
Photodegradation of Riboflavin under Alkaline Conditions: What Can Gas-Phase Photolysis Tell Us about What Happens in Solution?


Abstract
:
1. Introduction
2. Results
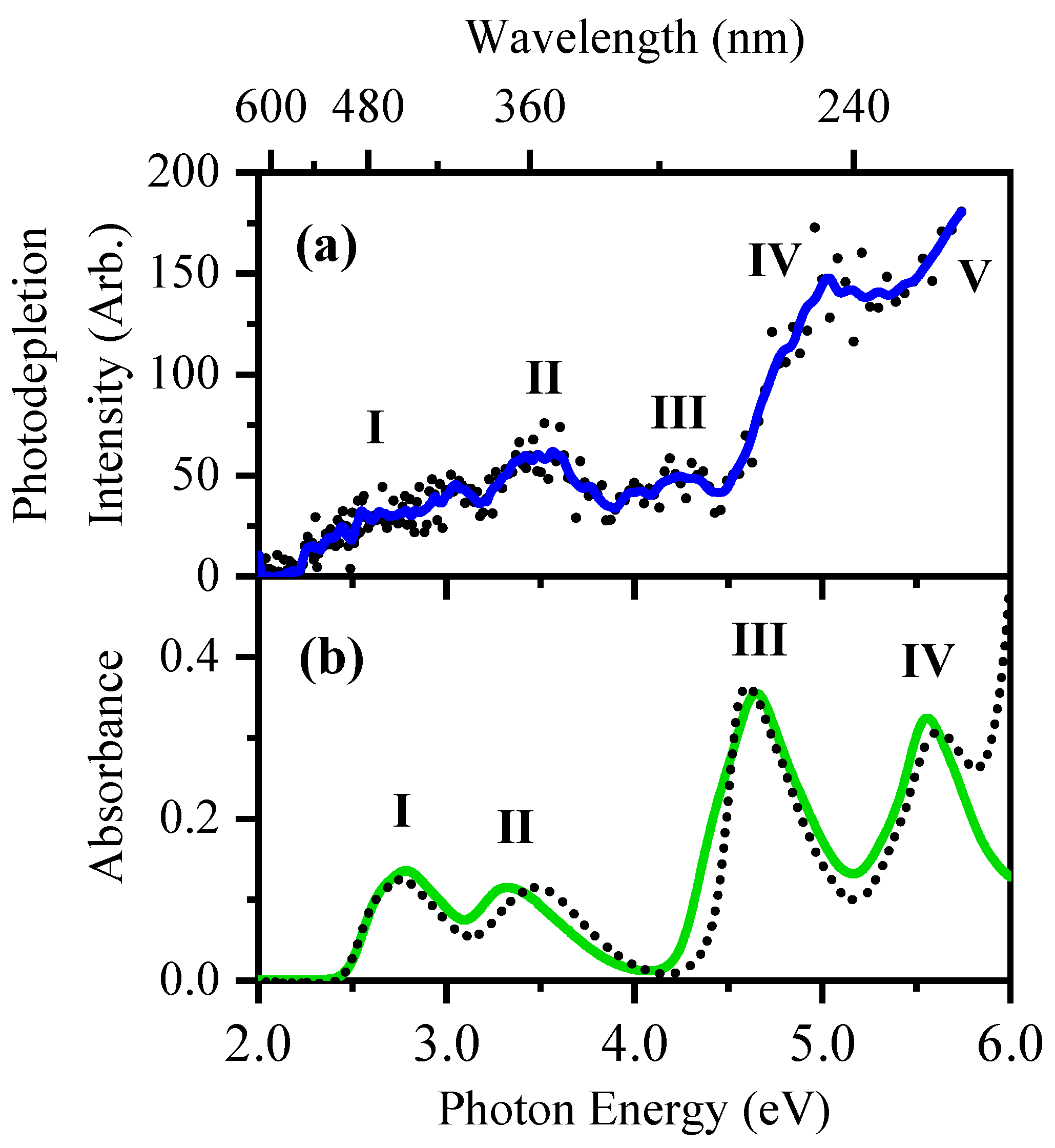
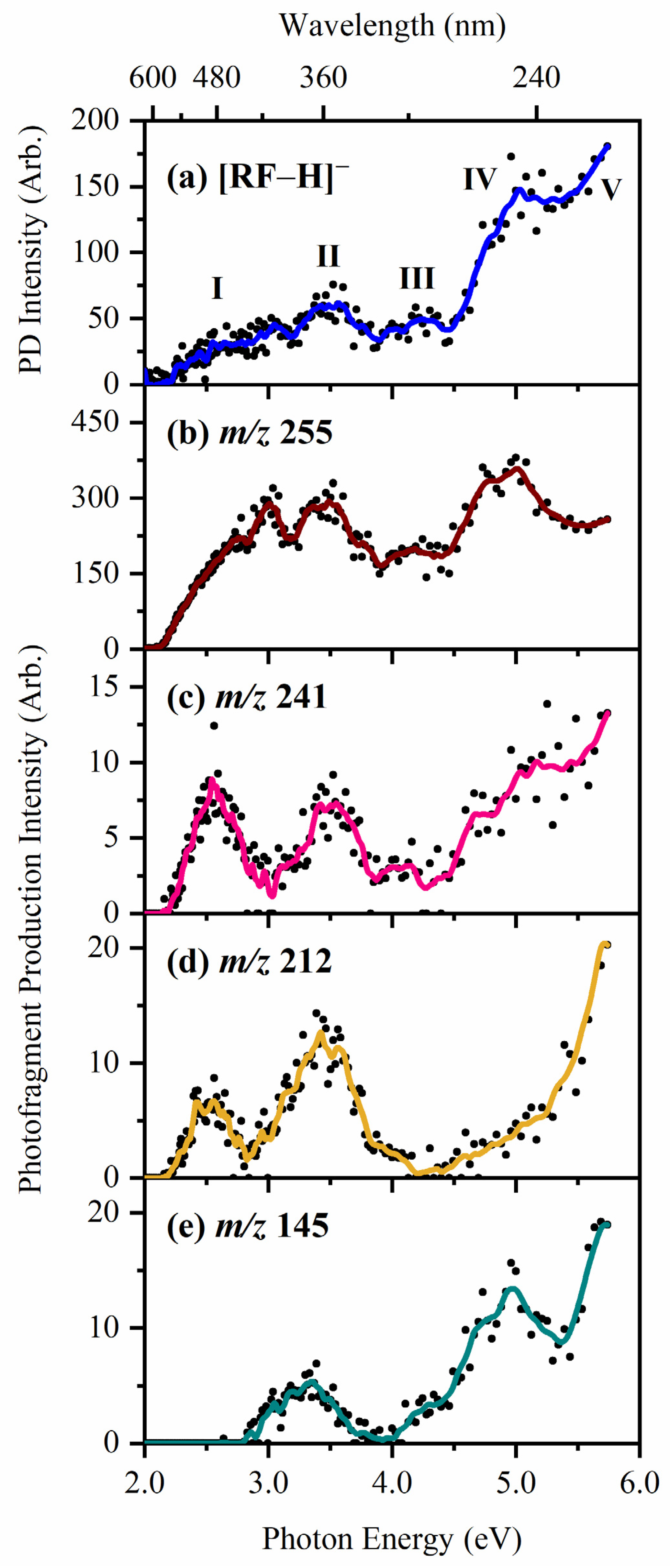
2.1. Gas-Phase Absorption Spectroscopy of [RF − H]−
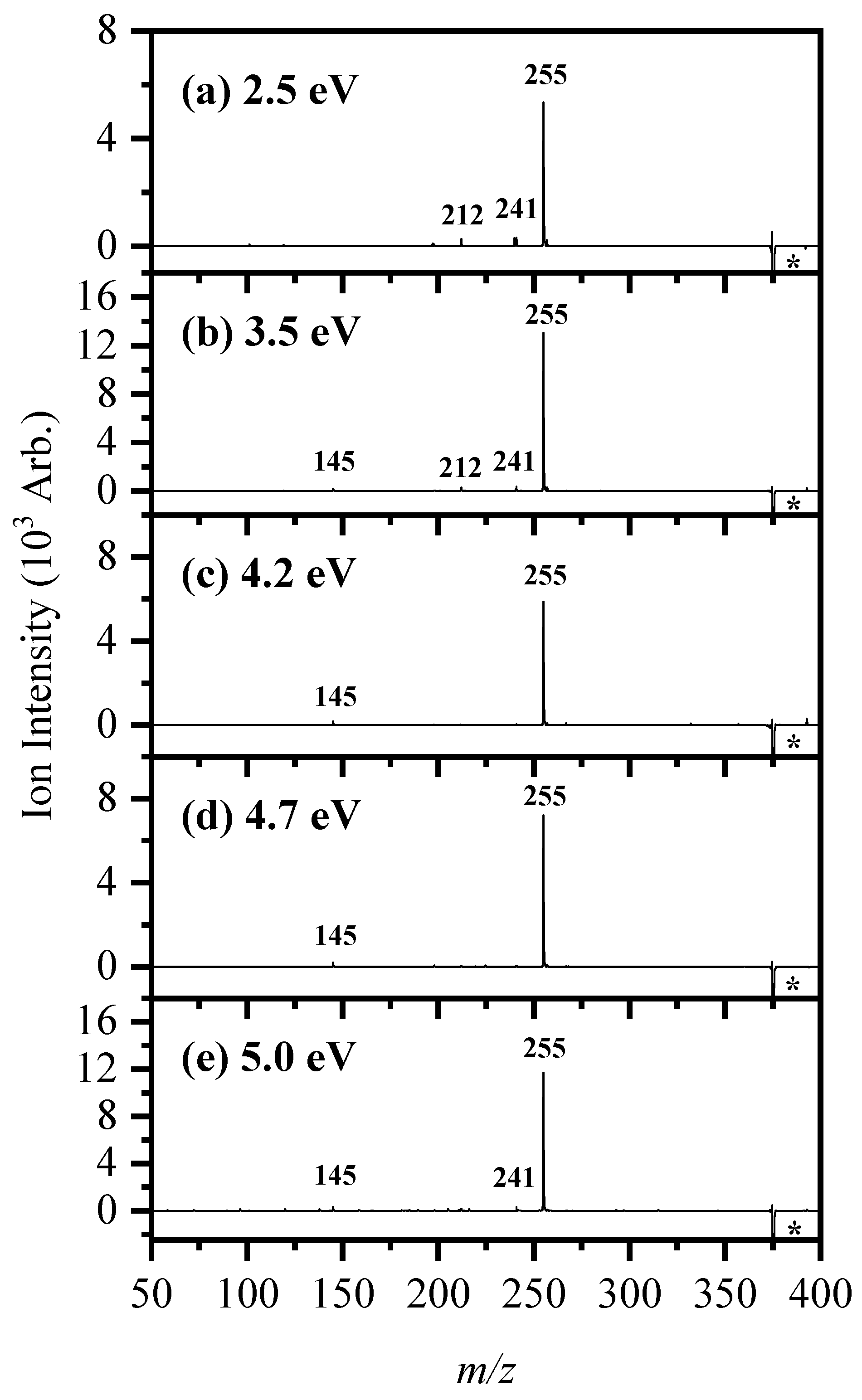
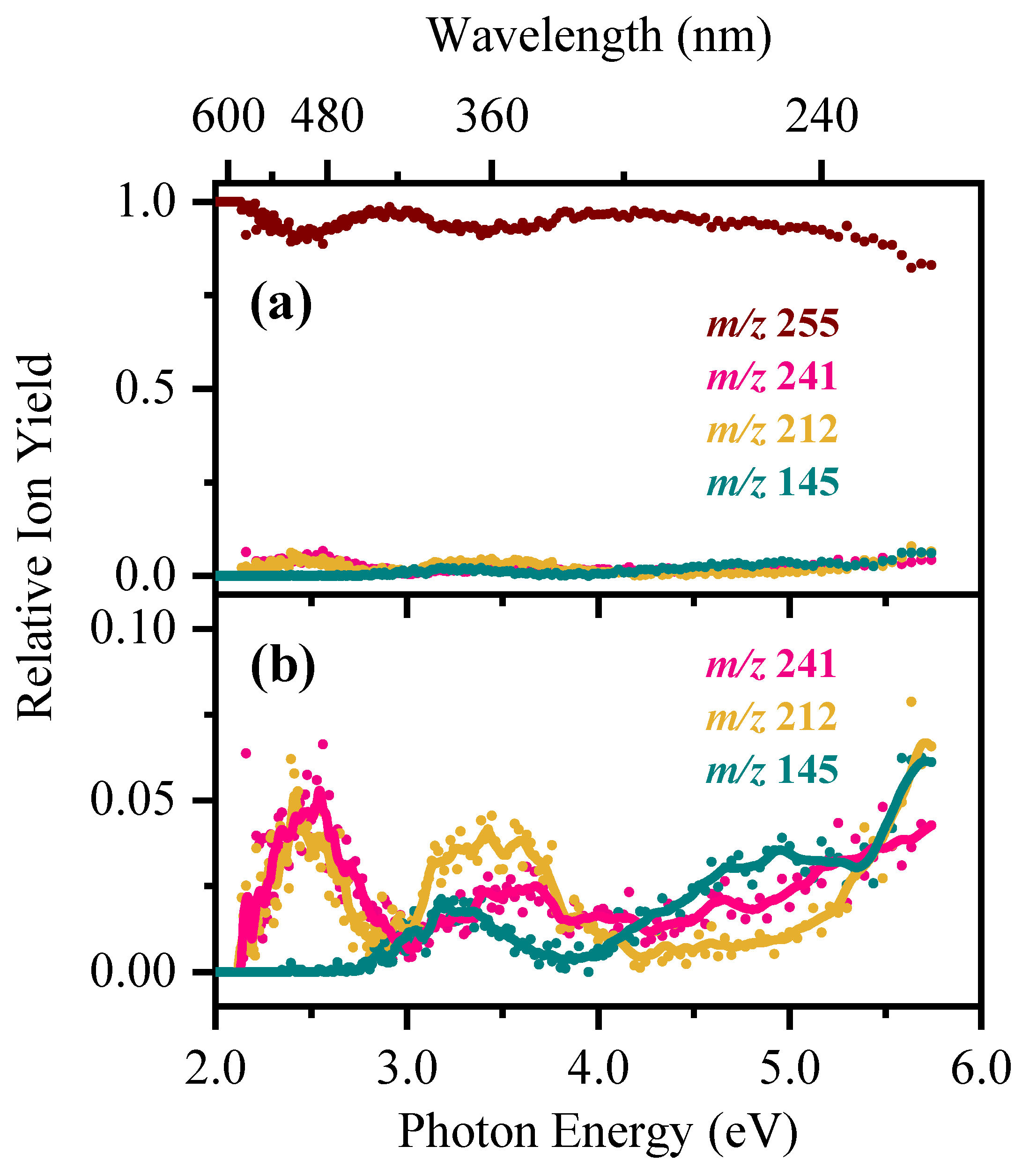
2.2. Gas-Phase Photofragmentation of [RF − H]−
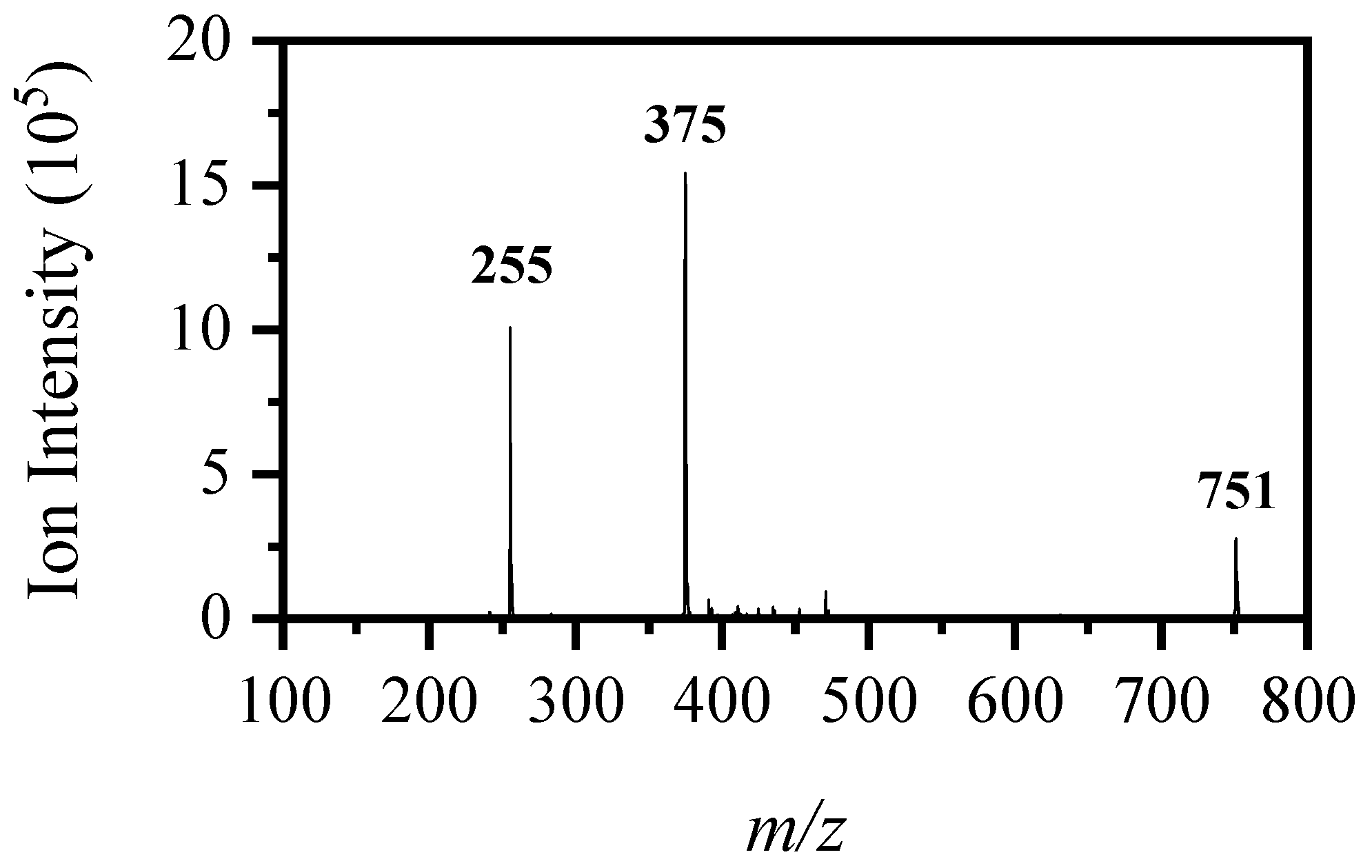
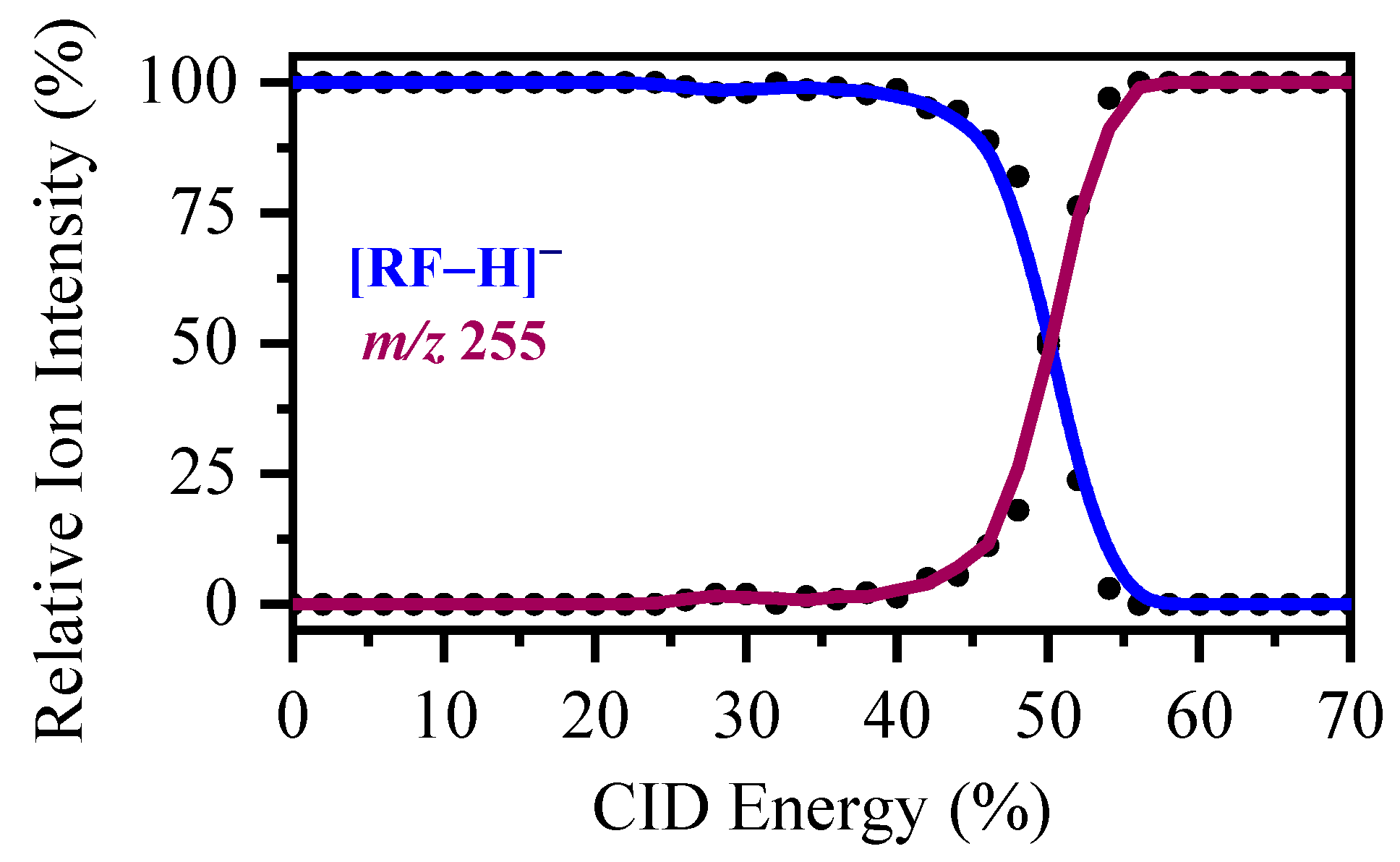
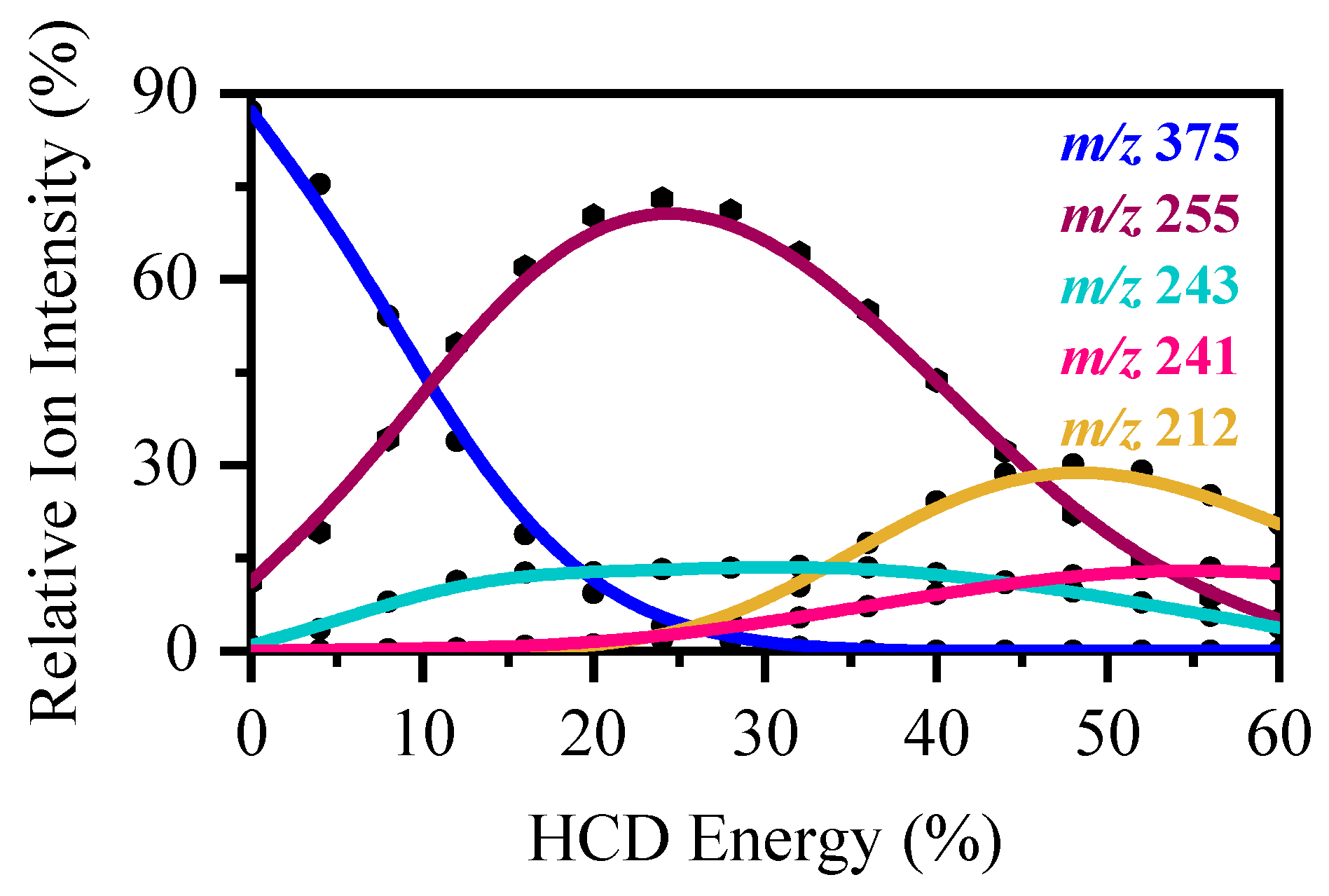
2.3. Collision-Induced Dissociation of [RF − H]−
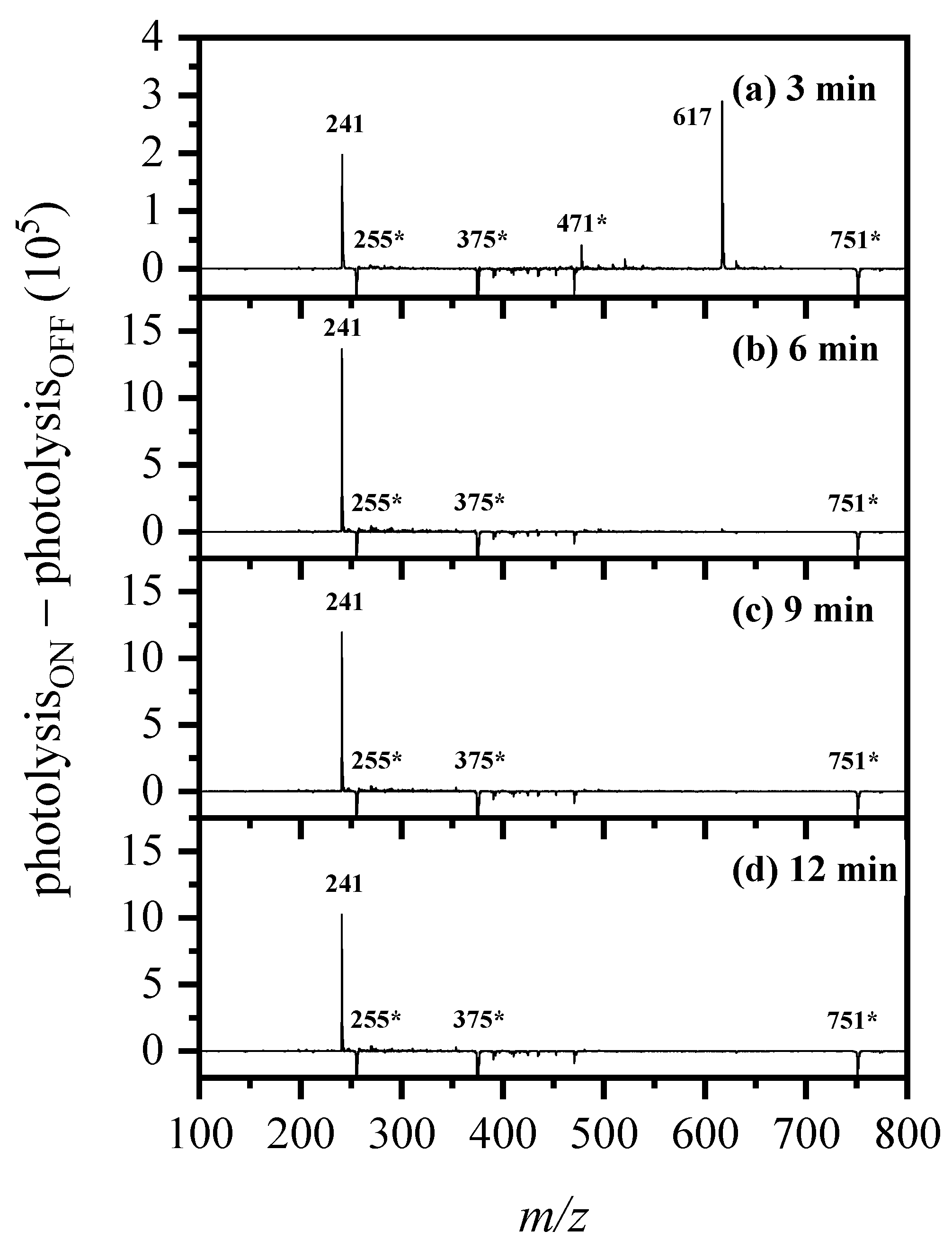
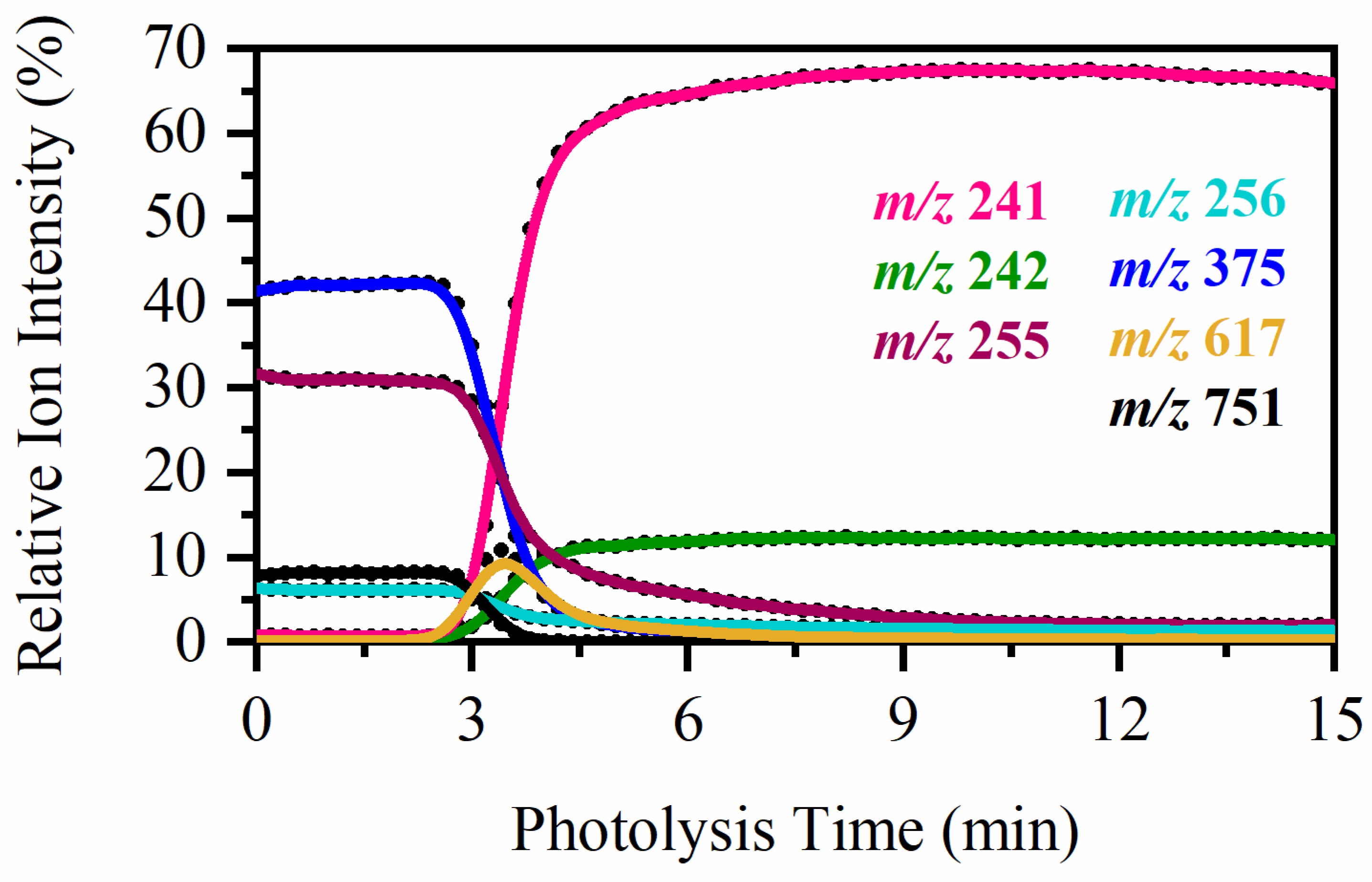
2.4. On-Line Solution-Phase Photolysis of [RF − H]−
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Sheraz, M.A.; Kazi, S.H.; Ahmed, S.; Anwar, Z.; Ahmad, I. Photo, Thermal and Chemical Degradation of Riboflavin. Beilstein J. Org. Chem. 2014, 10, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Mirza, T.; Musharraf, S.G.; Anwar, Z.; Sheraz, M.A.; Ahmed, S.; Ejaz, M.A.; Khurshid, A. Photolysis of Carboxymethylflavin in Aqueous and Organic Solvent: A Kinetic Study. RSC Adv. 2019, 9, 26559–26571. [Google Scholar] [CrossRef] [Green Version]
- Knak, A.; Regensburger, J.; Maisch, T.; Bäumler, W. Exposure of Vitamins to UVB and UVA Radiation Generates Singlet Oxygen. Photochem. Photobiol. Sci. 2014, 13, 820–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, I.; Fasihullah, Q.; Noor, A.; Ansari, I.A.; Ali, Q.N.M. Photolysis of Riboflavin in Aqueous Solution: A Kinetic Study. Int. J. Pharm. 