
On the Interactions of Melatonin/β-Cyclodextrin Inclusion Complex: A Novel Approach Combining Efficient Semiempirical Extended Tight-Binding (xTB) Results with Ab Initio Methods

 , , , , ,
, , , , ,
Abstract
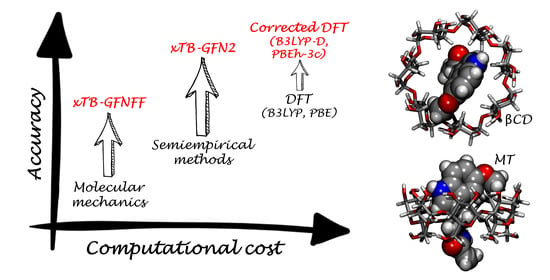
:
1. Introduction
2. Results and Discussion
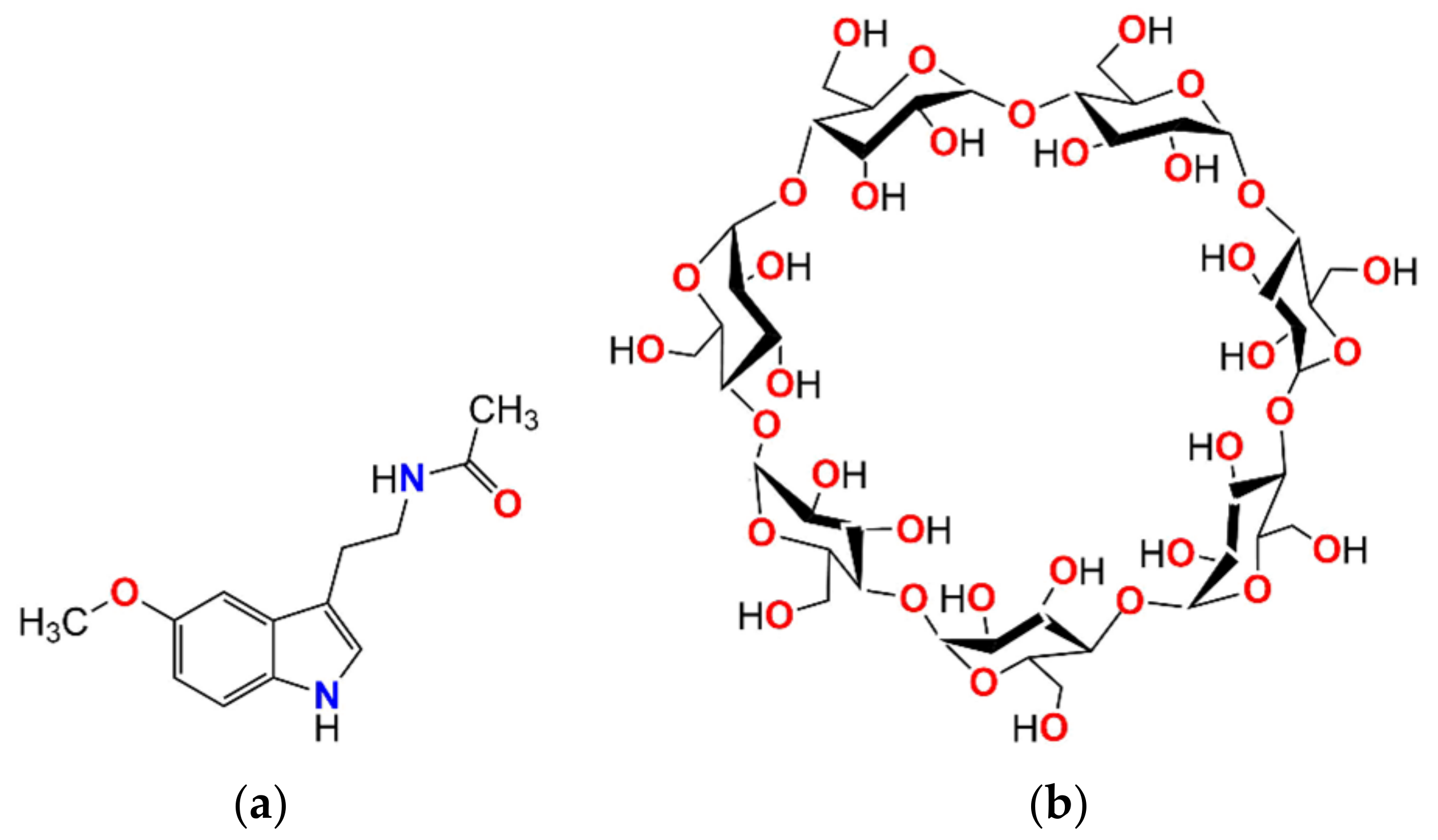
2.1. Melatonin: From the Isolated Molecule to the Crystal Structure
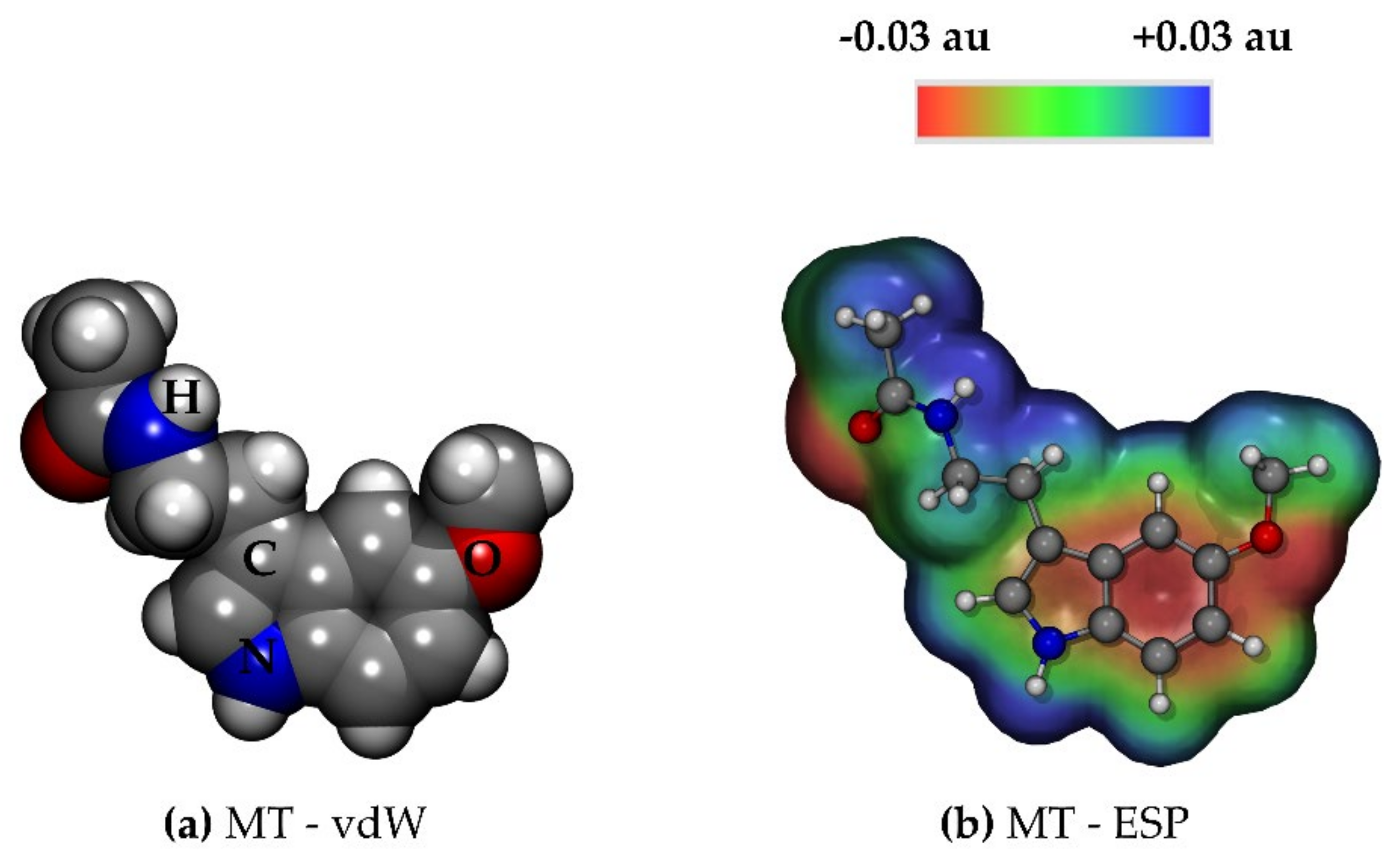
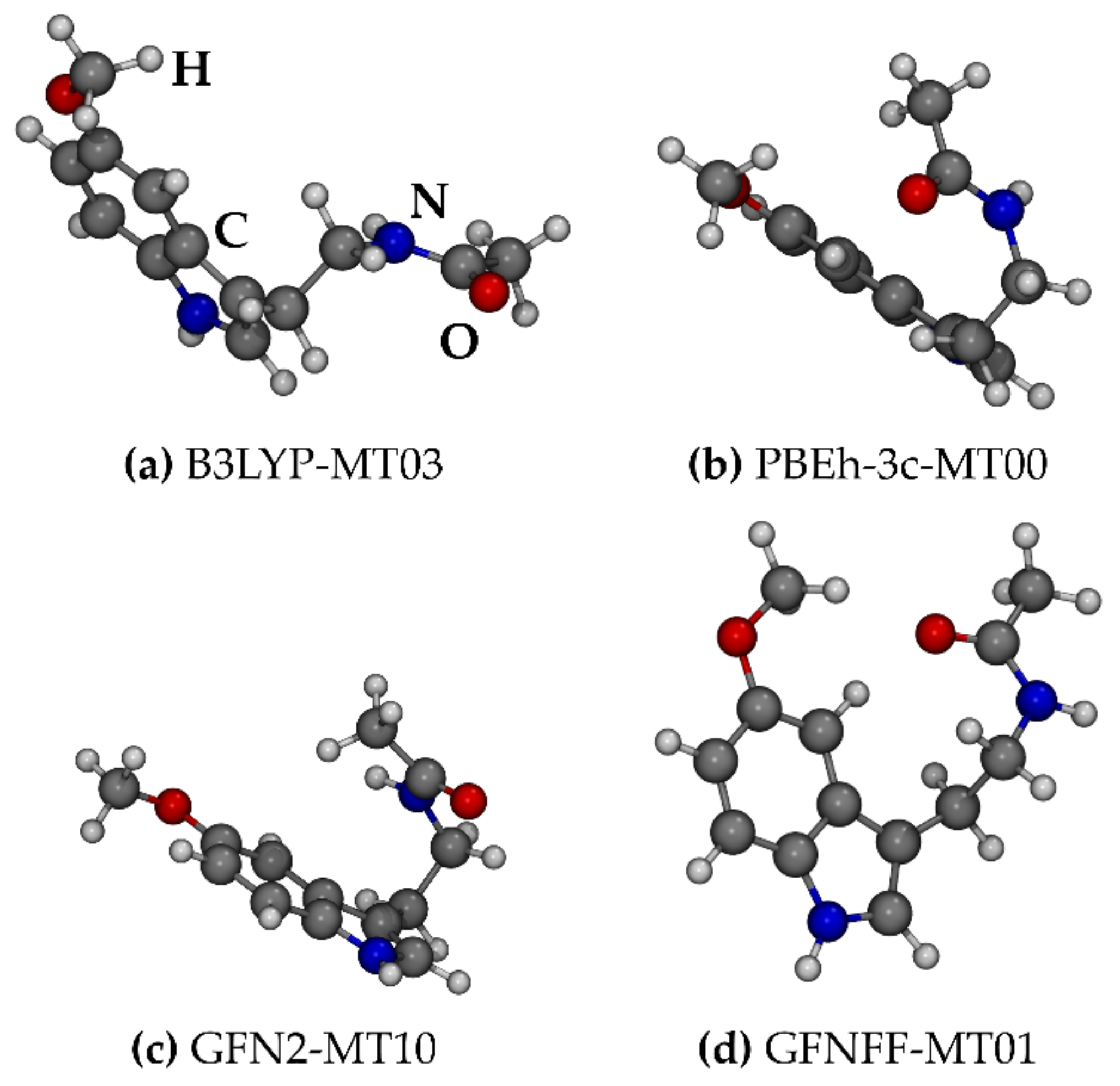
2.1.1. Melatonin: Conformational Analysis
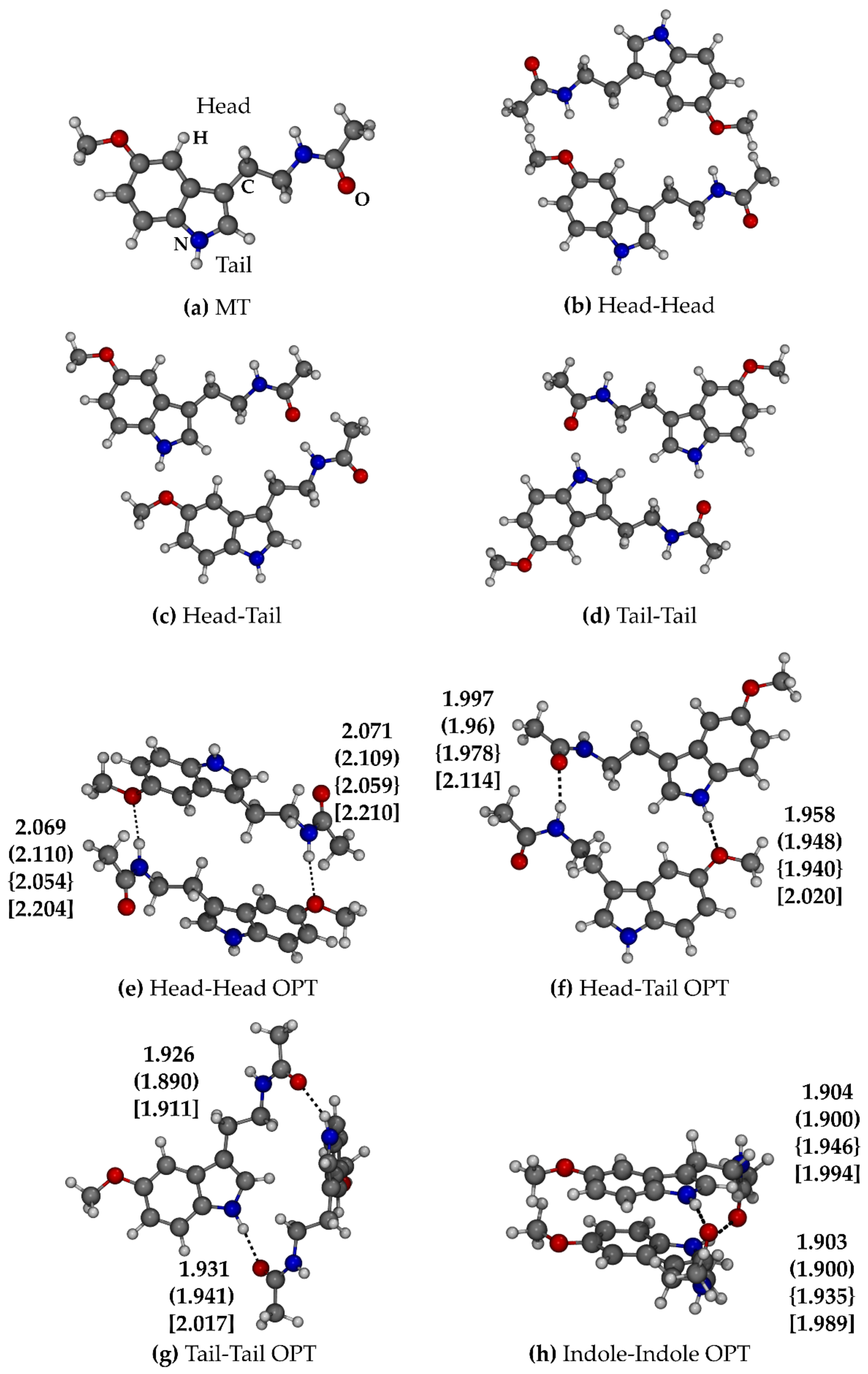
2.1.2. Melatonin: Dimer
- Head–Head (HH): Both the amidic N-H groups of the two melatonin molecules donate a hydrogen bond to the methoxy groups. The hydrogen bonds