
Volumetric Absorptive Microsampling of Blood for Untargeted Lipidomics


 , and
, and 
Abstract
:
1. Introduction
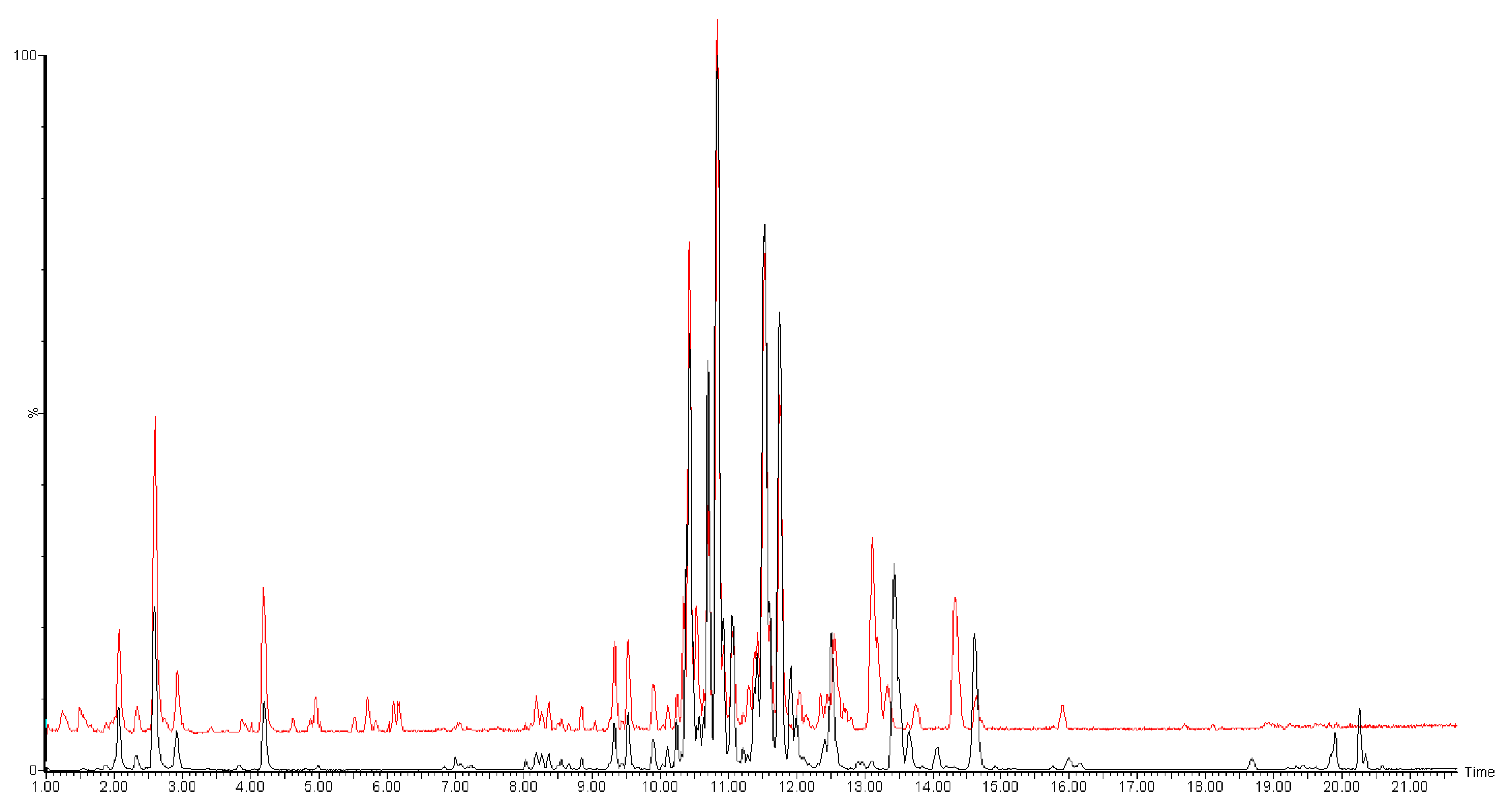
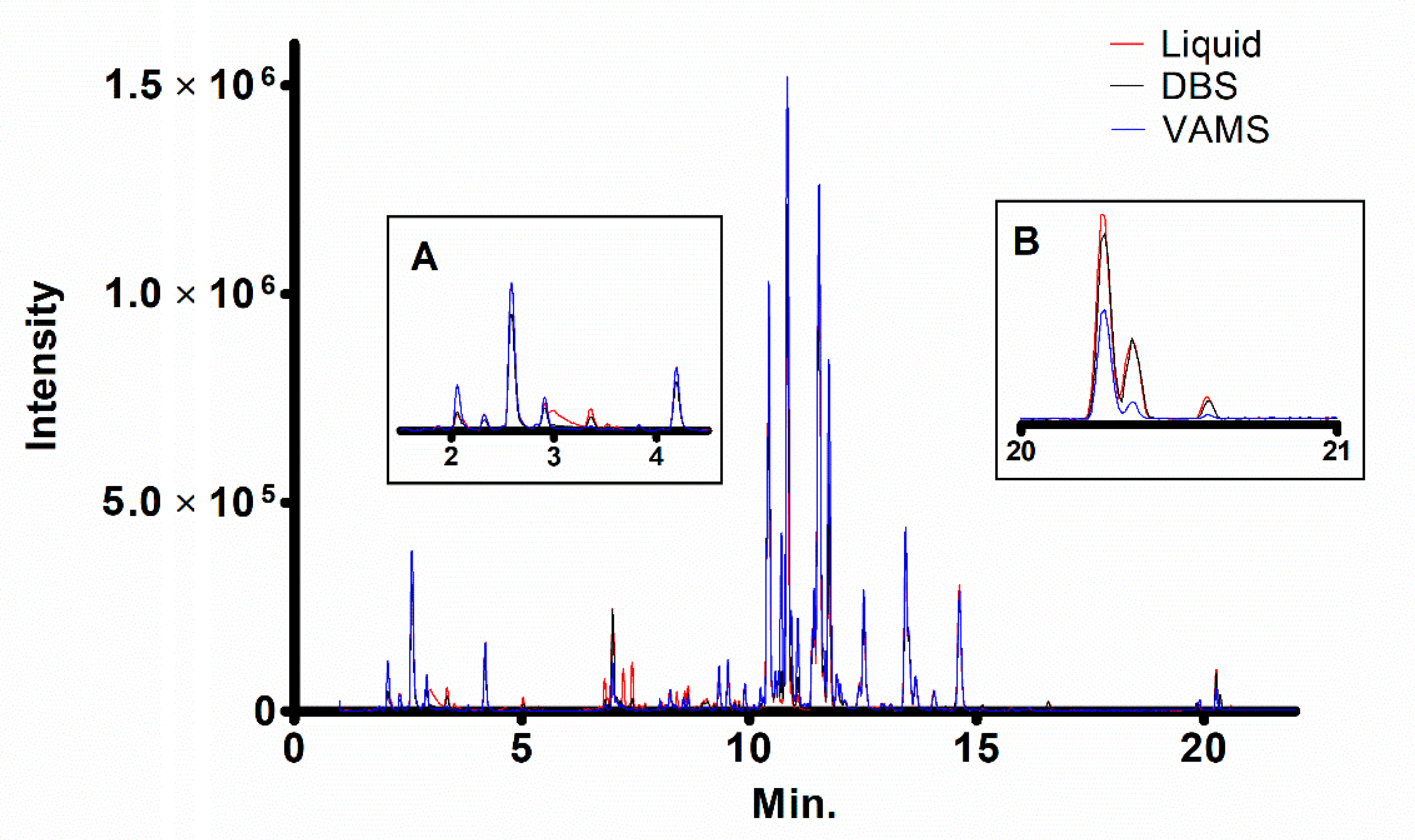
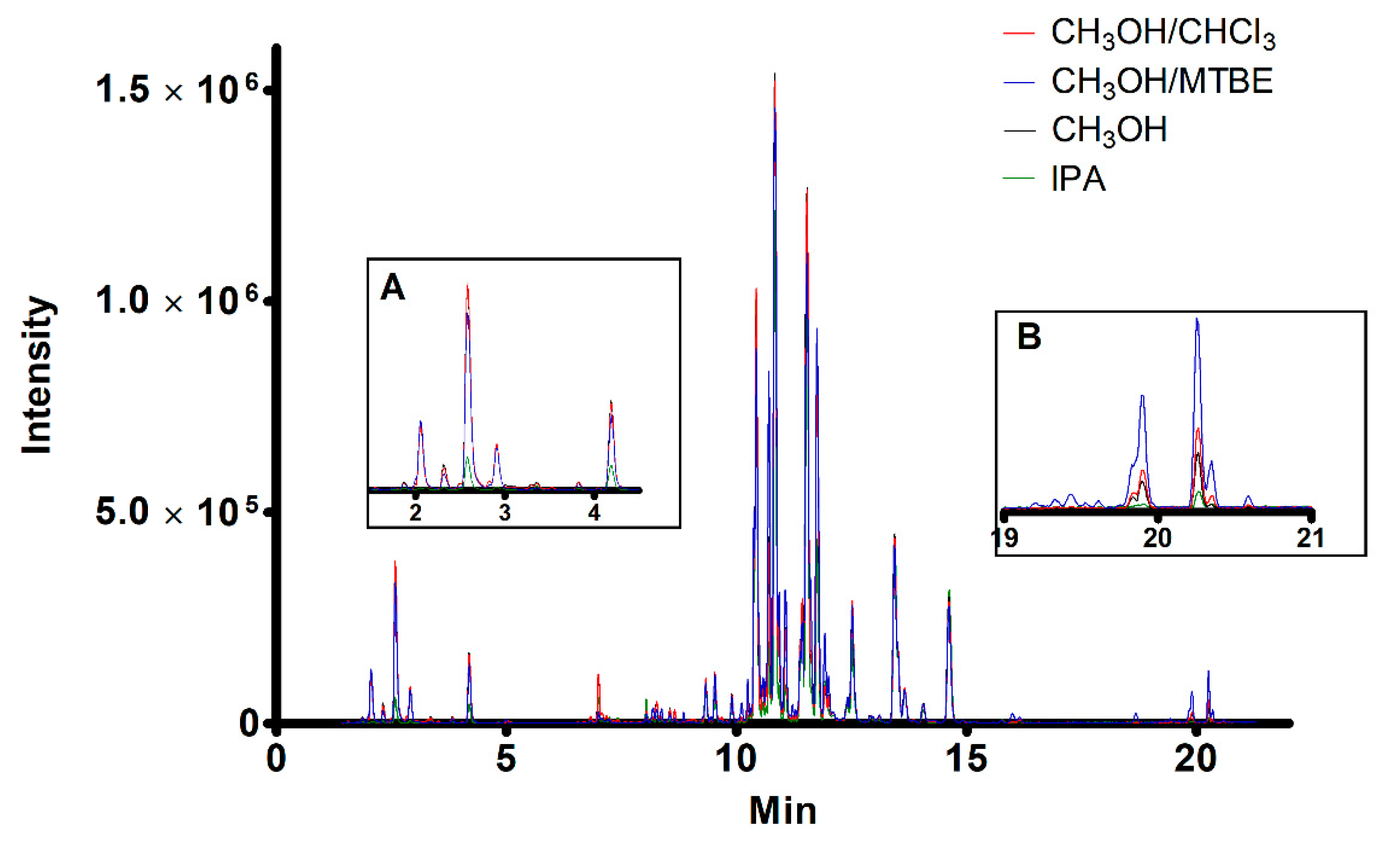
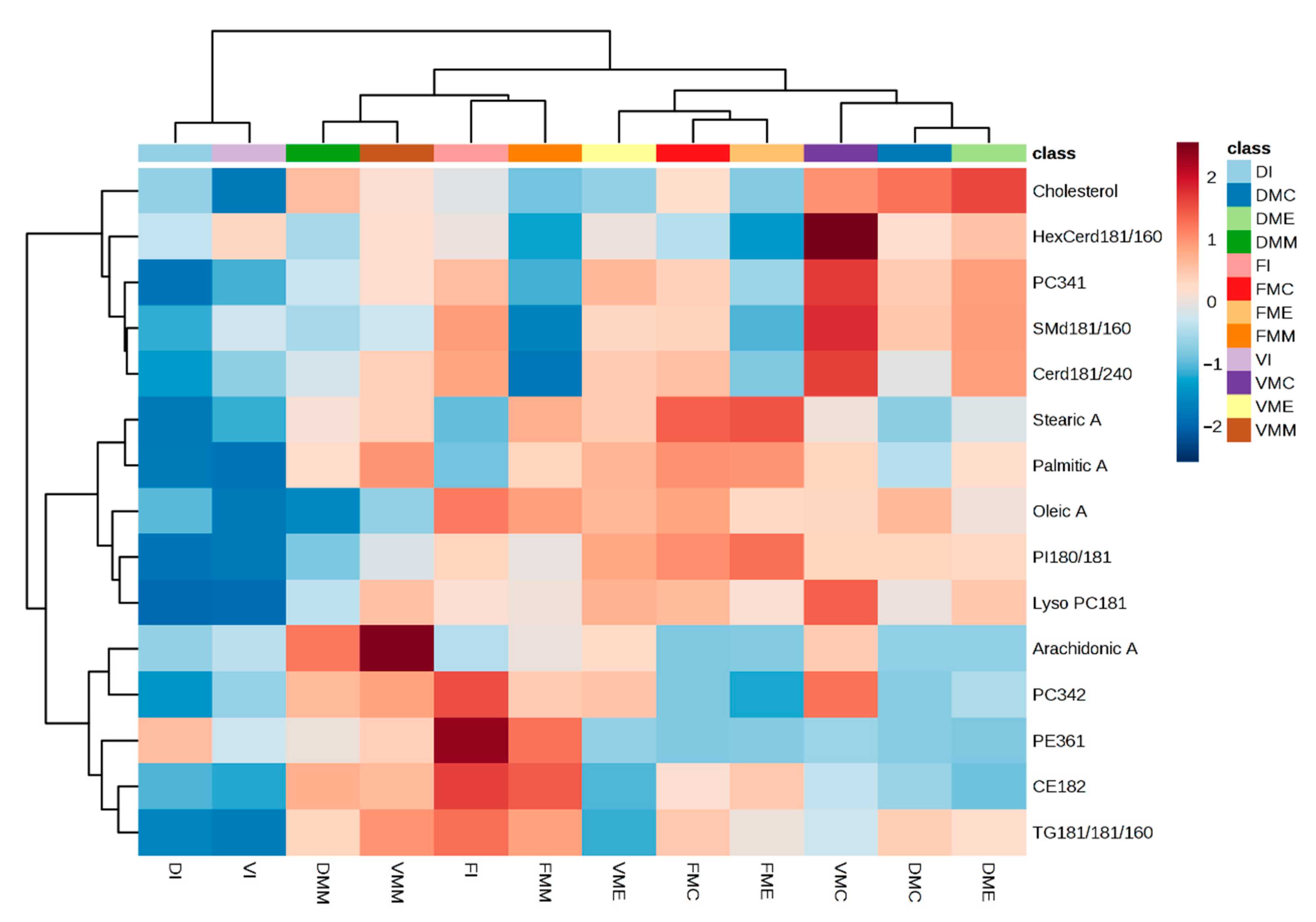
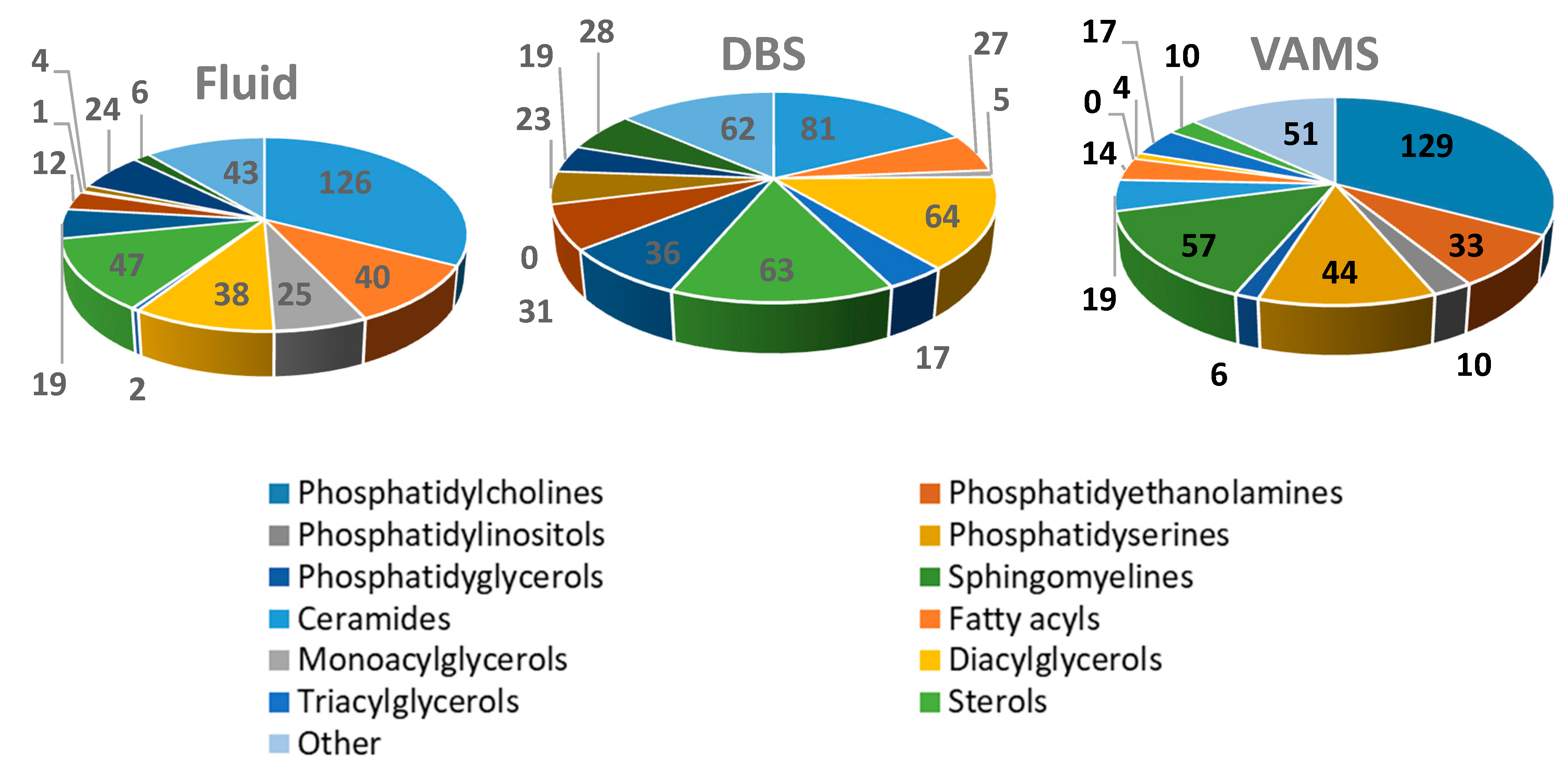
2. Results and Discussion
3. Materials and Methods
3.1. Solvents and Instrumentation
3.2. Biological Sample Collection
3.3. Extraction of Lipids from Blood Samples
3.4. LC-MS/MS Analysis
3.5. Data Analysis
3.6. Data Availability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Wenk, M.R. The emerging field of lipidomics. Nat. Rev. Drug Discov. 2005, 4, 594–610. [Google Scholar] [PubMed]
- Afshinnia, F.; Rajendiran, T.M.; Wernisch, S.; Soni, T.; Jadoon, A.; Karnovsky, A.; Michailidis, G.; Pennathur, S. Lipidomics and Biomarker Discovery in Kidney Disease. Semin. Nephrol. 2018, 38, 127–141. [Google Scholar] [PubMed]
- Herzog, K.; Pras-Raves, M.L.; Ferdinandusse, S.; Vervaart, M.A.T.; Luyf, A.C.M.; Van Kampen, A.H.C.; Wanders, R.J.A.; Waterham, H.R.; Vaz, F.M. Plasma lipidomics as a diagnostic tool for peroxisomal disorders. J. Inherit. Metab. Dis. 2017, 41, 489–498. [Google Scholar] [PubMed] [Green Version]
- Wood, P.L.; Cebak, J.E. Lipidomics biomarker studies: Errors, limitations, and the future. Biochem. Biophys. Res. Commun. 2018, 504, 569–575. [Google Scholar]
- Yan, F.; Zhao, H.; Zeng, Y. Lipidomics: A promising cancer biomarker. Clin. Transl. Med. 2018, 7, 21. [Google Scholar]