2004, 280, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Astanov, S.K.; Kasimova, G.K.; Kurtaliev, E.N.; Nizomov, N.N.; Jumabaev, A. Electronic Nature and Structure of Aggregates of Riboflavin Molecules. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2021, 248, 119177. [Google Scholar] [CrossRef] [PubMed]
- Matthews, E.; Dessent, C.E.H. Observation of Near-Threshold Resonances in the Flavin Chromophore Anions Alloxazine and Lumichrome. J. Phys. Chem. Lett. 2018, 9, 6124–6130. [Google Scholar] [CrossRef] [Green Version]
- Matthews, E.; Cercola, R.; Dessent, C. Protomer-Dependent Electronic Spectroscopy and Photochemistry of the Model Flavin Chromophore Alloxazine. Molecules 2018, 23, 2036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bull, J.N.; Carrascosa, E.; Giacomozzi, L.; Bieske, E.J.; Stockett, M.H. Ion Mobility Action Spectroscopy of Flavin Dianions Reveals Deprotomer-Dependent Photochemistry. Phys. Chem. Chem. Phys. 2018, 20, 19672–19681. [Google Scholar] [CrossRef] [Green Version]
- Stockett, M.H. Photo-Induced Proton-Coupled Electron Transfer and Dissociation of Isolated Flavin Adenine Dinucleotide Mono-Anions. Phys. Chem. Chem. Phys. 2017, 19, 25829–25833. [Google Scholar] [CrossRef]
- Günther, A.; Nieto, P.; Müller, D.; Sheldrick, A.; Gerlich, D.; Dopfer, O. BerlinTrap: A New Cryogenic 22-Pole Ion Trap Spectrometer. J. Mol. Spectrosc. 2017, 332, 8–15. [Google Scholar] [CrossRef]
