
Development of an In-Line Enzyme Reactor Integrated into a Capillary Electrophoresis System


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
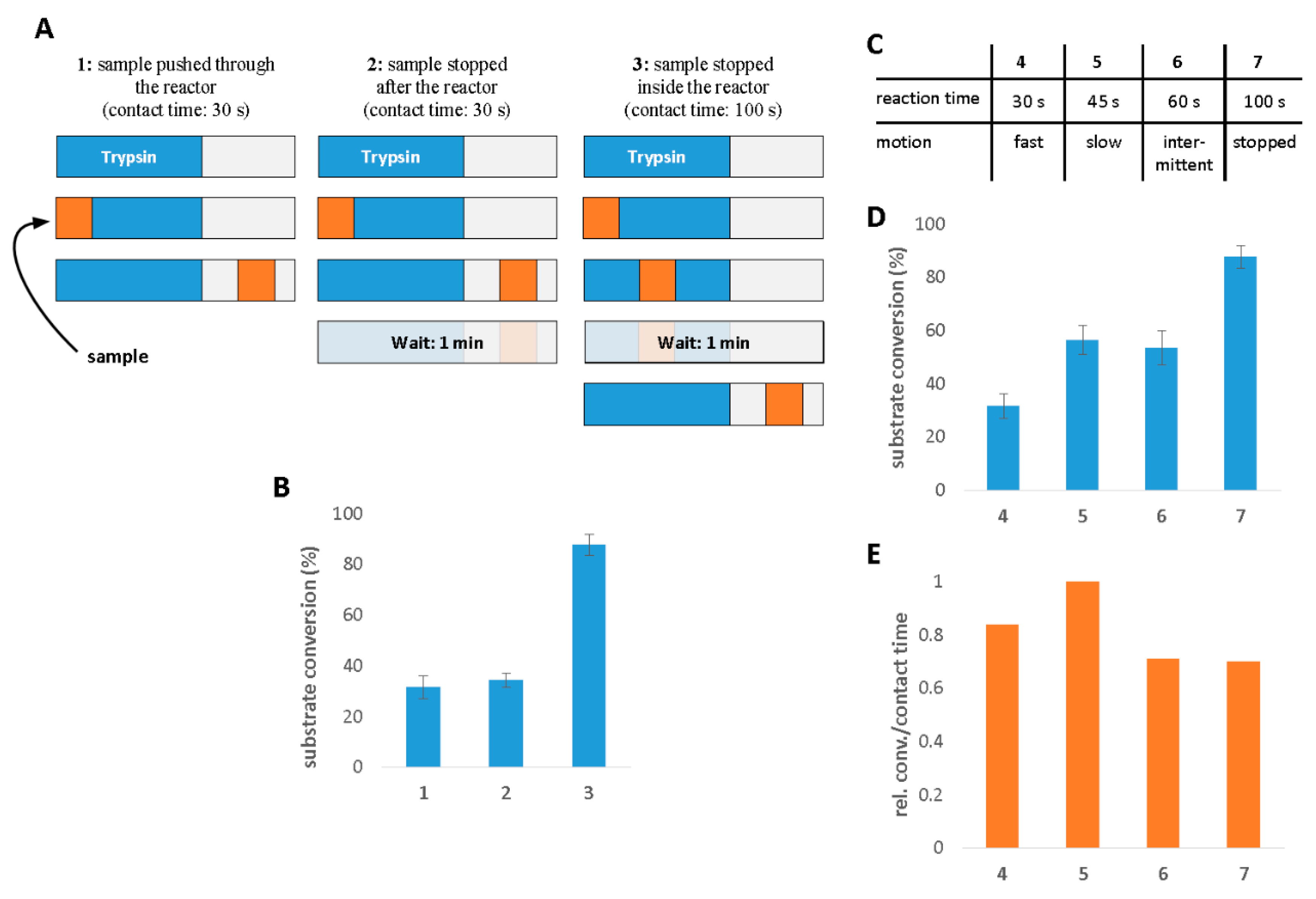
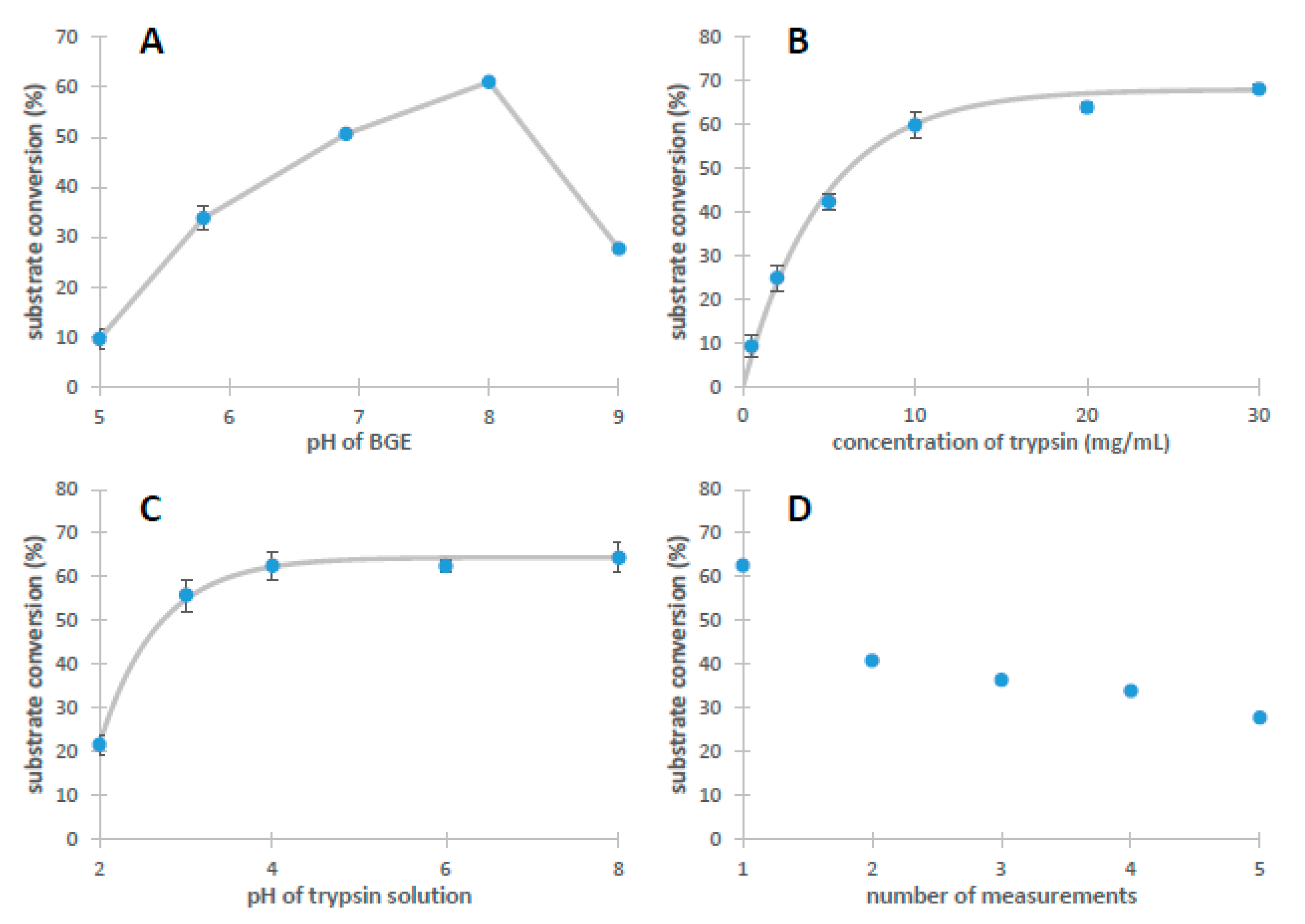
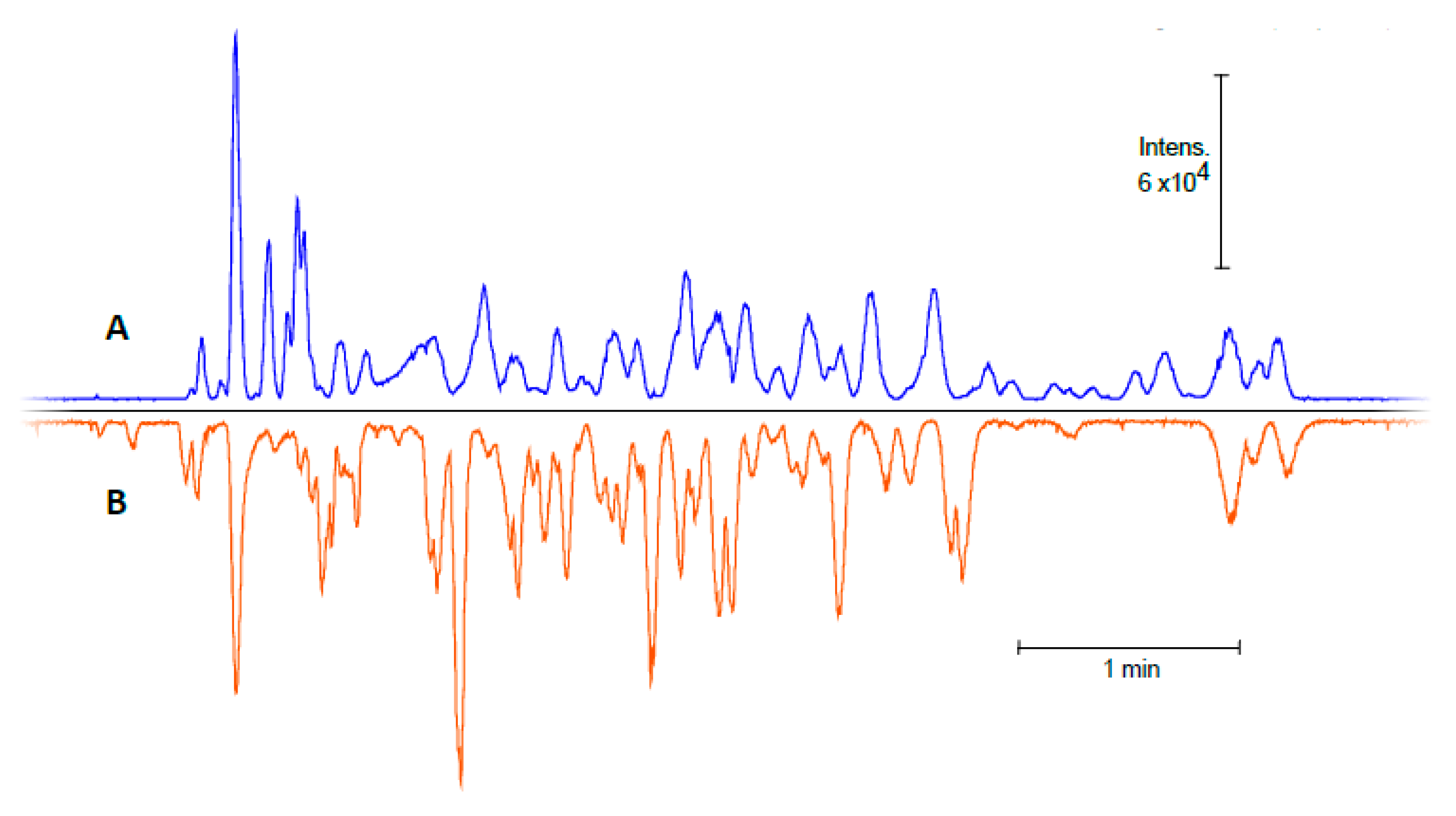
2.1. The Characterization of IMER Efficiency
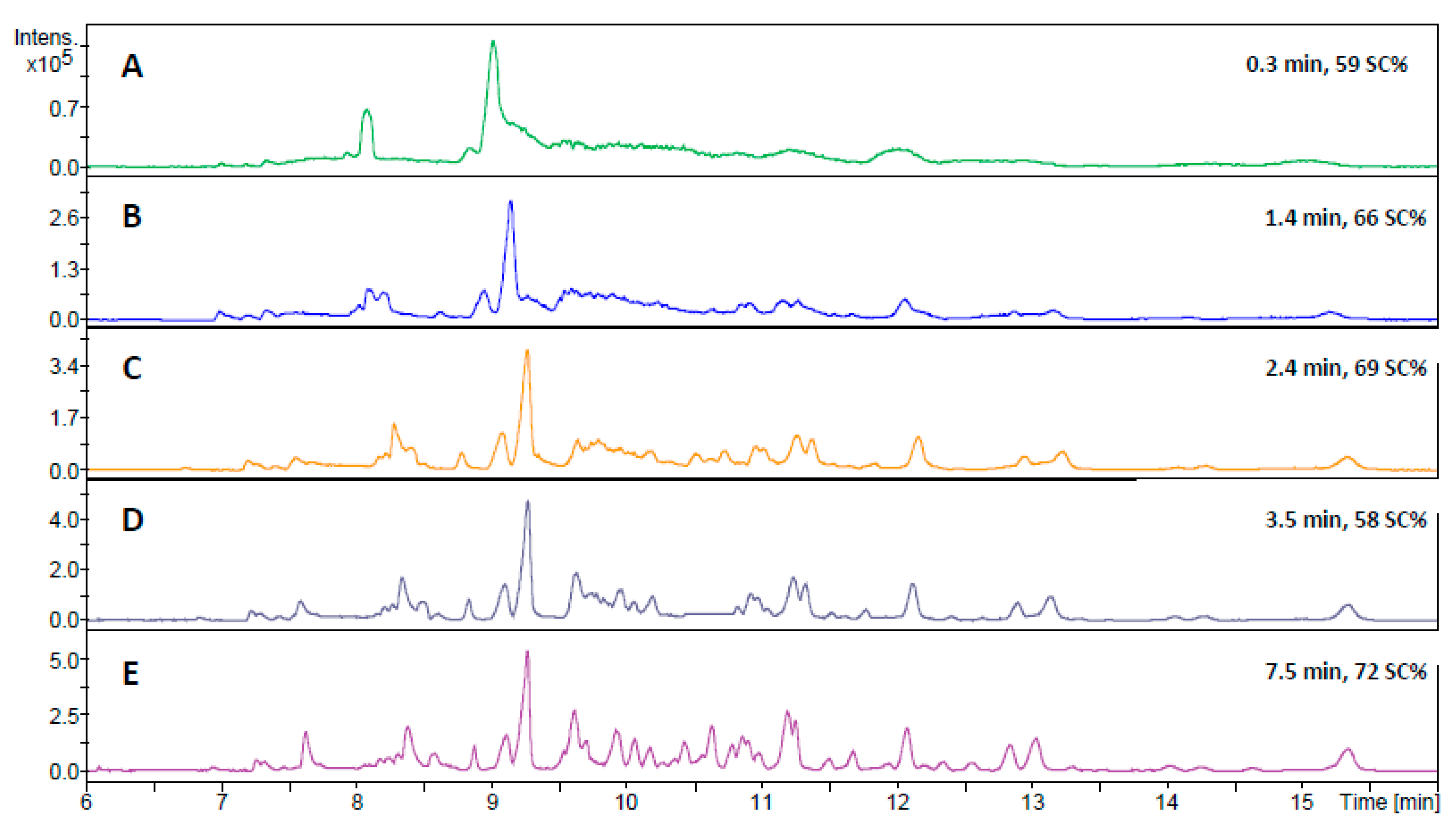
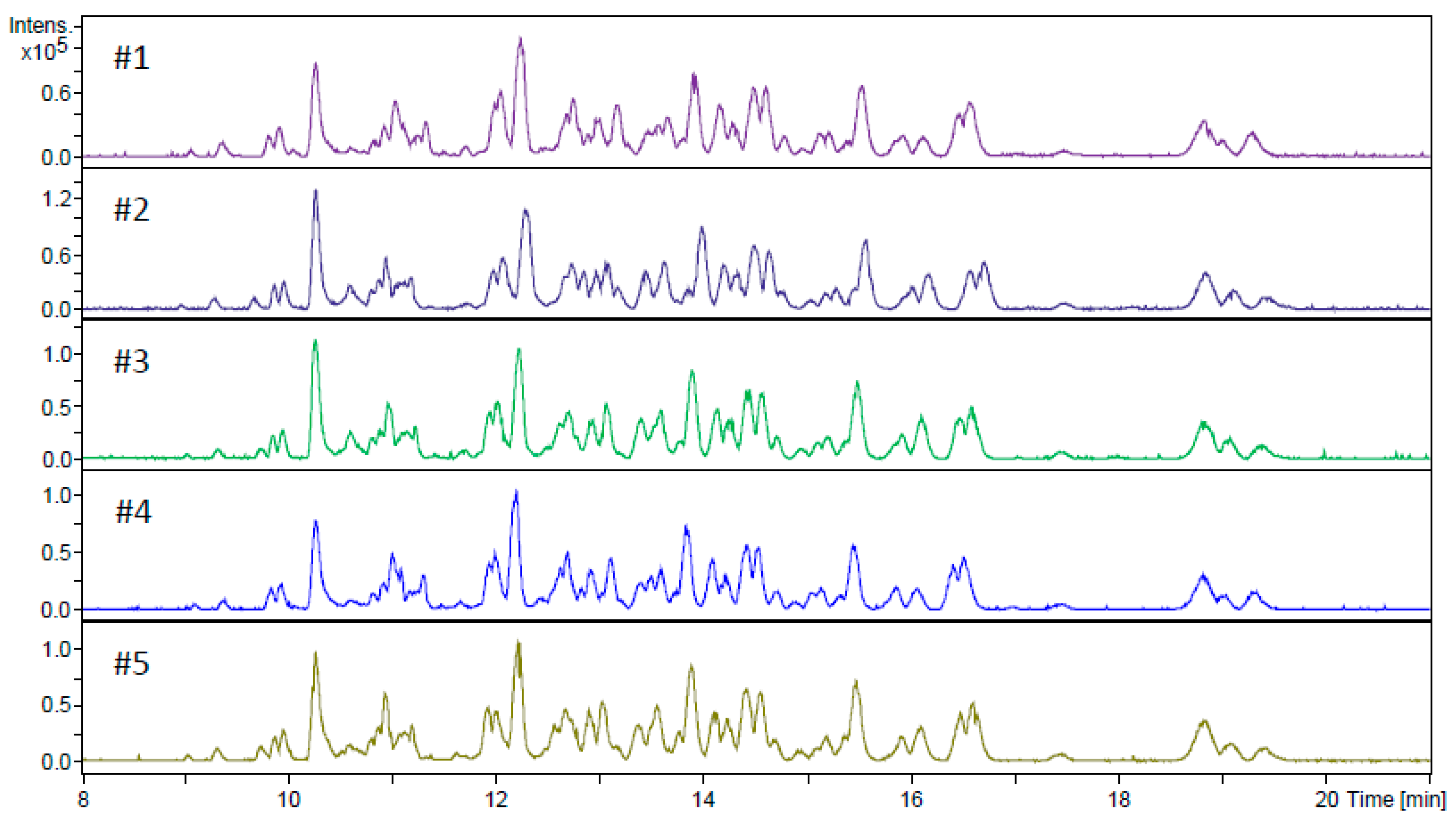
2.2. Digestion of HSA Using CE-MS
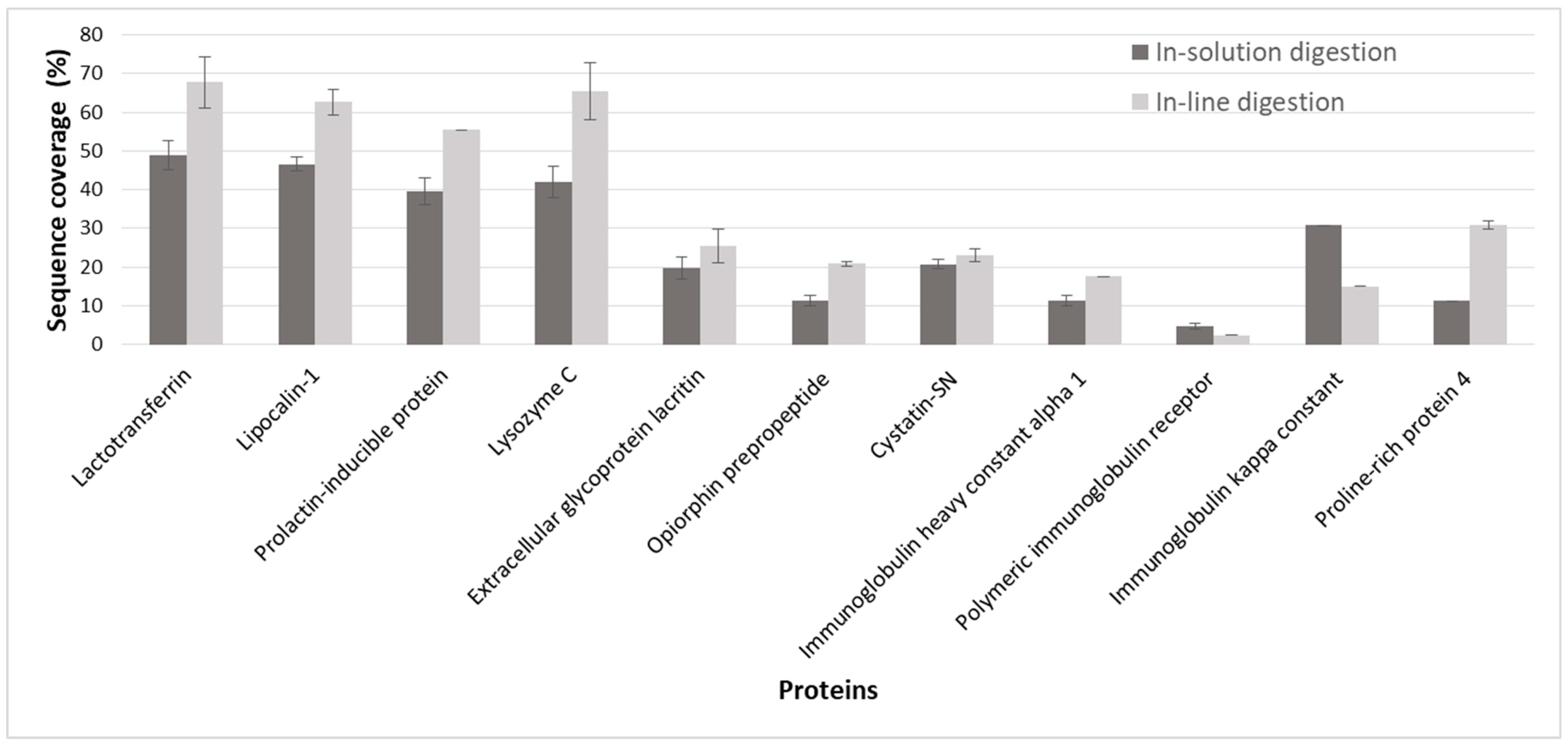
2.3. Application of the In-Line Microreactor for CE-MS
3. Materials and Methods
3.1. Reagents and Solutions
3.2. Instrumentation, Software
3.3. Preparation and Operation of the Microreactor in the CE Capillary
3.4. Operation of the μIMER-CE-MS System
3.5. CZE Separations
3.6. Enzymatic Digestion of Protein Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Washburn, M.P.; Wolters, D.; Yates, J.R. Large-Scale Analysis of the Yeast Proteome by Multidimensional Protein Identification Technology. Nat. Biotechnol. 