- Burla, B.; Arita, M.; Arita, M.; Bendt, A.K.; Gassiot, A.C.; Dennis, E.A.; Ekroos, K.; Han, X.; Ikeda, K.; Liebisch, G.; et al. MS-based lipidomics of human blood plasma: A community-initiated position paper to develop accepted guidelines. J. Lipid Res. 2018, 59, 2001–2017. [Google Scholar]
- Hu, T.; Zhang, J. Mass-spectrometry-based lipidomics. J. Sep. Sci. 2018, 41, 351–372. [Google Scholar]
- Zalloua, P.A.; Kadar, H.; Hariri, E.; Farraj, L.A.; Brial, F.; Hedjazi, L.; Le Lay, A.; Colleu, A.; Dubus, J.; Touboul, D.; et al. Untargeted Mass Spectrometry Lipidomics identifies correlation between serum sphingomyelins and plasma cholesterol. Lipids Health Dis. 2019, 18, 38. [Google Scholar]
- Cajka, T.; Fiehn, O. Comprehensive analysis of lipids in biological systems by liquid chromatography-mass spectrometry. TrAC Trends Anal. Chem. 2014, 61, 192–206. [Google Scholar]
- Baglai, A.; Gargano, A.F.; Jordens, J.; Mengerink, Y.; Honing, M.; van der Wal, S.; Schoenmakers, P.J. Comprehensive lipidomic analysis of human plasma using multidimensional liquid- and gas-phase separations: Two-dimensional liquid chromatography-mass spectrometry vs. liquid chromatography-trapped-ion-mobility-mass spectrometry. J. Chromatogr. A 2017, 1530, 90–103. [Google Scholar]
- Rampler, E.; Criscuolo, A.; Zeller, M.; El Abiead, Y.; Schoeny, H.; Hermann, G.; Sokol, E.; Cook, K.; Peake, D.A.; Delanghe, B.; et al. A Novel Lipidomics Workflow for Improved Human Plasma Identification and Quantification Using RPLC-MSn Methods and Isotope Dilution Strategies. Anal. Chem. 2018, 90, 6494–6501. [Google Scholar] [PubMed]
