Differential Protein Expression in Exponential and Stationary Growth Phases of Mycobacterium avium subsp. hominissuis 104


Abstract
:1. Introduction
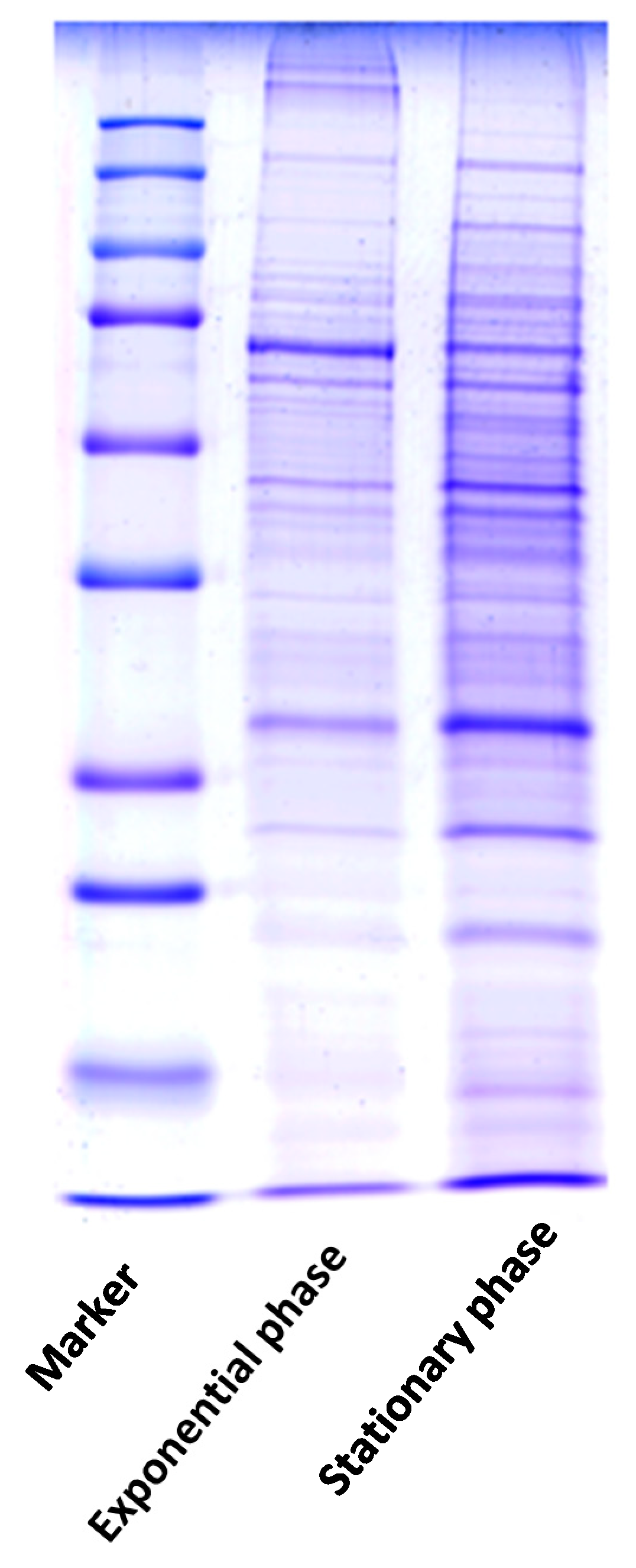
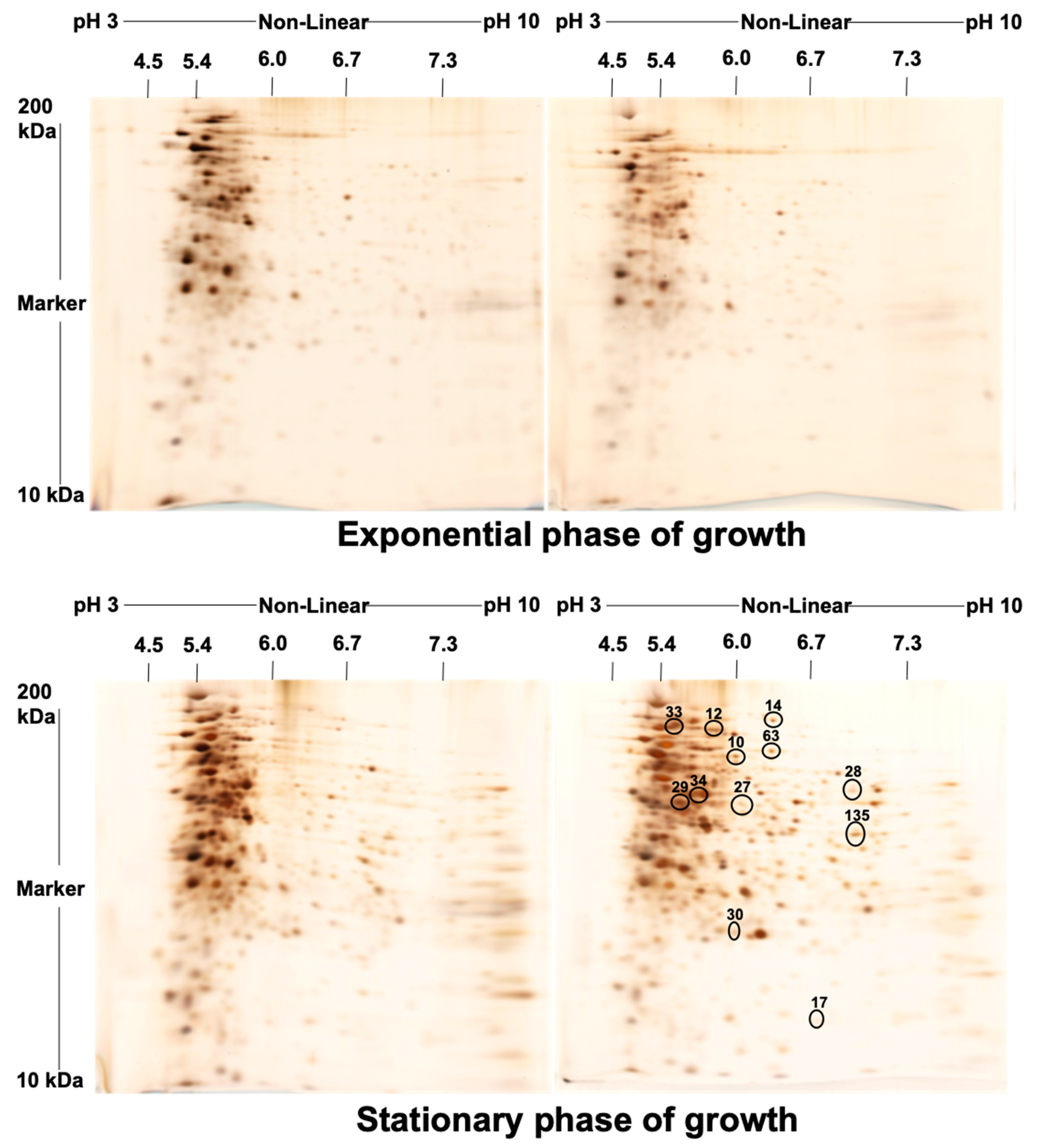
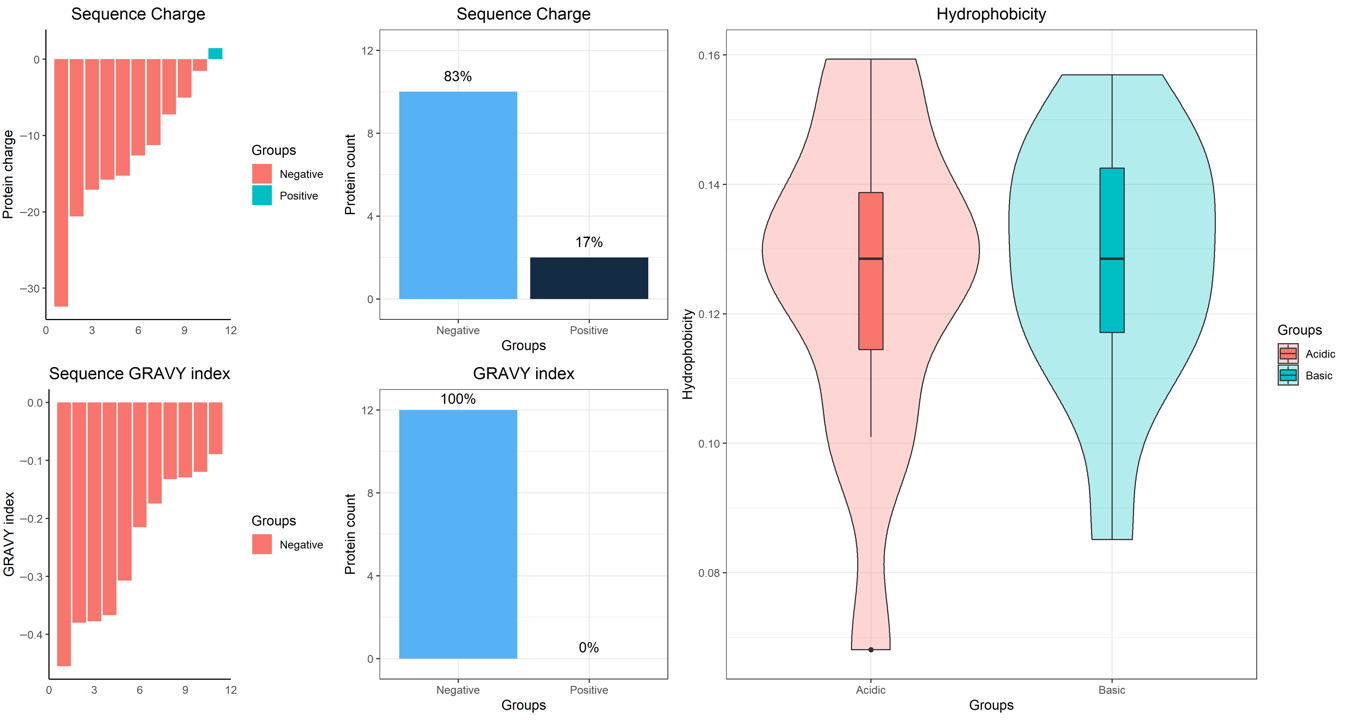
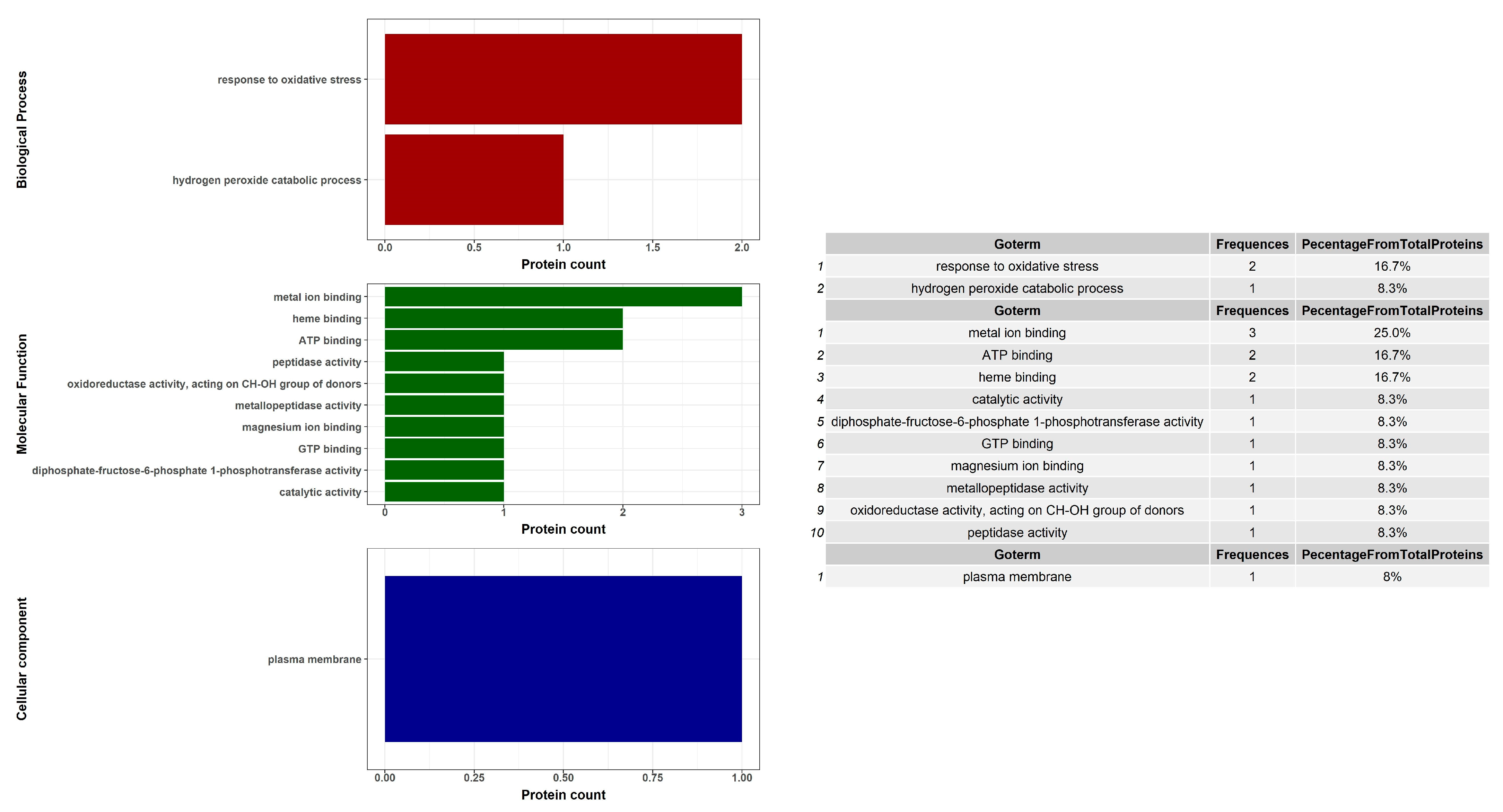
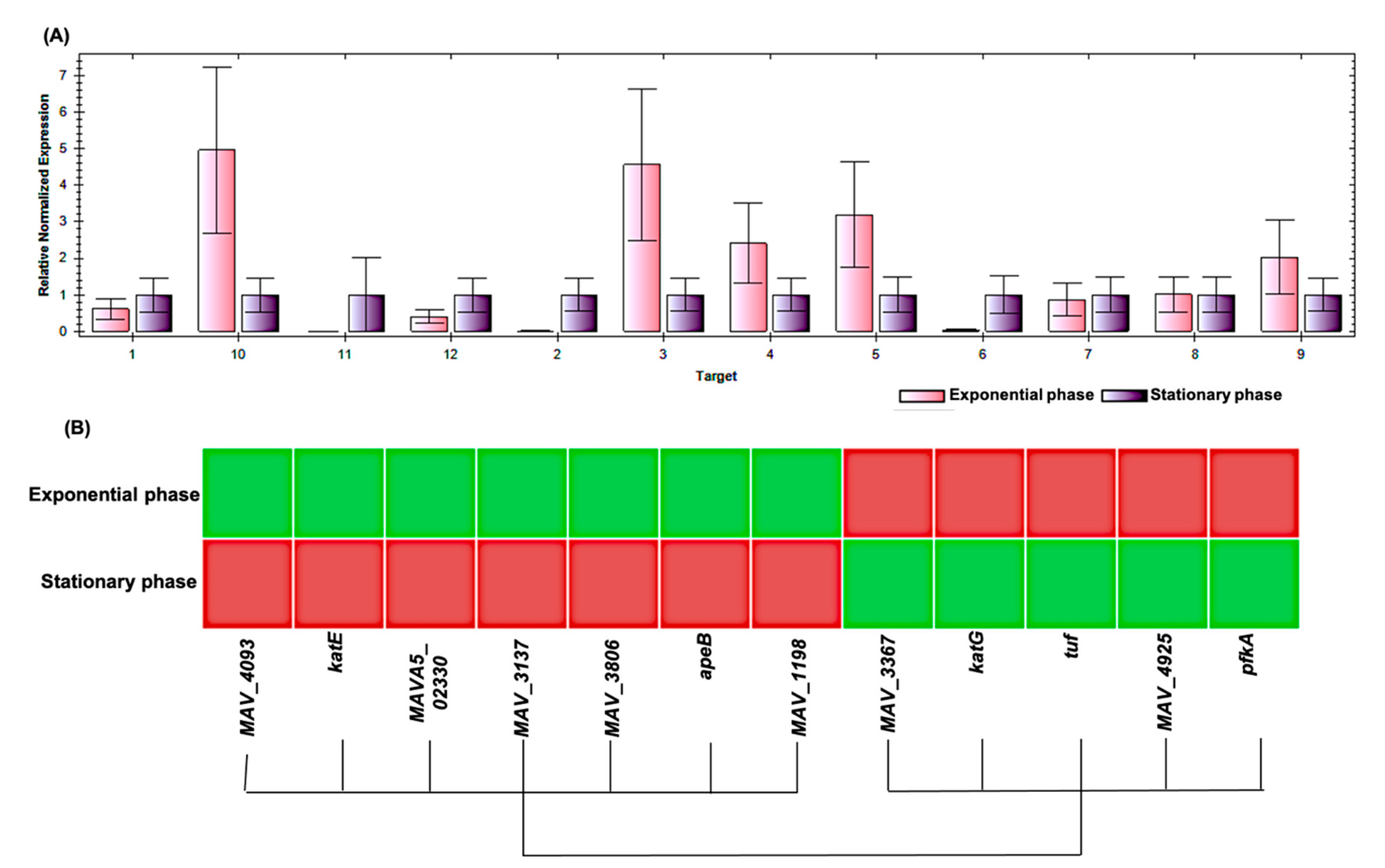
2. Results
3. Discussion
4. Methods
4.1. Bacterial Strains, Culture Media, and Growth Conditions
4.2. Bacterial Protein Preparation
4.3. SDS-PAGE
4.4. Isoelectric Focusing (IEF) and Second Dimension Gel Electrophoresis (2-DE)
4.5. Staining of the Gel and Visualization of the Spots
4.6. Processing of the Spot and Mass Identification (TripleTOF MS/MS)
4.7. Bioinformatic Analysis
4.8. Traditional PCR Reaction
4.9. RNA Extraction
4.10. Quantitative Reverse Transcription Real-Time PCR (qRT-PCR)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Park, S.C.; Kang, M.J.; Han, C.H.; Lee, S.M.; Kim, C.J.; Lee, J.M.; Kang, Y.A. Prevalence, incidence, and mortality of nontuberculous mycobacterial infection in Korea: A nationwide population-based study. BMC Pulm. Med. 2019, 19, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namkoong, H.; Kurashima, A.; Morimoto, K.; Hoshino, Y.; Hasegawa, N.; Ato, M.; Mitarai, S. Epidemiology of Pulmonary Nontuberculous Mycobacterial Disease, Japan. Emerg. Infect. Dis. 2016, 22, 1116–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivero-Lezcano, O.M.; Gonzalez-Cortes, C.; Mirsaeidi, M. The unexplained increase of nontuberculous mycobacteriosis. Int. J. Mycobacteriol. 2019, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Kuronuma, K.; Saito, A.; Ikeda, K.; Ariki, S.; Saitou, A.; Otsuka, M.; Chiba, H.; Takahashi, S.; Takahashi, M.; et al. Insufficient serum L-ficolin is associated with disease presence and extent of pulmonary Mycobacterium avium complex disease. Respir. Res. 2019, 20, 224. [Google Scholar] [CrossRef] [PubMed]