2001, 19, 242–247. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, H.; Zhang, J.; Brasier, A.R.; Zhao, Y. Quantitative Assessment of the Effects of Trypsin Digestion Methods on Affinity Purification-Mass Spectrometry-Based Protein-Protein Interaction Analysis. J. Proteome Res. 2017, 16, 3068–3082. [Google Scholar] [CrossRef] [PubMed]
- Ehrfeld, W.; Hessel, V.; Löwe, H. Microreactors: New Technology for Modern Chemistry; Wiley-VCH: Weinheim, Germany, 2000. [Google Scholar]
- Kecskemeti, A.; Gaspar, A. Particle-Based Immobilized Enzymatic Reactors in Microfluidic Chips. Talanta 2018, 180, 211–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; Wang, W.H.; Tan, Y.J.; Bruening, M.L. Facile Trypsin Immobilization in Polymeric Membranes for Rapid, Efficient Protein Digestion. Anal. Chem. 2010, 82, 10045–10051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaspar, A.; Gomez, F.A. Application of Surface Plasmon Resonance Spectroscopy for Adsorption Studies of Different Types of Components on Poly(Dimethylsiloxane). Anal. Chim. Acta 2013, 777, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Křenková, J.; Klepárník, K.; Foret, F. Capillary Electrophoresis Mass Spectrometry Coupling with Immobilized Enzyme Electrospray Capillaries. J. Chromatogr. A 2007, 1159, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Amankwa, L.N.; Kuhr, W.G. Trypsin-Modified Fused-Silica Capillary Microreactor for Peptide Mapping by Capillary Zone Electrophoresis. Anal. Chem. 1992, 64, 1610–1613. [Google Scholar] [CrossRef]
- Kecskemeti, A.; Bako, J.; Csarnovics, I.; Csosz, E.; Gaspar, A. Development of an Enzymatic Reactor Applying Spontaneously Adsorbed Trypsin on the Surface of a PDMS Microfluidic Device. Anal. Bioanal. Chem. 2017, 409, 3573–3585. [Google Scholar] [CrossRef] [PubMed]
- Nagy, C.; Huszank, R.; Gaspar, A. Study of the Geometry of Open Channels in a Layer-Bed-Type Microfluidic Immobilized Enzyme Reactor. Anal. Bioanal. Chem. 2021. [Google Scholar] [CrossRef] [PubMed]