- Song, J.; Liu, X.; Wu, J.; Meehan, M.J.; Blevitt, J.M.; Dorrestein, P.C.; Milla, M.E. A highly efficient, high-throughput lipidomics platform for the quantitative detection of eicosanoids in human whole blood. Anal. Biochem. 2013, 433, 181–188. [Google Scholar] [PubMed]
- Yin, P.; Lehmann, R.; Xu, G. Effects of pre-analytical processes on blood samples used in metabolomics studies. Anal. Bioanal. Chem. 2015, 407, 4879–4892. [Google Scholar] [PubMed] [Green Version]
- Becker, S.; Röhnike, S.; Empting, S.; Haas, D.; Mohnike, K.; Beblo, S.; Mütze, U.; Husain, R.A.; Thiery, J.; Ceglarek, U. LC-MS/MS-based quantification of cholesterol and related metabolites in dried blood for the screening of inborn errors of sterol metabolism. Anal. Bioanal. Chem. 2015, 407, 5227–5233. [Google Scholar] [PubMed]
- Jacob, M.; Malkawi, A.; Albast, N.; Al Bougha, S.; Lopata, A.; Dasouki, M.; Rahman, A.M.A. A targeted metabolomics approach for clinical diagnosis of inborn errors of metabolism. Anal. Chim. Acta 2018, 1025, 141–153. [Google Scholar]
- Koulman, A.; Prentice, P.; Wong, M.C.Y.; Matthews, L.; Bond, N.J.; Eiden, M.; Griffin, J.L.; Dunger, D.B. The development and validation of a fast and robust dried blood spot based lipid profiling method to study infant metabolism. Metabolomics 2014, 10, 1018–1025. [Google Scholar]
- Dittakavi, S.; Jat, R.K.; Mullangi, R. Quantitative analysis of enasidenib in dried blood spots of mouse blood using an increased-sensitivity LC-MS/MS method: Application to a pharmacokinetic study. Biomed. Chromatogr. 2019, 33, e4491. [Google Scholar] [PubMed]
- Björkesten, J.; Enroth, S.; Shen, Q.; Wik, L.; Hougaard, D.M.; Cohen, A.S.; Sörensen, L.; Giedraitis, V.; Ingelsson, M.; Larsson, A.; et al. Stability of Proteins in Dried Blood Spot Biobanks. Mol. Cell. Proteom. 2017, 16, 1286–1296. [Google Scholar]