calculated by PBEh-3c are weaker (2.20–2.21 Å), and, accordingly, the interaction energy is lower (−2.0 kcal/mol, see Table 2) with respect to the other methods (2.05–2.11 Å, −4.0 < E < −7.5 kcal/mol of interaction energy).
- Head–Tail (HT): In this case, the indole -NH- donates a hydrogen bond to the methoxy group, while the amidic -NH- to the amidic -CO- (see Figure 3f). In this case, the structural (1.94–2.02 Å and 1.97–2.11 Å are the ranges of the two hydrogen bonds) and energetic accordance (−3.9 < E < −8.6 kcal/mol) among the methods is good. B3LYP shortens the hydrogen bonds and presents the highest interaction energy (−8.6 kcal/mol).
- Tail–Tail (TT): The hydrogen bond donors are the indole -NH- groups, while the acceptors are the amidic -CO- (see Figure 3g). The steric hindrance of -CH- and -CH2- groups led to a conformation with the two planes of the indoles in perpendicular orientation with one another. In this way, the two melatonin molecules can maximize their interactions; indeed, the hydrogen bonds are shorter (1.89–2.02 Å) with respect the previous cases, and the TT adduct is more stable with respect to both HH and HT (−9.0 < E < −11.3 kcal/mol). B3LYP is the only method which does not find a minimum in the structure described above, leading the two indole rings to interact with each other (Indole–Indole) through the π-electrons of the aromatic rings (π-π interaction), keeping the -NH∙∙∙OC- hydrogen bonds (see Figure 3h).