- Langer, J.; Günther, A.; Seidenbecher, S.; Berden, G.; Oomens, J.; Dopfer, O. Probing Protonation Sites of Isolated Flavins Using IR Spectroscopy: From Lumichrome to the Cofactor Flavin Mononucleotide. ChemPhysChem 2014, 15, 2550–2562. [Google Scholar] [CrossRef]
- Zhang, T.; Papson, K.; Ochran, R.; Ridge, D.P. Stability of Flavin Semiquinones in the Gas Phase: The Electron Affinity, Proton Affinity, and Hydrogen Atom Affinity of Lumiflavin. J. Phys. Chem. A 2013, 117, 11136–11141. [Google Scholar] [CrossRef]
- Guyon, L.; Tabarin, T.; Thuillier, B.; Antoine, R.; Broyer, M.; Boutou, V.; Wolf, J.-P.; Dugourd, P. Femtosecond Pump-Probe Experiments on Trapped Flavin: Optical Control of Dissociation. J. Chem. Phys. 2008, 128, 075103. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, A.; Müller, D.; Günther, A.; Nieto, P.; Dopfer, O. Optical Spectroscopy of Isolated Flavins: Photodissociation of Protonated Lumichrome. Phys. Chem. Chem. Phys. 2018, 20, 7407–7414. [Google Scholar] [CrossRef] [PubMed]
- Cercola, R.; Wong, N.G.K.; Rhodes, C.; Olijnyk, L.; Mistry, N.S.; Hall, L.M.; Berenbeim, J.A.; Lynam, J.M.; Dessent, C.E.H. A “One Pot” Mass Spectrometry Technique for Characterizing Solution- and Gas-Phase Photochemical Reactions by Electrospray Mass Spectrometry. RSC Adv. 2021, 11, 19500–19507. [Google Scholar] [CrossRef]
- Mehara, J.; Roithová, J. Identifying Reactive Intermediates by Mass Spectrometry. Chem. Sci. 2020, 11, 11960–11972. [Google Scholar] [CrossRef] [PubMed]