- Henao, J.J.A.; Metherel, A.H.; Smith, R.W.; Stark, K.D. Tailored Extraction Procedure Is Required to Ensure Recovery of the Main Lipid Classes in Whole Blood When Profiling the Lipidome of Dried Blood Spots. Anal. Chem. 2016, 88, 9391–9396. [Google Scholar]
- Cozma, C.; Iurașcu, M.-I.; Eichler, S.; Hovakimyan, M.; Brandau, O.; Zielke, S.; Böttcher, T.; Giese, A.-K.; Lukas, J.; Rolfs, A. C26-Ceramide as highly sensitive biomarker for the diagnosis of Farber Disease. Sci. Rep. 2017, 7, 1–13. [Google Scholar]
- Kyle, J.E.; Casey, C.P.; Stratton, K.G.; Zink, E.M.; Kim, Y.-M.; Zheng, X.; Monroe, M.E.; Weitz, K.K.; Bloodsworth, K.J.; Orton, D.J.; et al. Comparing identified and statistically significant lipids and polar metabolites in 15-year old serum and dried blood spot samples for longitudinal studies. Rapid Commun. Mass Spectrom. 2017, 31, 447–456. [Google Scholar] [PubMed] [Green Version]
- Gao, F.; McDaniel, J.; Chen, E.Y.; Rockwell, H.E.; Drolet, J.; Vishnudas, V.K.; Tolstikov, V.; Sarangarajan, R.; Narain, N.R.; Kiebish, M.A. Dynamic and temporal assessment of human dried blood spot MS/MS(ALL) shotgun lipidomics analysis. Nutr. Metab. 2017, 14, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denniff, P.; Spooner, N. Volumetric Absorptive Microsampling: A Dried Sample Collection Technique for Quantitative Bioanalysis. Anal. Chem. 2014, 86, 8489–8495. [Google Scholar] [CrossRef]
