox State of Bloodstream T. brucei
3. Conclusions
4. Materials and Methods
4.1. Protein and Ligand Preparation
4.2. Binding Site Detection and Docking
4.3. Reagents
4.4. Expression and Purification of Recombinant Proteins
4.5. G6PDH Enzymatic Assay
4.6. Generation of T. cruzi Cell Line with Inducible Expression of G6PDH
4.7. Western Blot
4.8. Indirect Immunofluorescence
4.9. Cytochemical Assay for G6PDH Enzymatic Activity
5. Cell Viability Assays
Redox Reporter Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Health Topics. Available online: https://www.who.int/health-topics (accessed on 31 October 2020).
- Croft, S.L.; Barrett, M.P.; Urbina, J.A. Chemotherapy of trypanosomiases and leishmaniasis. Trends Parasitol. 2005, 21, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Barrett, M.P.; Croft, S.L. Management of trypanosomiasis and leishmaniasis. Br. Med. Bull. 2012, 104, 175–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuart, K.; Brun, R.; Croft, S.; Fairlamb, A.; Gürtler, R.E.; McKerrow, J.; Reed, S.; Tarleton, R. Kinetoplastids: Related protozoan pathogens, different diseases. J. Clin. Investig. 2008, 118, 1301–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comini, M.A.; Ortiz, C.; Cazzulo, J.J. Drug Targets in Trypanosomal and Leishmanial Pentose Phosphate Pathway. In Trypanosomatids Diseases, Molecular Routes to Drug Discovery; Jäger, T., Koch, O., Flohé, L., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2013; ISBN 978-3-527-33255-7. [Google Scholar]
- Barrett, M.P. The pentose phosphate pathway and parasitic protozoa. Parasitol. Today 1997, 13, 11–16. [Google Scholar] [CrossRef]
- Igoillo-Esteve, M.; Maugeri, D.; Stern, A.L.; Beluardi, P.; Cazzulo, J.J. The pentose phosphate pathway in Trypanosoma cruzi: A potential target for the chemotherapy of Chagas disease. An. Acad. Bras. Cienc. 2007, 79, 649–663. [Google Scholar] [CrossRef] [Green Version]
- Loureiro, I.; Faria, J.; Santarem, N.; Smith, T.K.; Tavares, J.; Cordeiro-da-Silva, A. Potential Drug Targets in the Pentose Phosphate Pathway of Trypanosomatids. Curr. Med. Chem. 2018, 25, 5239–5265. [Google Scholar] [CrossRef]
- Kovářová, J.; Barrett, M.P. The Pentose Phosphate Pathway in Parasitic Trypanosomatids. Trends Parasitol. 2016, 32, 622–634. [Google Scholar] [CrossRef]
- Irigoín, F.; Cibils, L.; Comini, M.A.; Wilkinson, S.R.; Flohé, L.; Radi, R. Insights into the redox biology of Trypanosoma cruzi: Trypanothione metabolism and oxidant detoxification. Free Radic. Biol. Med. 2008, 45, 733–742. [Google Scholar] [CrossRef]
- Cordeiro, A.T. NADPH Producing Enzymes as Promising Drug Targets for Chagas Disease. Curr. Med. Chem. 2019, 26, 6564–6571. [Google Scholar] [CrossRef]
- Kovářová, J.; Nagar, R.; Faria, J.; Ferguson, M.A.J.; Barrett, M.P.; Horn, D. Gluconeogenesis using glycerol as a substrate in bloodstream-form Trypanosoma brucei. PLoS Pathog. 2018, 14, e1007475. [Google Scholar] [CrossRef]
- Allmann, S.; Morand, P.; Ebikeme, C.; Gales, L.; Biran, M.; Hubert, J.; Brennand, A.; Mazet, M.; Franconi, J.-M.; Michels, P.A.M.; et al. Cytosolic NADPH homeostasis in glucose-starved procyclic Trypanosoma brucei relies on malic enzyme and the pentose phosphate pathway fed by gluconeogenic flux. J. Biol. Chem. 2013, 288, 18494–18505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordeiro, A.T.; Thiemann, O.H.; Michels, P.A.M. Inhibition of Trypanosoma brucei glucose-6-phosphate dehydrogenase by human steroids and their effects on the viability of cultured parasites. Bioorg. Med. Chem. 2009, 17, 2483–2489. [Google Scholar] [CrossRef] [PubMed]