- Insińska-Rak, M.; Prukała, D.; Golczak, A.; Fornal, E.; Sikorski, M. Riboflavin Degradation Products; Combined Photochemical and Mass Spectrometry Approach. J. Photochem. Photobiol. A Chem. 2020, 403, 112837. [Google Scholar] [CrossRef]
- Pahl, M.; Mayer, M.; Schneider, M.; Belder, D.; Asmis, K.R. Joining Microfluidics with Infrared Photodissociation: Online Monitoring of Isomeric Flow-Reaction Intermediates. Anal. Chem. 2019, 91, 3199–3203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Wan, Q.; Badu-Tawiah, A.K. Picomole-Scale Real-Time Photoreaction Screening: Discovery of the Visible-Light-Promoted Dehydrogenation of Tetrahydroquinolines under Ambient Conditions. Angew. Chem. Int. Ed. 2016, 55, 9345–9349. [Google Scholar] [CrossRef]
- Insińska-Rak, M.; Golczak, A.; Sikorski, M. Photochemistry of Riboflavin Derivatives in Methanolic Solutions. J. Phys. Chem. A 2012, 116, 1199–1207. [Google Scholar] [CrossRef]
- Matthews, E.; Dessent, C.E.H. Locating the Proton in Nicotinamide Protomers via Low-Resolution UV Action Spectroscopy of Electrosprayed Solutions. J. Phys. Chem. A 2016, 120, 9209–9216. [Google Scholar] [CrossRef]
- Matthews, E.; Sen, A.; Yoshikawa, N.; Bergström, E.; Dessent, C.E.H. UV Laser Photoactivation of Hexachloroplatinate Bound to Individual Nucleobases: In Vacuo as Molecular Level Probes of a Model Photopharmaceutical. Phys. Chem. Chem. Phys. 2016, 18, 15143–15152. [Google Scholar] [CrossRef] [Green Version]