- Velghe, S.; Stove, C.P. Volumetric absorptive microsampling as an alternative tool for therapeutic drug monitoring of first-generation anti-epileptic drugs. Anal. Bioanal. Chem. 2018, 410, 2331–2341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verougstraete, N.; Lapauw, B.; Van Aken, S.; Delanghe, J.; Stove, C.; Stove, V. Volumetric absorptive microsampling at home as an alternative tool for the monitoring of HbA(1c) in diabetes patients. Clin. Chem. Lab. Med. 2017, 55, 462–469. [Google Scholar] [PubMed]
- Protti, M.; Mandrioli, R.; Mercolini, L. Tutorial: Volumetric absorptive microsampling (VAMS). Anal. Chim. Acta 2019, 1046, 32–47. [Google Scholar] [CrossRef]
- Kita, K.; Noritake, K.; Mano, Y. Application of a Volumetric Absorptive Microsampling Device to a Pharmacokinetic Study of Tacrolimus in Rats: Comparison with Wet Blood and Plasma. Eur. J. Drug Metab. Pharmacokinet. 2018, 44, 91–102. [Google Scholar] [CrossRef]
- Protti, M.; Catapano, M.C.; Dekel, B.G.S.; Rudge, J.; Gerra, G.; Somaini, L.; Mandrioli, R.; Mercolini, L. Determination of oxycodone and its major metabolites in haematic and urinary matrices: Comparison of traditional and miniaturised sampling approaches. J. Pharm. Biomed. Anal. 2018, 152, 204–214. [Google Scholar] [CrossRef]
- Protti, M.; Rudge, J.; Sberna, A.E.; Gerra, G.; Mercolini, L. Dried haematic microsamples and LC-MS/MS for the analysis of natural and synthetic cannabinoids. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1044, 77–86. [Google Scholar]
- Mercolini, L.; Protti, M.; Catapano, M.C.; Rudge, J.; Sberna, A.E. LC-MS/MS and volumetric absorptive microsampling for quantitative bioanalysis of cathinone analogues in dried urine, plasma and oral fluid samples. J. Pharm. Biomed. Anal. 2016, 123, 186–194. [Google Scholar] [CrossRef]