- Indole–Indole (II) was then reoptimized with the other methods for consistency. For all the methods, the hydrogen bonds are the shortest (1.90–1.99 Å); moreover, the π-π interaction leads to an extra-stabilization which makes this dimer the most stable one (−11.9 < E < −18.8 kcal/mol).
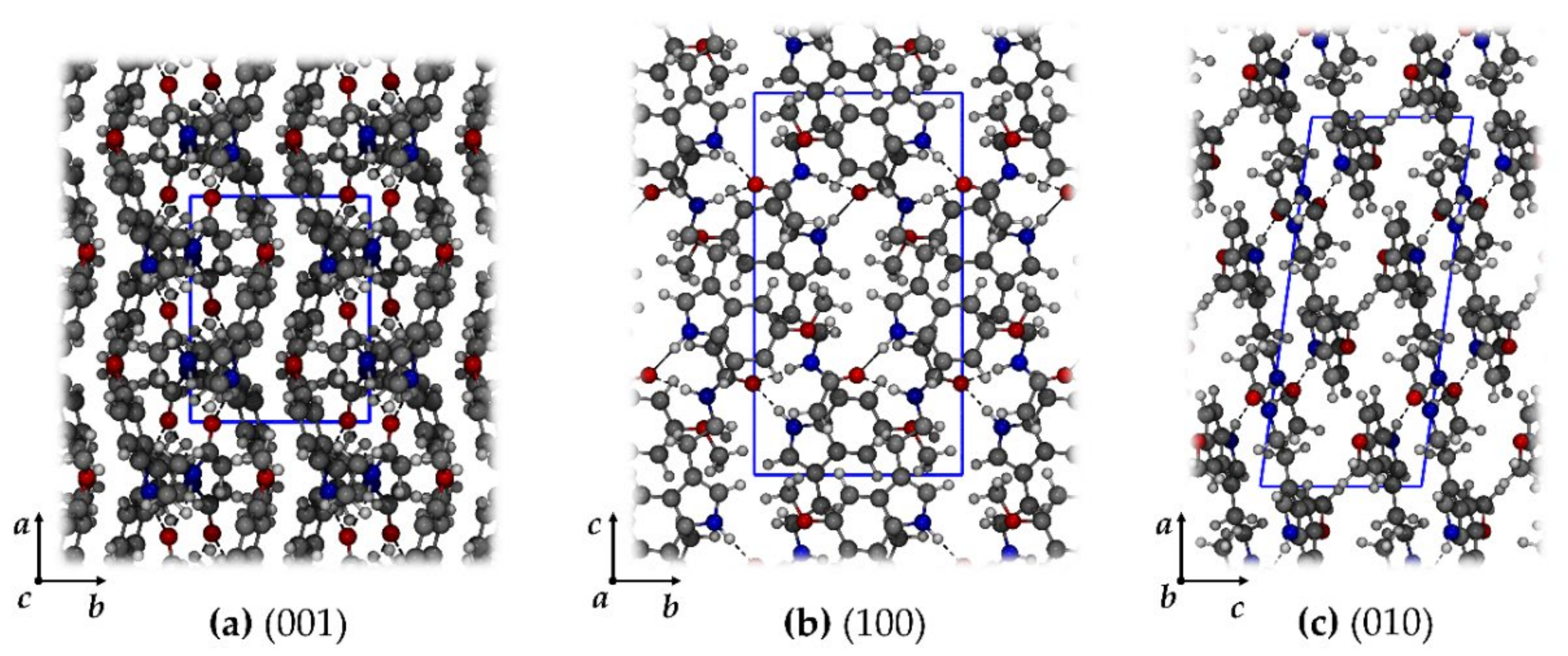
2.1.3. Melatonin: Crystal
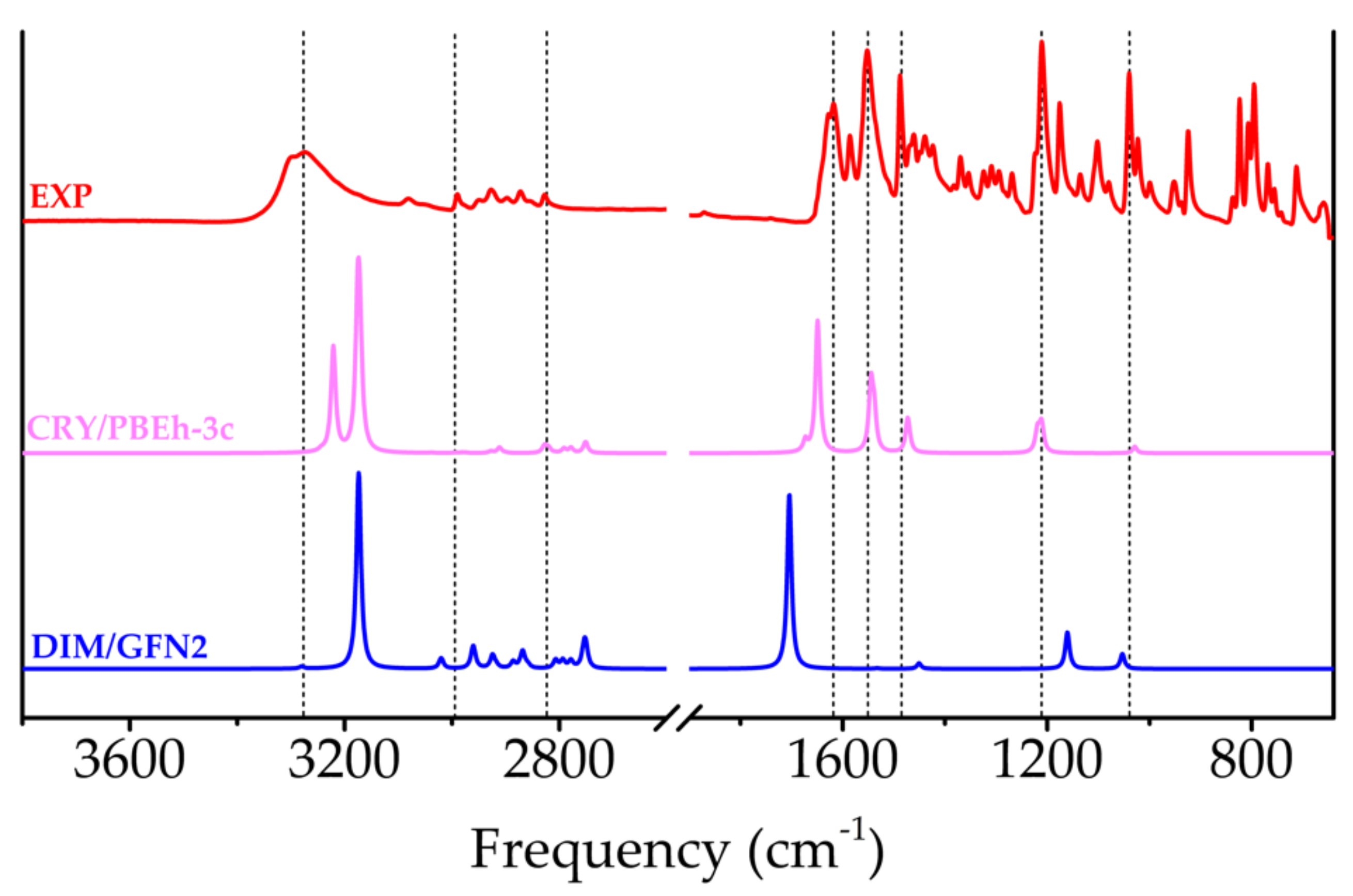
2.1.4. FTIR Spectroscopy
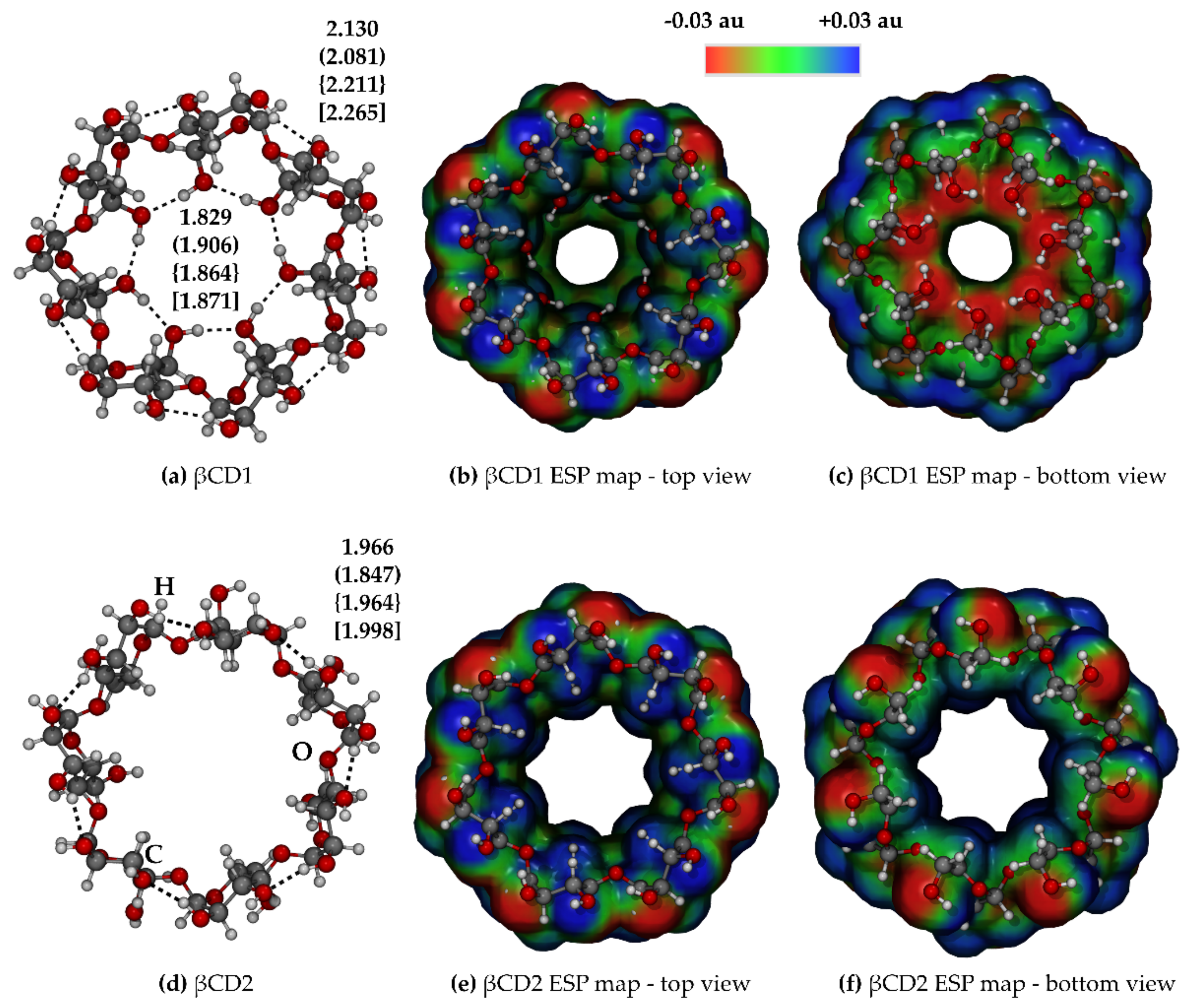
2.2. β-Cyclodextrin
2.3. Inclusion Complex
2.3.1. Extended Melatonin Inclusion Complex: Method Benchmark
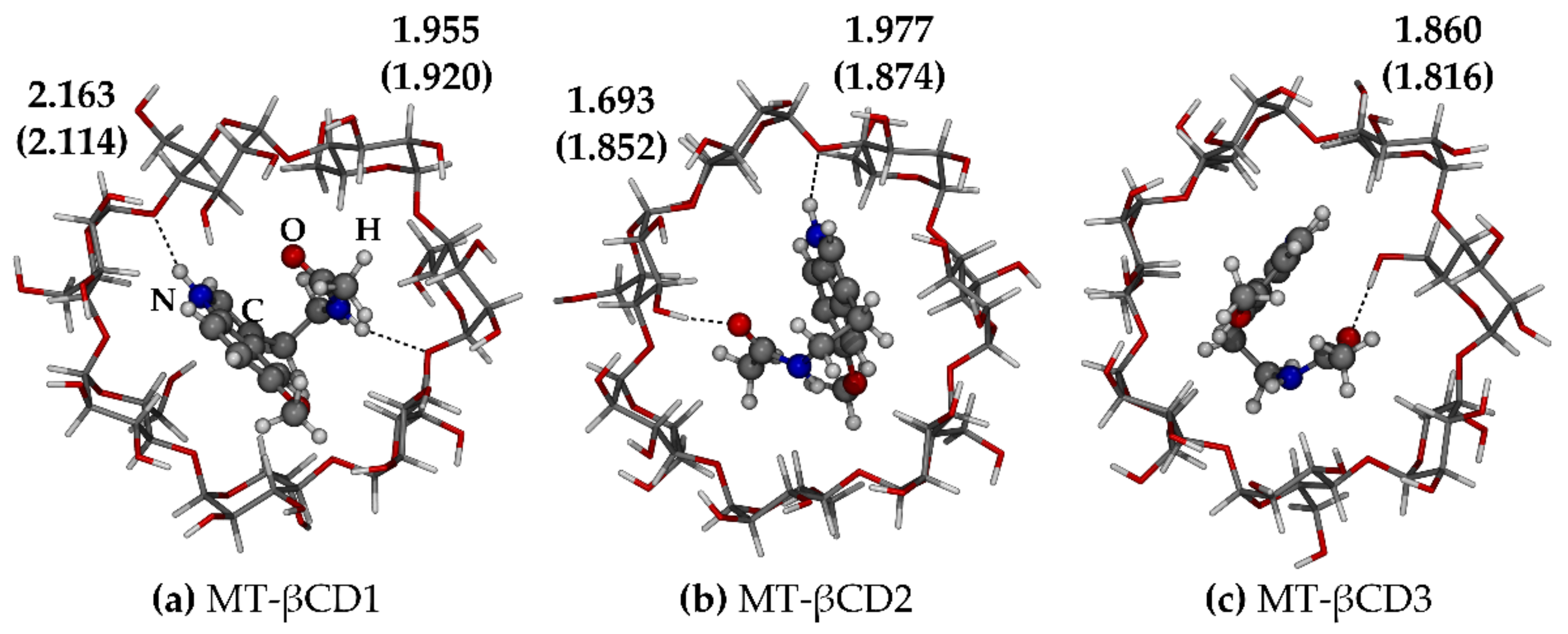
2.3.2. Melatonin/β-Cyclodextrin Complexes
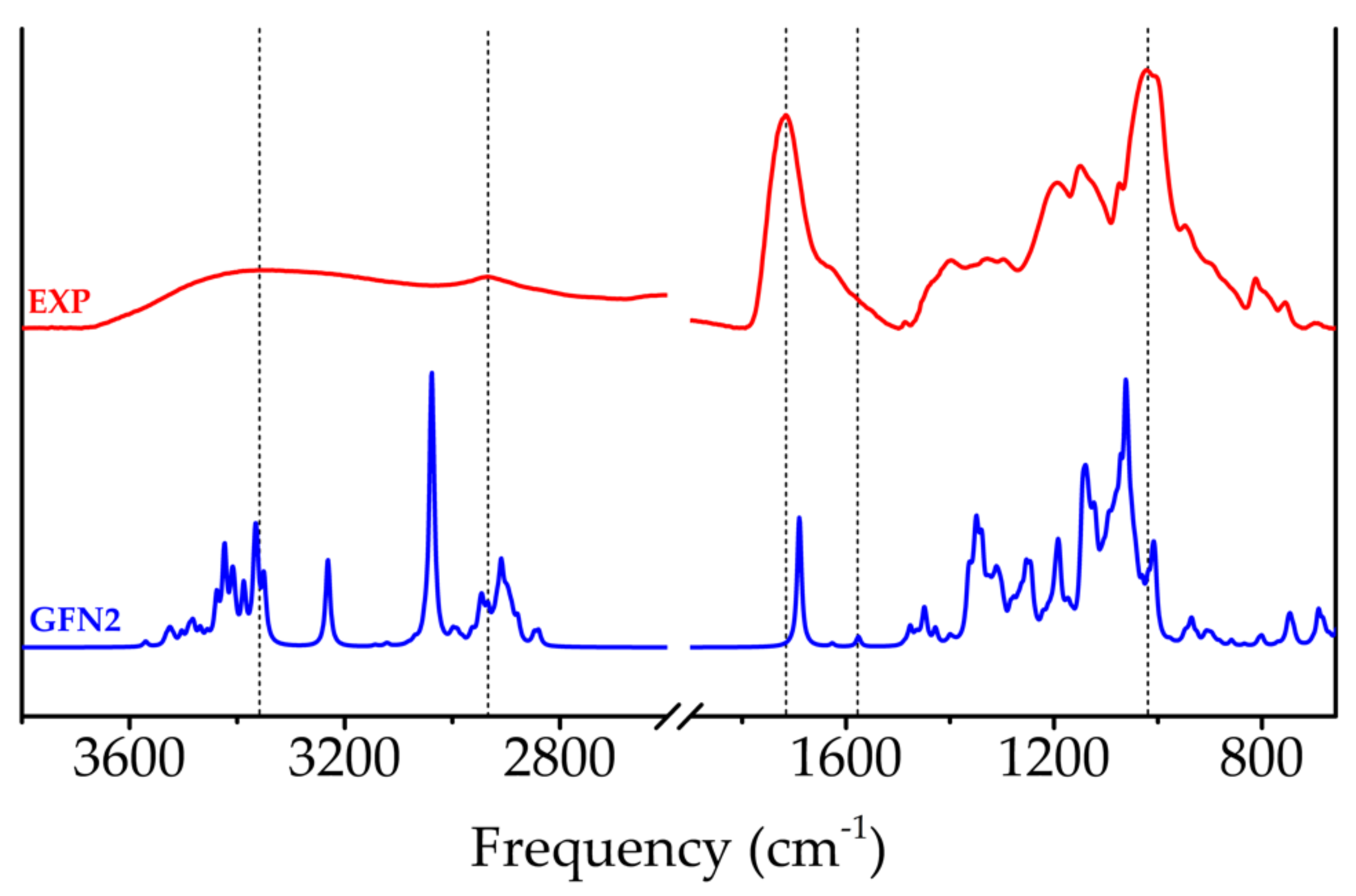
2.3.3. FTIR Spectroscopy
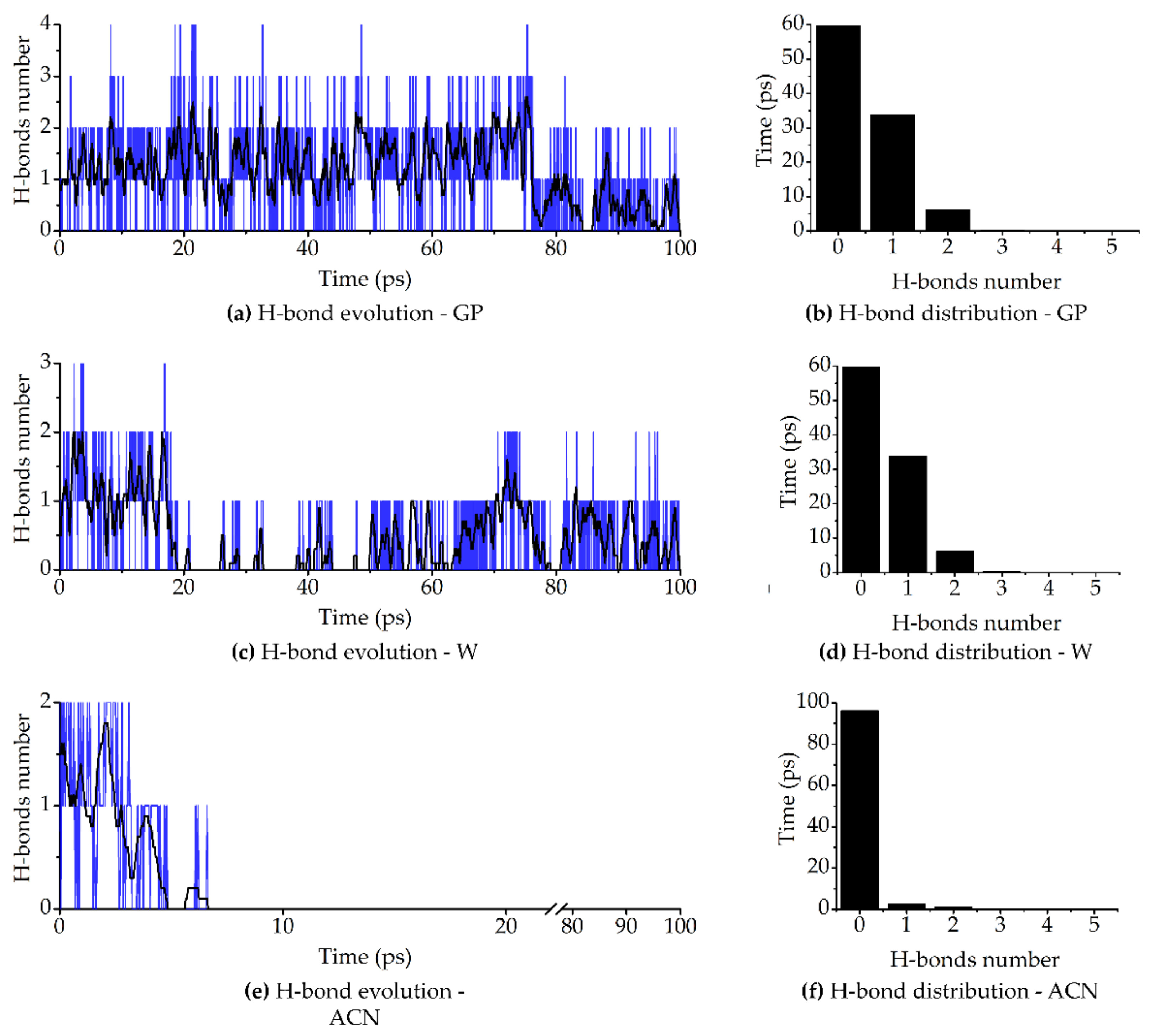
2.3.4. Molecular Dynamics Simulations
3. Materials and Methods
3.1. Computational Details
3.1.1. xTB
3.1.2. ORCA
3.1.3. CRYSTAL
3.1.4. Gaussian
3.2. Experimental Details
3.2.1. Synthesis of Inclusion Complex
3.2.2. FTIR Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nowak, J.Z.; Zawilska, J.B. Melatonin and its physiological and therapeutic properties. Pharm. World Sci. 1998, 20, 18–27. [Google Scholar] [CrossRef]