- Wong, N.G.K.; Berenbeim, J.A.; Hawkridge, M.; Matthews, E.; Dessent, C.E.H. Mapping the Intrinsic Absorption Properties and Photodegradation Pathways of the Protonated and Deprotonated Forms of the Sunscreen Oxybenzone. Phys. Chem. Chem. Phys. 2019, 21, 14311–14321. [Google Scholar] [CrossRef]
- Sen, A.; Luxford, T.F.M.; Yoshikawa, N.; Dessent, C.E.H. Solvent Evaporation versus Proton Transfer in Nucleobase–Pt(CN)4,62− Dianion Clusters: A Collisional Excitation and Electronic Laser Photodissociation Spectroscopy Study. Phys. Chem. Chem. Phys. 2014, 16, 15490–15500. [Google Scholar] [CrossRef]
- Antoine, R.; Dugourd, P. Visible and Ultraviolet Spectroscopy of Gas Phase Protein Ions. Phys. Chem. Chem. Phys. 2011, 13, 16494–16509. [Google Scholar] [CrossRef]
- Wellman, S.M.; Jockusch, R.A. Moving in on the Action: An Experimental Comparison of Fluorescence Excitation and Photodissociation Action Spectroscopy. J. Phys. Chem. A 2015, 119, 6333–6338. [Google Scholar] [CrossRef] [PubMed]
- Budavari, S.; Smith, A.; Heckelman, P.E.; O’Neil, M.J. The Merck Index, 15th ed.; Merck: Rahway, NJ, USA, 1989. [Google Scholar]
- Drössler, P.; Holzer, W.; Penzkofer, A.; Hegemann, P. pH Dependence of the Absorption and Emission Behaviour of Riboflavin in Aqueous Solution. Chem. Phys. 2002, 282, 429–439. [Google Scholar] [CrossRef]