- Sakatani, M. The non-tuberculous mycobacteriosis. Kekkaku [Tuberculosis] 2005, 80, 25–30. [Google Scholar]
- Uchiya, K.; Takahashi, H.; Yagi, T.; Moriyama, M.; Inagaki, T.; Ichikawa, K.; Nakagawa, T.; Nikai, T.; Ogawa, K. Comparative genome analysis of Mycobacterium avium revealed genetic diversity in strains that cause pulmonary and disseminated disease. PLoS ONE 2013, 8, e71831. [Google Scholar] [CrossRef]
- Horan, K.L.; Freeman, R.; Weigel, K.; Semret, M.; Pfaller, S.; Covert, T.C.; van Soolingen, D.; Leao, S.C.; Behr, M.A.; Cangelosi, G.A. Isolation of the genome sequence strain Mycobacterium avium 104 from multiple patients over a 17-year period. J. Clin. Microbiol. 2006, 44, 783–789. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Bannantine, J.P.; Zhang, Q.; Amonsin, A.; May, B.J.; Alt, D.; Banerji, N.; Kanjilal, S.; Kapur, V. The complete genome sequence of Mycobacterium avium subspecies paratuberculosis. Proc. Natl. Acad. Sci. USA 2005, 102, 12344–12349. [Google Scholar] [CrossRef] [Green Version]
- Ignatov, D.; Malakho, S.; Majorov, K.; Skvortsov, T.; Apt, A.; Azhikina, T. RNA-Seq analysis of Mycobacterium avium non-coding transcriptome. PLoS ONE 2013, 8, e74209. [Google Scholar] [CrossRef]
- Janagama, H.K.; Lamont, E.A.; George, S.; Bannantine, J.P.; Xu, W.W.; Tu, Z.J.; Wells, S.J.; Schefers, J.; Sreevatsan, S. Primary transcriptomes of Mycobacterium avium subsp. paratuberculosis reveal proprietary pathways in tissue and macrophages. BMC Genom. 2010, 11, 561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannantine, J.P.; Talaat, A.M. Genomic and transcriptomic studies in Mycobacterium avium subspecies paratuberculosis. Vet. Immunol. Immunopathol. 