- Kerkhoven, E.J.; Achcar, F.; Alibu, V.P.; Burchmore, R.; Gilbert, I.H.; Trybiło, M.; Driessen, N.N.; Gilbert, D.; Breitling, R.; Bakker, B.M.; et al. Handling Uncertainty in Dynamic Models: The Pentose Phosphate Pathway in Trypanosoma brucei. PLoS Comput. Biol. 2013, 9, e1003371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, R.D.; Ebert, F. Evidence for NADH- and NADPH-linked glutamate dehydrogenases in Trypanosoma cruzi epimastigotes. J. Protozool. 1979, 26, 653–656. [Google Scholar] [CrossRef]
- Barderi, P.; Campetella, O.; Frasch, A.C.; Santomé, J.A.; Hellman, U.; Pettersson, U.; Cazzulo, J.J. The NADP+-linked glutamate dehydrogenase from Trypanosoma cruzi: Sequence, genomic organization and expression. Biochem. J. 1998, 330, 951–958. [Google Scholar] [CrossRef] [Green Version]
- Igoillo Esteve, M.; Cazzulo, J.J. The 6-phosphogluconate dehydrogenase from Trypanosoma cruzi: The absence of two inter-subunit salt bridges as a reason for enzyme instability. Mol. Biochem. Parasitol. 2004, 133, 197–207. [Google Scholar] [CrossRef]
- Igoillo-Esteve, M.; Cazzulo, J.J. The glucose-6-phosphate dehydrogenase from Trypanosoma cruzi: Its role in the defense of the parasite against oxidative stress. Mol. Biochem. Parasitol. 2006, 149, 170–181. [Google Scholar] [CrossRef]
- Leroux, A.E.; Maugeri, D.A.; Opperdoes, F.R.; Cazzulo, J.J.; Nowicki, C. Comparative studies on the biochemical properties of the malic enzymes from Trypanosoma cruzi and Trypanosoma brucei. FEMS Microbiol. Lett. 2011, 314, 25–33. [Google Scholar] [CrossRef]
- Leroux, A.E.; Maugeri, D.A.; Cazzulo, J.J.; Nowicki, C. Functional characterization of NADP-dependent isocitrate dehydrogenase isozymes from Trypanosoma cruzi. Mol. Biochem. Parasitol. 2011, 177, 61–64. [Google Scholar] [CrossRef]
- Ortíz, C.; Botti, H.; Buschiazzo, A.; Comini, M.A. Glucose-6-Phosphate Dehydrogenase from the Human Pathogen Trypanosoma cruzi Evolved Unique Structural Features to Support Efficient Product Formation. J. Mol. Biol. 2019, 431, 2143–2162. [Google Scholar] [CrossRef]
- Bringaud, F.; Rivière, L.; Coustou, V. Energy metabolism of trypanosomatids: Adaptation to available carbon sources. Mol. Biochem. Parasitol. 2006, 149, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, D.A.; Cazzulo, J.J. The pentose phosphate pathway in Trypanosoma cruzi. FEMS Microbiol. Lett. 2004, 234, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Finzi, J.K.; Chiavegatto, C.W.M.; Corat, K.F.; Lopez, J.A.; Cabrera, O.G.; Mielniczki-Pereira, A.A.; Colli, W.; Alves, M.J.M.; Gadelha, F.R. Trypanosoma cruzi response to the oxidative stress generated by hydrogen peroxide. Mol. Biochem. Parasitol. 2004, 133, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Mercaldi, G.F.; Ranzani, A.T.; Cordeiro, A.T. Discovery of new uncompetitive inhibitors of glucose-6-phosphate dehydrogenase. J. Biomol. Screen. 2014, 19, 1362–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, N.M.; Dawson, M.; Fairweather, E.E.; Hamilton, N.S.; Hitchin, J.R.; James, D.I.; Jones, S.D.; Jordan, A.M.; Lyons, A.J.; Small, H.F.; et al. Novel steroid inhibitors of glucose 6-phosphate dehydrogenase. J. Med. Chem. 2012, 55, 4431–4445. [Google Scholar] [CrossRef] [PubMed]
- Mele, L.; Paino, F.; Papaccio, F.; Regad, T.; Boocock, D.; Stiuso, P.; Lombardi, A.; Liccardo, D.; Aquino, G.; Barbieri, A.; et al. A new inhibitor of glucose-6-phosphate dehydrogenase blocks pentose phosphate pathway and suppresses malignant proliferation and metastasis in vivo. Cell Death Dis. 2018, 9, 572. [Google Scholar] [CrossRef] [Green Version]