- Cipolla-Neto, J.; Do Amaral, F.G. Melatonin as a Hormone: New Physiological and Clinical Insights. Endocr. Rev. 2018, 39, 990–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, V.; Spence, D.W.; Pandi-Perumal, S.R.; Trakht, I.; Cardinali, D.P. Jet lag: Therapeutic use of melatonin and possible application of melatonin analogs. Travel Med. Infect. Dis. 2008, 6, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Pandi-Perumal, S.R.; Zisapel, N.; Srinivasan, V.; Cardinali, D.P. Melatonin and sleep in aging population. Exp. Gerontol. 2005, 40, 911–925. [Google Scholar] [CrossRef] [PubMed]
- Tordjman, S.; Chokron, S.; Delorme, R.; Charrier, A.; Bellissant, E.; Jaafari, N.; Fougerou, C.; del Valle Bessone, C.; Fajreldines, H.D.; de Barboza, G.E.D.; et al. Protective role of melatonin on retinal ganglionar cell: In vitro an in vivo evidences. Life Sci. 2019, 15, 1689–1699. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Rosales-Corral, S.; Galano, A.; Zhou, X.J.; Xu, B. Mitochondria: Central organelles for melatonins antioxidant and anti-Aging actions. Molecules 2018, 23, 509. [Google Scholar] [CrossRef] [Green Version]
- Chrustek, A.; Olszewska-Słonina, D. Melatonin as a powerful antioxidant. Acta Pharm. 2021, 71, 335–354. [Google Scholar] [CrossRef]