2010, 138, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Rojony, R.; Martin, M.; Campeau, A.; Wozniak, J.M.; Gonzalez, D.J.; Jaiswal, P.; Danelishvili, L.; Bermudez, L.E. Quantitative analysis of Mycobacterium avium subsp. hominissuis proteome in response to antibiotics and during exposure to different environmental conditions. Clin. Proteom. 2019, 16, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinison, J.J.; Danelishvili, L.; Gupta, R.; Rose, S.J.; Babrak, L.M.; Bermudez, L.E. Identification of Mycobacterium avium subsp. hominissuis secreted proteins using an in vitro system mimicking the phagosomal environment. BMC Microbiol. 2016, 16, 270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radosevich, T.J.; Reinhardt, T.A.; Lippolis, J.D.; Bannantine, J.P.; Stabel, J.R. Proteome and differential expression analysis of membrane and cytosolic proteins from Mycobacterium avium subsp. paratuberculosis strains K-10 and 187. J. Bacteriol. 2007, 189, 1109–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Z.; De Buck, J. Localization of proteins in the cell wall of Mycobacterium avium subsp. paratuberculosis K10 by proteomic analysis. Proteome Sci. 2010, 8, 21. [Google Scholar] [CrossRef] [Green Version]
- Lange, R.; Hengge-Aronis, R. Identification of a central regulator of stationary-phase gene expression in Escherichia coli. Mol. Microbiol. 1991, 5, 49–59. [Google Scholar] [CrossRef]
- Phillips, Z.E.; Strauch, M.A. Bacillus subtilis sporulation and stationary phase gene expression. Cell. Mol. Life Sci. CMLS 2002, 59, 392–402. [Google Scholar] [CrossRef]
- Bernhardt, J.; Weibezahn, J.; Scharf, C.; Hecker, M. Bacillus subtilis during feast and famine: Visualization of the overall regulation of protein synthesis during glucose starvation by proteome analysis. Genome Res. 2003, 13, 224–237. [Google Scholar] [CrossRef] [Green Version]