- Marks, P.A.; Banks, J. Inhibition of mammalian glucose-6-phosphate dehydrogenase by steroids. Proc. Natl. Acad. Sci. USA 1960, 46, 447–452. [Google Scholar] [CrossRef] [Green Version]
- Raineri, R.; Levy, H.R. On the specificity of steroid interaction with mammary glucose 6-phosphate dehydrogenase. Biochemistry 1970, 9, 2233–2243. [Google Scholar] [CrossRef]
- Gordon, G.; Mackow, M.C.; Levy, H.R. On the mechanism of interaction of steroids with human glucose 6-phosphate dehydrogenase. Arch. Biochem. Biophys. 1995, 318, 25–29. [Google Scholar] [CrossRef]
- Gupta, S.; Cordeiro, A.T.; Michels, P.A.M. Glucose-6-phosphate dehydrogenase is the target for the trypanocidal action of human steroids. Mol. Biochem. Parasitol. 2011, 176, 112–115. [Google Scholar] [CrossRef]
- Ortiz, C.; Moraca, F.; Medeiros, A.; Botta, M.; Hamilton, N.; Comini, M.A. Binding Mode and Selectivity of Steroids towards Glucose-6-phosphate Dehydrogenase from the Pathogen Trypanosoma cruzi. Molecules 2016, 21, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordeiro, A.T.; Thiemann, O.H. 16-bromoepiandrosterone, an activator of the mammalian immune system, inhibits glucose 6-phosphate dehydrogenase from Trypanosoma cruzi and is toxic to these parasites grown in culture. Bioorg. Med. Chem. 2010, 18, 4762–4768. [Google Scholar] [CrossRef] [PubMed]
- Au, S.W.; Naylor, C.E.; Gover, S.; Vandeputte-Rutten, L.; Scopes, D.A.; Mason, P.J.; Luzzatto, L.; Lam, V.M.; Adams, M.J. Solution of the structure of tetrameric human glucose 6-phosphate dehydrogenase by molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 1999, 55, 826–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Au, S.W.; Gover, S.; Lam, V.M.; Adams, M.J. Human glucose-6-phosphate dehydrogenase: The crystal structure reveals a structural NADP(+) molecule and provides insights into enzyme deficiency. Structure 2000, 8, 293–303. [Google Scholar] [CrossRef] [Green Version]
- Kotaka, M.; Gover, S.; Vandeputte-Rutten, L.; Au, S.W.N.; Lam, V.M.S.; Adams, M.J. Structural studies of glucose-6-phosphate and NADP+ binding to human glucose-6-phosphate dehydrogenase. Acta Crystallogr. D Biol. Crystallogr. 2005, 61, 495–504. [Google Scholar] [CrossRef] [Green Version]
- Mercaldi, G.F.; Dawson, A.; Hunter, W.N.; Cordeiro, A.T. The structure of a Trypanosoma cruzi glucose-6-phosphate dehydrogenase reveals differences from the mammalian enzyme. FEBS Lett. 2016, 590, 2776–2786. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.-B.; Liu, Y.; Yao, Y. Computational determination of binding structures and free energies of glucose 6-phosphate dehydrogenase with novel steroid inhibitors. J. Mol. Graph. Model. 2014, 51, 168–172. [Google Scholar] [CrossRef]
- Rangel-Aldao, R.; Comach, G.; Mendoza, A. In vitro translation of Trypanosoma cruzi antigens recognized by human chagasic sera. J. Parasitol. 1987, 73, 855–857. [Google Scholar] [CrossRef]
- Roldán, A.; Comini, M.A.; Crispo, M.; Krauth-Siegel, R.L. Lipoamide dehydrogenase is essential for both bloodstream and procyclic Trypanosoma brucei. Mol. Microbiol. 2011, 81, 623–639. [Google Scholar] [CrossRef]
- Ramos, T.C.; Freymüller-Haapalainen, E.; Schenkman, S. Three-dimensional reconstruction of Trypanosoma cruzi epimastigotes and organelle distribution along the cell division cycle. Cytometry A 2011, 79, 538–544. [Google Scholar] [CrossRef]