- Kostoglou-Athanassiou, I. Therapeutic applications of melatonin. Ther. Adv. Endocrinol. Metab. 2013, 4, 13–24. [Google Scholar] [CrossRef]
- Harpsøe, N.G.; Andersen, L.P.H.; Gögenur, I.; Rosenberg, J. Clinical pharmacokinetics of melatonin: A systematic review. Eur. J. Clin. Pharmacol. 2015, 71, 901–909. [Google Scholar] [CrossRef]
- Zafra-Roldán, A.; Corona-Avendaño, S.; Montes-Sánchez, R.; Palomar-Pardavé, M.; Romero-Romo, M.; Ramírez-Silva, M.T. New insights on the spectrophotometric determination of melatonin pKa values and melatonin-βCD inclusion complex formation constant. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 190, 442–449. [Google Scholar] [CrossRef]
- Bongiorno, D.; Ceraulo, L.; Mele, A.; Panzeri, W.; Selva, A.; Turco Liveri, V. Structural and physicochemical characterization of the inclusion complexes of cyclomaltooligosaccharides (cyclodextrins) with melatonin. Carbohydr. Res. 2002, 337, 743–754. [Google Scholar] [CrossRef]
- Topal, B.; Çetin Altindal, D.; Gümüsderelioglu, M. Melatonin/HPβCD complex: Microwave synthesis, integration with chitosan scaffolds and inhibitory effects on MG-63CELLS. Int. J. Pharm. 2015, 496, 801–811. [Google Scholar] [CrossRef]