- Ntolosi, B.A.; Betts, J.; Zappe, H.; Powles, R.; Steyn, L.M. Growth phase-associated changes in protein expression in Mycobacterium smegmatis identify a new low molecular weight heat shock protein. Tuberculosis 2001, 81, 279–289. [Google Scholar] [CrossRef]
- Yuan, Y.; Crane, D.D.; Barry, C.E., 3rd. Stationary phase-associated protein expression in Mycobacterium tuberculosis: Function of the mycobacterial alpha-crystallin homolog. J. Bacteriol. 1996, 178, 4484–4492. [Google Scholar] [CrossRef] [Green Version]
- Jaishankar, J.; Srivastava, P. Molecular Basis of Stationary Phase Survival and Applications. Front. Microbiol. 2017, 8, 2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, K.L.; Jarocki, V.M.; Charles, I.G.; Djordjevic, S.P. The Diverse Functional Roles of Elongation Factor Tu (EF-Tu) in Microbial Pathogenesis. Front. Microbiol. 2019, 10, 2351. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.H.; Garrity, J.; Crosby, H.A.; Escalante-Semerena, J.C. In Salmonella enterica, the sirtuin-dependent protein acylation/deacylation system (SDPADS) maintains energy homeostasis during growth on low concentrations of acetate. Mol. Microbiol. 2011, 80, 168–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.H.; Han, M.J.; Lee, S.Y.; Jeong, K.J.; Yoo, J.S. Combined transcriptome and proteome analysis of Escherichia coli during high cell density culture. Biotechnol. Bioeng. 2003, 81, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.E.; Smalley, D.J.; Conway, T. Gene expression profiling of Escherichia coli growth transitions: An expanded stringent response model. Mol. Microbiol. 2002, 45, 289–306. [Google Scholar] [CrossRef]
- Mitchell, G.; Ge, L.; Huang, Q.; Chen, C.; Kianian, S.; Roberts, M.F.; Schekman, R.; Portnoy, D.A. Avoidance of autophagy mediated by PlcA or ActA is required for Listeria monocytogenes growth in macrophages. Infect. Immun. 2015, 83, 2175–2184. [Google Scholar] [CrossRef] [Green Version]