- Matthews, E.; Cercola, R.; Mensa-Bonsu, G.; Neumark, D.M.; Dessent, C.E.H. Photoexcitation of Iodide Ion-Pyrimidine Clusters above the Electron Detachment Threshold: Intracluster Electron Transfer versus Nucleobase-Centred Excitations. J. Chem. Phys. 2018, 148, 084304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, N.G.K.; Rankine, C.D.; Dessent, C.E.H. Measurement of the Population of Electrosprayed Deprotomers of Coumaric Acids Using UV-Vis Laser Photodissociation Spectroscopy. J. Phys. Chem. A 2021, 125, 6703–6714. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.G.K.; Rankine, C.D.; Dessent, C.E.H. Linking Electronic Relaxation Dynamics and Ionic Photofragmentation Patterns for the Deprotonated UV Filter Benzophenone-4. J. Phys. Chem. Lett. 2021, 12, 2831–2836. [Google Scholar] [CrossRef] [PubMed]
- Berenbeim, J.A.; Wong, N.G.K.; Cockett, M.C.R.; Berden, G.; Oomens, J.; Rijs, A.M.; Dessent, C.E.H. Unravelling the Keto–Enol Tautomer Dependent Photochemistry and Degradation Pathways of the Protonated UVA Filter Avobenzone. J. Phys. Chem. A 2020, 124, 2919–2930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boxford, W.E.; Dessent, C.E.H. Probing the Intrinsic Features and Environmental Stabilization of Multiply Charged Anions. Phys. Chem. Chem. Phys. 2006, 8, 5151–5165. [Google Scholar] [CrossRef]
- Cercola, R.; Matthews, E.; Dessent, C.E.H. Photoexcitation of Adenosine 5’-Triphosphate Anions in Vacuo: Probing the Influence of Charge State on the UV Photophysics of Adenine. J. Phys. Chem. B 2017, 121, 5553–5561. [Google Scholar] [CrossRef] [Green Version]