- Vlachou, M.; Papamichael, M.; Siamidi, A.; Fragouli, I.; Afroudakis, P.A.; Kompogennitaki, R.; Dotsikas, Y. Comparative in vitro controlled release studies on the chronobiotic hormone melatonin from cyclodextrins-containing matrices and cyclodextrin: Melatonin complexes. Int. J. Mol. Sci. 2017, 18, 1641. [Google Scholar] [CrossRef] [Green Version]
- Del Valle, E.M.M. Cyclodextrins and their uses: A review. Process Biochem. 2004, 39, 1033–1046. [Google Scholar] [CrossRef]
- Jambhekar, S.S.; Breen, P. Cyclodextrins in pharmaceutical formulations I: Structure and physicochemical properties, formation of complexes, and types of complex. Drug Discov. Today 2016, 21, 356–362. [Google Scholar] [CrossRef]
- Connors, K.A. The stability of cyclodextrin complexes in solution. Chem. Rev. 1997, 97, 1325–1357. [Google Scholar] [CrossRef]
- Jansook, P.; Ogawa, N.; Loftsson, T. Cyclodextrins: Structure, physicochemical properties and pharmaceutical applications. Int. J. Pharm. 2018, 535, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Barbiric, D.J.; Castro, E.A.; De Rossi, R.H. A molecular mechanics study of 1:1 complexes between azobenzene derivatives and β-cyclodextrin. J. Mol. Struct. THEOCHEM 2000, 532, 171–181. [Google Scholar] [CrossRef]