- Van den Broek, I.; Fu, Q.; Kushon, S.; Kowalski, M.P.; Millis, K.; Percy, A.; Holewinski, R.J.; Venkatraman, V.; Van Eyk, J.E. Application of volumetric absorptive microsampling for robust, high-throughput mass spectrometric quantification of circulating protein biomarkers. Clin. Mass Spectrom. 2017, 4, 25–33. [Google Scholar] [CrossRef]
- Kok, M.G.; Nix, C.; Nys, G.; Fillet, M. Targeted metabolomics of whole blood using volumetric absorptive microsampling. Talanta 2019, 197, 49–58. [Google Scholar] [CrossRef]
- Volani, C.; Caprioli, G.; Calderisi, G.; Sigurdsson, B.B.; Rainer, J.; Gentilini, I.; Hicks, A.A.; Pramstaller, P.P.; Weiss, G.; Smarason, S.V.; et al. Pre-analytic evaluation of volumetric absorptive microsampling and integration in a mass spectrometry-based metabolomics workflow. Anal. Bioanal. Chem. 2017, 409, 6263–6276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kale, N.S.; Haug, K.; Conesa, P.; Jayseelan, K.; Moreno, P.; Rocca-Serra, P.; Nainala, V.C.; Spicer, R.A.; Williams, M.; Philippe, R.-S.; et al. MetaboLights: An Open-Access Database Repository for Metabolomics Data. Curr. Protoc. Bioinform. 2016, 53, 14.13.1–14.13.18. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.A.; Heckert, A.; Ulmer, C.Z.; Jones, C.M.; Koelmel, J.P.; Abdullah, L.; Ahonen, L.; Alnouti, Y.; Armando, A.M.; Asara, J.M.; et al. Harmonizing lipidomics: NIST interlaboratory comparison exercise for lipidomics using SRM 1950-Metabolites in Frozen Human Plasma. J. Lipid Res. 2017, 58, 2275–2288. [Google Scholar] [CrossRef] [Green Version]