- Unnikrishnan, M.; Cohen, J.; Sriskandan, S. Growth-phase-dependent expression of virulence factors in an M1T1 clinical isolate of Streptococcus pyogenes. Infect. Immun. 1999, 67, 5495–5499. [Google Scholar] [CrossRef] [Green Version]
- Martinot, A.J.; Farrow, M.; Bai, L.; Layre, E.; Cheng, T.Y.; Tsai, J.H.; Iqbal, J.; Annand, J.W.; Sullivan, Z.A.; Hussain, M.M.; et al. Mycobacterial Metabolic Syndrome: LprG and Rv1410 Regulate Triacylglyceride Levels, Growth Rate and Virulence in Mycobacterium tuberculosis. PLoS Pathog. 2016, 12, e1005351. [Google Scholar] [CrossRef] [Green Version]
- Aguilar-Lopez, B.A.; Correa, F.; Moreno-Altamirano, M.M.B.; Espitia, C.; Hernandez-Longoria, R.; Oliva-Ramirez, J.; Padierna-Olivos, J.; Sanchez-Garcia, F.J. LprG and PE_PGRS33 Mycobacterium tuberculosis virulence factors induce differential mitochondrial dynamics in macrophages. Scand. J. Immunol. 2019, 89, e12728. [Google Scholar] [CrossRef] [Green Version]
- Viale, M.N.; Park, K.T.; Imperiale, B.; Gioffre, A.K.; Colombatti Olivieri, M.A.; Moyano, R.D.; Morcillo, N.; Santangelo, M.d.l.P.; Davis, W.; Romano, M.I. Characterization of a Mycobacterium avium subsp. Avium Operon Associated with Virulence and Drug Detoxification. BioMed Res. Int. 2014, 2014, 809585. [Google Scholar] [CrossRef] [PubMed]
- Lazdunski, A.M. Peptidases and proteases of Escherichia coli and Salmonella typhimurium. FEMS Microbiol. Rev. 1989, 5, 265–276. [Google Scholar] [CrossRef]
- Dragset, M.S.; Ioerger, T.R.; Loevenich, M.; Haug, M.; Sivakumar, N.; Marstad, A.; Cardona, P.J.; Klinkenberg, G.; Rubin, E.J.; Steigedal, M.; et al. Global Assessment of Mycobacterium avium subsp. hominissuis Genetic Requirement for Growth and Virulence. mSystems 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Manca, C.; Paul, S.; Barry, C.E., 3rd; Freedman, V.H.; Kaplan, G. Mycobacterium tuberculosis catalase and peroxidase activities and resistance to oxidative killing in human monocytes in vitro. Infect. Immun. 