- Suelter, C.H.; DeLuca, M. How to Prevent Losses of Protein by Adsorption to Glass and Plastic. Anal. Biochem. 1983, 135, 112–119. [Google Scholar] [CrossRef]
- Liu, X.; Yang, J.; Yang, L. Capillary Electrophoresis-Integrated Immobilized Enzyme Reactors. Rev. Anal. Chem. 2016, 35, 115–131. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, Z.Y.Y.; Wang, Y.Z.Z.; Zhou, D.-D.D.; Yang, F.Q.Q.; Li, D.Q.Q. On-Line Immobilized Trypsin Microreactor for Evaluating Inhibitory Activity of Phenolic Acids by Capillary Electrophoresis and Molecular Docking. Food Chem. 2020, 310, 125823. [Google Scholar] [CrossRef] [PubMed]
- Nicoli, R.; Gaud, N.; Stella, C.; Rudaz, S.; Veuthey, J.L. Trypsin Immobilization on Three Monolithic Disks for On-Line Protein Digestion. J. Pharm. Biomed. Anal. 2008, 48, 398–407. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Tait, N.; Regnier, F. Fabrication of Nanocolumns for Liquid Chromatography. Anal. Chem. 1998, 70, 3790–3797. [Google Scholar] [CrossRef] [PubMed]