- Fahy, E.; Sud, M.; Cotter, D.; Subramaniam, S. LIPID MAPS online tools for lipid research. Nucleic Acids Res. 2007, 35, W606–W612. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Wishart, D.S. Using MetaboAnalyst 3.0 for Comprehensive Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2016, 55, 14.10.1–14.10.91. [Google Scholar] [CrossRef]
- Chong, J.; Xia, J. MetaboAnalystR: An R package for flexible and reproducible analysis of metabolomics data. Bioinformatics 2018, 34, 4313–4314. [Google Scholar] [CrossRef] [Green Version]
- Goracci, L.; Tortorella, S.; Tiberi, P.; Pellegrino, R.M.; Di Veroli, A.; Valeri, A.; Cruciani, G. Lipostar, a Comprehensive Platform-Neutral Cheminformatics Tool for Lipidomics. Anal. Chem. 2017, 89, 6257–6264. [Google Scholar] [CrossRef]
- La Barbera, G.; Antonelli, M.; Cavaliere, C.; Cruciani, G.; Goracci, L.; Montone, C.M.; Piovesana, S.; Laganà, A.; Capriotti, A.L. Delving into the Polar Lipidome by Optimized Chromatographic Separation, High-Resolution Mass Spectrometry, and Comprehensive Identification with Lipostar: Microalgae as Case Study. Anal. Chem. 2018, 90, 12230–12238. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N | Category | Lipid | Molecular Formula | LogP | m/z | Adduct | Min. |
|---|---|---|---|---|---|---|---|
| 1 | Fatty Acid | Arachidonic Acid (20:4) | C20H31O2 | 6.22 | 303.23 | [M–H]− | 4.6 |
| 2 | Fatty Acid | Oleic Acid (18:1) | C18H33O2 | 6.11 | 281.24 | [M–H]− | 6.1 |
| 3 | Fatty Acid | Stearic Acid (18:0) | C18H35O2 | 6.33 | 283.20 | [M–H]− | 7.6 |
| 4 | Fatty Acid | Palmitic Acid (16:0) | C16H31O2 | 5.55 | 255.23 | [M–H]− | 5.8 |
| 5 | Phospholipid | PI (18:0/18:1) | C45H85O13P | 11.81 | 863.57 | [M–H]− | 11.2 |