1999, 67, 74–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phong, W.Y.; Lin, W.; Rao, S.P.; Dick, T.; Alonso, S.; Pethe, K. Characterization of phosphofructokinase activity in Mycobacterium tuberculosis reveals that a functional glycolytic carbon flow is necessary to limit the accumulation of toxic metabolic intermediates under hypoxia. PLoS ONE 2013, 8, e56037. [Google Scholar] [CrossRef]
- Muthusamy, S.; Lundin, D.; Mamede Branca, R.M.; Baltar, F.; Gonzalez, J.M.; Lehtio, J.; Pinhassi, J. Comparative proteomics reveals signature metabolisms of exponentially growing and stationary phase marine bacteria. Environ. Microbiol. 2017, 19, 2301–2319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bathke, J.; Konzer, A.; Remes, B.; McIntosh, M.; Klug, G. Comparative analyses of the variation of the transcriptome and proteome of Rhodobacter sphaeroides throughout growth. BMC Genom. 2019, 20, 358. [Google Scholar] [CrossRef] [Green Version]
- Lanigan, M.D.; Vaughan, J.A.; Shiell, B.J.; Beddome, G.J.; Michalski, W.P. Mycobacterial proteome extraction: Comparison of disruption methods. Proteomics 2004, 4, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Enany, S.; Yoshida, Y.; Yamamoto, T. Exploring extra-cellular proteins in methicillin susceptible and methicillin resistant Staphylococcus aureus by liquid chromatography-tandem mass spectrometry. World J. Microbiol. Biotechnol. 2014, 30, 1269–1283. [Google Scholar] [CrossRef]
- Magdeldin, S.; Enany, S.; Yoshida, Y.; Xu, B.; Zhang, Y.; Zureena, Z.; Lokamani, I.; Yaoita, E.; Yamamoto, T. Basics and recent advances of two dimensional- polyacrylamide gel electrophoresis. Clin. Proteom. 2014, 11, 16. [Google Scholar] [CrossRef] [Green Version]