- Furlan, C.; Berenbeim, J.A.; Dessent, C.E.H. Photoproducts of the Photodynamic Therapy Agent Verteporfin Identified via Laser Interfaced Mass Spectrometry. Molecules 2020, 25, 5280. [Google Scholar] [CrossRef]
- Wong, N.G.K.; Berenbeim, J.A.; Dessent, C.E.H. Direct Observation of Photochemical Free Radical Production from the Sunscreen 2-Phenylbenzimidazole-5-Sulfonic Acid via Laser-Interfaced Mass Spectrometry. ChemPhotoChem 2019, 3, 1231–1237. [Google Scholar] [CrossRef]
- Berenbeim, J.A.; Wong, N.G.K.; Cockett, M.C.R.; Berden, G.; Oomens, J.; Rijs, A.M.; Dessent, C.E.H. Sodium Cationization Can Disrupt the Intramolecular Hydrogen Bond That Mediates the Sunscreen Activity of Oxybenzone. Phys. Chem. Chem. Phys. 2020, 22, 19522–19531. [Google Scholar] [CrossRef] [PubMed]
- Luxford, T.F.M.M.; Milner, E.M.; Yoshikawa, N.; Bullivant, C.; Dessent, C.E.H. Complexation of Carboxylate Anions with the Arginine Gas-Phase Amino Acid: Effects of Chain Length on the Geometry of Extended Ion Binding. Chem. Phys. Lett. 2013, 577, 1–5. [Google Scholar] [CrossRef]
- Zhang, J.; Brodbelt, J.S.; Wang, J. Threshold Dissociation and Molecular Modeling of Transition Metal Complexes of Flavonoids. J. Am. Soc. Mass Spectrom. 2005, 16, 139–151. [Google Scholar] [CrossRef] [Green Version]
- Wan, K.X.; Gross, M.L.; Shibue, T. Gas-Phase Stability of Double-Stranded Oligodeoxynucleotides and Their Noncovalent Complexes with DNA-Binding Drugs as Revealed by Collisional Activation in an Ion Trap. J. Am. Soc. Mass Spectrom. 2000, 11, 450–457. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