| 6 | Phospholipid | Lyso PC (18:1) | C26H51NO7P | 6.56 | 520.34 | [M+H]+ | 2.0 |
| 7 | Phospholipid | PC (34:2) | C42H79NO8P | 12.37 | 756.55 | [M+H]+ | 10.3 |
| 8 | Phospholipid | PC (34:1) | C42H81NO8P | 12.59 | 758.57 | [M+H]+ | 10.8 |
| 9 | Phospholipid | PE (36:1) | C41H81NO8P | 13.43 | 746.57 | [M+H]+ | 11.0 |
| 10 | Sphingolipid | SM (d18:1/16:0) | C39H80N2O6P | 11.21 | 703.57 | [M+H]+ | 10.4 |
| 11 | Sphingolipid | HexCer (d18:1/16:0) | C40H78NO8 | 9.96 | 700.57 | [M+H]+ | 10.8 |
| 12 | Sphingolipid | Cer (d18:1/24:0) | C42H84NO3 | 13.54 | 650.64 | [M+H]+ | 16.1 |
| 13 | Sterol | Cholesterol | C27H44 | 7.68 | 369.35 | [M–H2O+H]+ | 10.3 |
| 14 | Sterol | CE (18:2) | C45H77O2 | 14.04 | 666.62 | [M+NH4]+ | 19.8 |
| 15 | Triacylglycerol | TG (16:0/18:1/18:1) | C55H106NO6 | 18.4 | 876.80 | [M+NH4]+ | 20.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marasca, C.; Arana, M.E.B.; Protti, M.; Cavalli, A.; Mercolini, L.; Armirotti, A. Volumetric Absorptive Microsampling of Blood for Untargeted Lipidomics. Molecules 2021, 26, 262. https://doi.org/10.3390/molecules26020262
Marasca C, Arana MEB, Protti M, Cavalli A, Mercolini L, Armirotti A. Volumetric Absorptive Microsampling of Blood for Untargeted Lipidomics. Molecules. 2021; 26(2):262. https://doi.org/10.3390/molecules26020262
Chicago/Turabian StyleMarasca, Camilla, Maria Encarnacion Blanco Arana, Michele Protti, Andrea Cavalli, Laura Mercolini, and Andrea Armirotti. 2021. "Volumetric Absorptive Microsampling of Blood for Untargeted Lipidomics" Molecules 26, no. 2: 262. https://doi.org/10.3390/molecules26020262