- Frederiks, W.M.; van Marle, J.; van Oven, C.; Comin-Anduix, B.; Cascante, M. Improved localization of glucose-6-phosphate dehydrogenase activity in cells with 5-cyano-2,3-ditolyl-tetrazolium chloride as fluorescent redox dye reveals its cell cycle-dependent regulation. J. Histochem. Cytochem. 2006, 54, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Nnadi, C.O.; Nwodo, N.J.; Kaiser, M.; Brun, R.; Schmidt, T.J. Steroid Alkaloids from Holarrhena africana with Strong Activity against Trypanosoma brucei rhodesiense. Molecules 2017, 22, 1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manta, B.; Comini, M.; Medeiros, A.; Hugo, M.; Trujillo, M.; Radi, R. Trypanothione: A unique bis-glutathionyl derivative in trypanosomatids. Biochim. Biophys. Acta 2013, 1830, 3199–3216. [Google Scholar] [CrossRef] [PubMed]
- Franco, J.; Sardi, F.; Szilágyi, L.; Kövér, K.E.; Fehér, K.; Comini, M.A. Diglycosyl diselenides alter redox homeostasis and glucose consumption of infective African trypanosomes. Int. J. Parasitol. Drugs Drug Resist. 2017, 7, 303–313. [Google Scholar] [CrossRef]
- Gutscher, M.; Pauleau, A.L.; Marty, L.; Brach, T.; Wabnitz, G.H.; Samstag, Y.; Meyer, A.J.; Dick, T.P. Real-time imaging of the intracellular glutathione redox potential. Nat. Methods 2008, 5, 553–559. [Google Scholar] [CrossRef]
- Franco, J.; Medeiros, A.; Benítez, D.; Perelmuter, K.; Serra, G.; Comini, M.A.; Scarone, L. In vitro activity and mode of action of distamycin analogues against African trypanosomes. Eur. J. Med. Chem. 2017, 126, 776–788. [Google Scholar] [CrossRef]
- Ebersoll, S.; Bogacz, M.; Günter, L.M.; Dick, T.P.; Krauth-Siegel, R.L. A tryparedoxin-coupled biosensor reveals a mitochondrial trypanothione metabolism in trypanosomes. eLife 2020, 9, e53227. [Google Scholar] [CrossRef]
- Gilge, J.L.; Fisher, M.; Chai, Y.-C. The effect of oxidant and the non-oxidant alteration of cellular thiol concentration on the formation of protein mixed-disulfides in HEK 293 cells. PLoS ONE 2008, 3, e4015. [Google Scholar] [CrossRef] [Green Version]
- Saudagar, P.; Dubey, V.K. Cloning, expression, characterization and inhibition studies on trypanothione synthetase, a drug target enzyme, from Leishmania donovani. Biol. Chem. 2011, 392, 1113–1122. [Google Scholar] [CrossRef]
- Comini, M.A.; Guerrero, S.A.; Haile, S.; Menge, U.; Lünsdorf, H.; Flohé, L. Validation of Trypanosoma brucei trypanothione synthetase as drug target. Free Radic. Biol. Med. 2004, 36, 1289–1302. [Google Scholar] [CrossRef]
- Mesías, A.C.; Sasoni, N.; Arias, D.G.; Pérez Brandán, C.; Orban, O.; Kunick, C.; Robello, C.; Comini, M.A.; Garg, N.J.; Zago, M.P. Trypanothione synthetase confers growth, survival advantage and resistance to anti-protozoal drugs in Trypanosoma cruzi. Free Radic. Biol. Med. 2019, 130, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Krieger, S.; Schwarz, W.; Ariyanayagam, M.R.; Fairlamb, A.H.; Krauth-Siegel, R.L.; Clayton, C. Trypanosomes lacking trypanothione reductase are avirulent and show increased sensitivity to oxidative stress. Mol. Microbiol. 2000, 35, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-2: LigPrep; Schrödinger, LLC: New York, NY, USA, 2020.
- Schrödinger Release 2020-2: SiteMap; Schrödinger, LLC: New York, NY, USA, 2020.
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Halgren, T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 2007, 69, 146–148. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-2: Glide; Schrödinger, LLC: New York, NY, USA, 2020.