- De Malsche, W.; Eghbali, H.; Clicq, D.; Vangelooven, J.; Gardeniers, H.; Desmet, G. Pressure-Driven Reverse-Phase Liquid Chromatography Separations in Ordered Nonporous Pillar Array Columns. Anal. Chem. 2007, 79, 5915–5926. [Google Scholar] [CrossRef]
- Nagy, C.; Kecskemeti, A.; Gaspar, A. Fabrication of Immobilized Enzyme Reactors with Pillar Arrays into Polydimethylsiloxane Microchip. Anal. Chim. Acta 2020, 1108, 70–78. [Google Scholar] [CrossRef]
- Licklider, L.; Kuhr, W.G. Optimization of On-Line Peptide Mapping by Capillary Zone Electrophoresis. Anal. Chem. 1994, 66, 4400–4407. [Google Scholar] [CrossRef]
- Duan, J.; Liang, Z.; Yang, C.; Zhang, J.; Zhang, L.; Zhang, W.; Zhang, Y. Rapid Protein Identification Using Monolithic Enzymatic Microreactor and LC-ESI-MS/MS. Proteomics 2006, 6, 412–419. [Google Scholar] [CrossRef]
- Gao, M.; Zhang, P.; Hong, G.; Guan, X.; Yan, G.; Deng, C.; Zhang, X. Novel Monolithic Enzymatic Microreactor Based on Single-Enzyme Nanoparticles for Highly