- O’Farrell, P.H. High resolution two-dimensional electrophoresis of proteins. J. Biol. Chem. 1975, 250, 4007–4021. [Google Scholar] [PubMed]
- Enany, S.; Yoshida, Y.; Tateishi, Y.; Ozeki, Y.; Nishiyama, A.; Savitskaya, A.; Yamaguchi, T.; Ohara, Y.; Yamamoto, T.; Ato, M.; et al. Mycobacterial DNA-binding protein 1 is critical for long term survival of Mycobacterium smegmatis and simultaneously coordinates cellular functions. Sci. Rep. 2017, 7, 6810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arenas, N.E.; Salazar, L.M. Steps and Tools for PCR-Based Technique Design. In Biotechnology and Bioengineering; Jacob-Lopes, E., Zepka, L.Q., Eds.; IntechOpen: London, UK, 2019; pp. 1–16. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spot No. | Fold Change (+/−) * | ANOVA (p) | Average Normalized Volumes | Accession No. | Protein Name | Protein Score | No. Identified Peptides | Gene Name | No. (qRT-PCR) | Molecular Function | Regulation at Transcriptomic Level * | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Exponential Growth Phase | Stationary Growth Phase | |||||||||||
| 10 | +4.4 | 0.018 | 11,896.756 | 52,584.334 | A0A0H2ZX69 | Pyruvate decarboxylase | 655 | 25 | MAV_4093 | 1 | Catalytic activity | + |
| 12 | +4.3 | 0.038 | 4334.274 | 18,578.945 | A0A0H2ZZB0 | Catalase | 2211 | 21 | katE | 2 | Catalase activity | + |
| 14 | +4 | 0.048 | 8140.139 | 32,853.588 | A0A0E2WBQ0 | ATP-dependent Clp protease ATP-binding protein | 1176 | 24 | MAVA5_02330 | 3 | ATP binding and Chaperone | − |
| 17 | −3.2 | 0.035 | 33,720.648 | 10,671.303 | A0A0H3A465 | Universal stress protein family protein | 348 | 24 | MAV_3137 | 4 | Response to stress | − |
| 27 | +2.2 | 0.003 | 7519.716 | 16,866.019 | A0A0H2ZYK5 | Dibenzothiophene desulfurization enzyme C | 353 | 21 | MAV_3806 | 5 | Oxidoreductase activity | − |