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision Glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shaw, D.E.; Shelley, M.; et al. Glide: A New approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Urban, I.; Santurio, L.B.; Chidichimo, A.; Yu, H.; Chen, X.; Mucci, J.; Agüero, F.; Buscaglia, C.A. Molecular diversity of the Trypanosoma cruzi TcSMUG family of mucin genes and proteins. Biochem. J. 2011, 438, 303–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazzulo, J.J.; Franke de Cazzulo, B.M.; Engel, J.C.; Cannata, J.J. End products and enzyme levels of aerobic glucose fermentation in trypanosomatids. Mol. Biochem. Parasitol. 1985, 16, 329–343. [Google Scholar] [CrossRef]
- Wirtz, E.; Leal, S.; Ochatt, C.; Cross, G.A. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 1999, 99, 89–101. [Google Scholar] [CrossRef]
- Taylor, M.C.; Kelly, J.M. pTcINDEX: A stable tetracycline-regulated expression vector for Trypanosoma cruzi. BMC Biotechnol. 2006, 6, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piacenza, L.; Peluffo, G.; Alvarez, M.N.; Kelly, J.M.; Wilkinson, S.R.; Radi, R. Peroxiredoxins play a major role in protecting Trypanosoma cruzi against macrophage- and endogenously-derived peroxynitrite. Biochem. J. 2008, 410, 359–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Chaumont, F.; Dallongeville, S.; Chenouard, N.; Hervé, N.; Pop, S.; Provoost, T.; Meas-Yedid, V.; Pankajakshan, P.; Lecomte, T.; Le Montagner, Y.; et al. Icy: An open bioimage informatics platform for extended reproducible research. Nat. Methods 2012, 9, 690–696. [Google Scholar] [CrossRef]
- Benítez, D.; Dibello, E.; Bonilla, M.; Comini, M.A. A simple, robust, and affordable bioluminescent assay for drug discovery against infective African trypanosomes. Drug Dev. Res. 2020. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (µM) | |||
|---|---|---|---|
| Compound | T. cruzi | H. sapiens | Selectivity Index b |
| EA | 3.0 ± 0.4 c | 9.4 ± 1.0 a | 3 |
| 1 | 0.053 ± 0.002 | 20.8 ± 4.4 a | 392 |
| 2 | 0.11 ± 0.02 | >100 | >909 |
| 3 | 1.5 ± 0.2 | >100 | >67 |
| DHEA | 21.5 ± 0.5 d | 11.3 ± 2.2 a | 0.5 |
| 4 | 3.3 ± 0.7 | 48.0 ± 2.6 | 14 |
| 5 | 0.079 ± 0.01 | >100 | >1266 |
| 6 | >50 | >100 | >2 |
| 7 | >50 | 15.8 ± 3.3 a | 0.32 |
| Conessine | >100 | >100 | ND |
| Compound | % Cytotoxicity (50 µM) a | EC50 (µM) a | CC50 (µM) a | Selectivity Index b |
|---|---|---|---|---|
| T. Cruzi Epimastigote Form | T. brucei Bloodstream Form | Murine Macrophage (Cell Line J774) | ||
| EA | 3.7 ± 0.7 | 36.5 ± 3.6 c | >100 | >2.7 |
| 1 | 14.5 ± 5.3 | 1.9 ± 0.2 | 8.8 ± 1.0 | 4.6 |
| 2 | 64.0 ± 4.1 | 5.3 ± 0.4 | 42.0 ± 1.0 | 7.9 |
| 3 | 73.9 ± 3.4 | 4.8 ± 0.5 | 1.0 ± 0.1 | 0.2 |
| DHEA | n.d. | 43.8 ± 2.0 c | 25.0 ± 13.0 d | 0.6 |
| 4 | 6.4 ± 1.6 | 7.5 ± 0.3 | 19.0 ± 3.0 | 2.5 |
| 5 | 30.9 ± 2.5 | 18.0 ± 3.0 | 39.0 ± 3.0 | 2.2 |
| 6 | 76.2 ± 0.8 | 5.3 ± 0.5 | 4.5 ± 0.4 | 0.9 |
| 7 | 3.9 ± 0.7 | >24 | >24 | n.d |
| Conessine | n.d. | 0.42 ± 0.01 e | 61.2 e | 145.7 |
| Nifurtimox | 46.0 ± 1.9 (at 24 µM) | 6.0 ± 0.4 | 140.0 ± 2.0 | 23.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortíz, C.; Moraca, F.; Laverriere, M.; Jordan, A.; Hamilton, N.; Comini, M.A. Glucose 6-Phosphate Dehydrogenase from Trypanosomes: Selectivity for Steroids and Chemical Validation in Bloodstream Trypanosoma brucei. Molecules 2021, 26, 358. https://doi.org/10.3390/molecules26020358
Ortíz C, Moraca F, Laverriere M, Jordan A, Hamilton N, Comini MA. Glucose 6-Phosphate Dehydrogenase from Trypanosomes: Selectivity for Steroids and Chemical Validation in Bloodstream Trypanosoma brucei. Molecules. 2021; 26(2):358. https://doi.org/10.3390/molecules26020358
Chicago/Turabian StyleOrtíz, Cecilia, Francesca Moraca, Marc Laverriere, Allan Jordan, Niall Hamilton, and Marcelo A. Comini. 2021. "Glucose 6-Phosphate Dehydrogenase from Trypanosomes: Selectivity for Steroids and Chemical Validation in Bloodstream Trypanosoma brucei" Molecules 26, no. 2: 358. https://doi.org/10.3390/molecules26020358