Efficient Proteolysis and Its Application in Multidimensional Liquid Chromatography. J. Chromatogr. A 2009, 1216, 7472–7477. [Google Scholar] [CrossRef]
- Meller, K.; Pomastowski, P.; Szumski, M.; Buszewski, B. Preparation of an Improved Hydrophilic Monolith to Make Trypsin-Immobilized Microreactors. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1043, 128–137. [Google Scholar] [CrossRef]
- Peterson, D.S.; Rohr, T.; Svec, F.; Fréchet, J.M.J. High-Throughput Peptide Mass Mapping Using a Microdevice Containing Trypsin Immobilized on a Porous Polymer Monolith Coupled to MALDI TOF and ESI TOF Mass Spectrometers. J. Proteome Res. 2002, 1, 563–568. [Google Scholar] [CrossRef]
- Wu, N.; Wang, S.; Yang, Y.; Song, J.; Su, P.; Yang, Y. DNA-Directed Trypsin Immobilization on a Polyamidoamine Dendrimer-Modified Capillary to Form a Renewable Immobilized Enzyme Microreactor. Int. J. Biol. Macromol. 2018, 113, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhu, X.; Camara, M.A.; Qu, Q.; Shan, Y.; Yang, L. Surface Modification with Highly-Homogeneous Porous Silica Layer for Enzyme Immobilization in Capillary Enzyme Microreactors. Talanta 2019, 197, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Sakai-Kato, K.; Kato, M.; Toyo’oka, T. On-Line Trypsin-Encapsulated Enzyme Reactor by the Sol-Gel Method Integrated into Capillary Electrophoresis. Anal. Chem. 2002, 74, 2943–2949. [Google Scholar] [CrossRef] [PubMed]
- Bossi, A.; Guizzardi, L.; D’Acunto, M.R.; Righetti, P.G. Controlled Enzyme-Immobilisation on Capillaries for Microreactors for Peptide Mapping. Anal. Bioanal. Chem. 2004, 378, 1722–1728. [Google Scholar] [CrossRef]
- Xu, X.; Wang, X.; Liu, Y.; Liu, B.; Wu, H.; Yang, P. Trypsin Entrapped in Poly(Diallyldimethylammonium Chloride) Silica Sol-Gel Microreactor Coupled to Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry. Rapid Commun. Mass Spectrom. 2008, 22, 1257–1264. [Google Scholar] [CrossRef] [PubMed]
- Kecskemeti, A.; Nagy, C.; Csosz, E.; Kallo, G.; Gaspar, A. The Application of a Microfluidic Reactor Including Spontaneously Adsorbed Trypsin for Rapid Protein Digestion of Human Tear Samples. Proteom. Clin. Appl. 2017, 11, 1700055. [Google Scholar] [CrossRef] [PubMed]
- Berta, A. Collection of Tear Samples with or without Stimulation. Am. J. Ophthalmol. 1983, 96, 115–116. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagy, C.; Szabo, R.; Gaspar, A. Development of an In-Line Enzyme Reactor Integrated into a Capillary Electrophoresis System. Molecules 2021, 26, 5902. https://doi.org/10.3390/molecules26195902
Nagy C, Szabo R, Gaspar A. Development of an In-Line Enzyme Reactor Integrated into a Capillary Electrophoresis System. Molecules. 2021; 26(19):5902. https://doi.org/10.3390/molecules26195902
Chicago/Turabian StyleNagy, Cynthia, Ruben Szabo, and Attila Gaspar. 2021. "Development of an In-Line Enzyme Reactor Integrated into a Capillary Electrophoresis System" Molecules 26, no. 19: 5902. https://doi.org/10.3390/molecules26195902