| 28 | +2.2 | 0.045 | 9994.532 | 21,965.532 | A0A0H2ZU78 | aspartyl aminopeptidase | 419 | 26 | apeB | 6 | Aminopeptidase, Hydrolase, Metalloprotease, and Aminopeptidase activity | + |
| 29 | −2.2 | 0.046 | 530,650.636 | 244,551.945 | A0A0H2ZT78 | Acetyl-CoA acetyltransferase | 1532 | 26 | MAV_1198 | 7 | Transferase activity | + |
| 30 | +2.1 | 0.029 | 19,961.352 | 42,368.898 | A0A0H3A4W6 | LprG protein | 323 | 26 | MAV_3367 | 8 | Phosphatidylinositol binding | + |
| 33 | −2 | 0.035 | 449,155.112 | 221,267.396 | A0QGA4 | Catalase-peroxidase | 2066 | 25 | katG | 9 | Catalase activity | − |
| 34 | −2 | 0.015 | 150,556.978 | 75,806.224 | A0QL35 | Elongation factor Tu | 2605 | 20 | tuf | 10 | GTPase activity + translation elongation factor activity | − |
| 63 | +5.1 | 0.050 | 6531.054 | 33,156.744 | A0A0H2ZRA7 | Glucose-methanol-choline | 1014 | 26 | MAV_4925 | 11 | Choline dehydrogenase activity | + |
| 135 | +2.2 | 0.050 | 35,082.949 | 77,486.389 | A0A0H3A360 | 6-phosphofructokinase 1 | 800 | 23 | pfkA | 12 | 6-phosphofructokinase activity | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Enany, S.; Ato, M.; Matsumoto, S. Differential Protein Expression in Exponential and Stationary Growth Phases of Mycobacterium avium subsp. hominissuis 104. Molecules 2021, 26, 305. https://doi.org/10.3390/molecules26020305
Enany S, Ato M, Matsumoto S. Differential Protein Expression in Exponential and Stationary Growth Phases of Mycobacterium avium subsp. hominissuis 104. Molecules. 2021; 26(2):305. https://doi.org/10.3390/molecules26020305
Chicago/Turabian StyleEnany, Shymaa, Manabu Ato, and Sohkichi Matsumoto. 2021. "Differential Protein Expression in Exponential and Stationary Growth Phases of Mycobacterium avium subsp. hominissuis 104" Molecules 26, no. 2: 305. https://doi.org/10.3390/molecules26020305