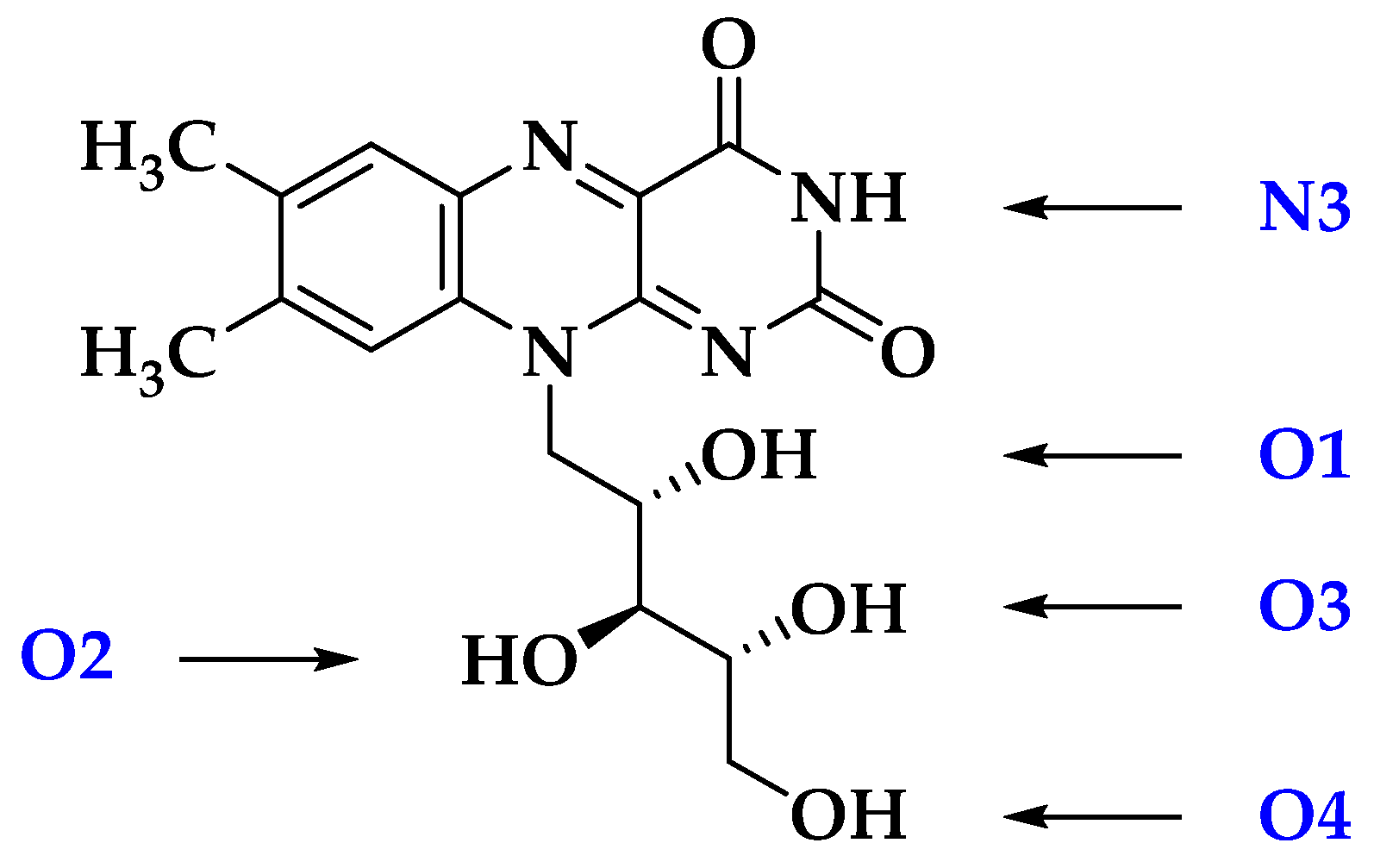
| Ionic Fragment Mass (m/z) 1 | Proposed Structure of Fragment | Mass of Accompanying Neutral Mass Lost | Observed in CID (via H2O) 1,2 | Observed in HCD (via EtOH) | Observed in Laser UV–Visible Photoexcitation 1,2 | Observed via Solution-Phase On-Line UV-A Photolysis 1,2 | ||
|---|---|---|---|---|---|---|---|---|
| 25% | 50% | 3 min | 6 min | |||||
| 255 |  | 120 | (vs) | (s) | (m) | (s) | (w) 4 | (vw) |
| 241 |  | 134 | - | - | (w) | (w) | (w) | (vs) |
| 212 |  | 163 | - | (w) | (m) | (w) | (w) | (w) |
| 145 |  | 230 | - | (w) | (w) | (w) | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, N.G.K.; Rhodes, C.; Dessent, C.E.H. Photodegradation of Riboflavin under Alkaline Conditions: What Can Gas-Phase Photolysis Tell Us about What Happens in Solution? Molecules 2021, 26, 6009. https://doi.org/10.3390/molecules26196009
Wong NGK, Rhodes C, Dessent CEH. Photodegradation of Riboflavin under Alkaline Conditions: What Can Gas-Phase Photolysis Tell Us about What Happens in Solution? Molecules. 2021; 26(19):6009. https://doi.org/10.3390/molecules26196009
Chicago/Turabian StyleWong, Natalie G. K., Chris Rhodes, and Caroline E. H. Dessent. 2021. "Photodegradation of Riboflavin under Alkaline Conditions: What Can Gas-Phase Photolysis Tell Us about What Happens in Solution?" Molecules 26, no. 19: 6009. https://doi.org/10.3390/molecules26196009