- Nasongkla, N.; Wiedmann, A.F.; Bruening, A.; Beman, M.; Ray, D.; Bornmann, W.G.; Boothman, D.A.; Gao, J. Enhancement of Solubility and Bioavailability of β-Lapachone Using Cyclodextrin Inclusion Complexes. Pharm. Res. 2003, 20, 1626–1633. [Google Scholar] [CrossRef] [PubMed]
- Nora, M.; Ismahan, L.; Abdelkrim, G.; Mouna, C.; Leila, N.; Fatiha, M.; Nada, B.; Brahim, H. Interactions in inclusion complex of β-cyclodextrin/l-Metheonine: DFT computational studies. J. Incl. Phenom. Macrocycl. Chem. 2020, 96, 43–54. [Google Scholar] [CrossRef]
- Al-Jaber, A.S.; Bani-Yaseen, A.D. On the encapsulation of Olsalazine by β-cyclodextrin: A DFT-based computational and spectroscopic investigations. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 214, 531–536. [Google Scholar] [CrossRef]
- Paulino, P.H.S.; de Sousa, S.M.R.; Da Silva, H.C.; De Almeida, W.B.; Ferrari, J.L.; Guimarães, L.; Nascimento, C.S. A theoretical investigation on the encapsulation process of mepivacaine into β-cyclodextrin. Chem. Phys. Lett. 2020, 740, 137060. [Google Scholar] [CrossRef]
- Azayez, M.; Fergoug, T.; Meddah-Araibi, N.; Zelmat, C.; Bouhadda, Y. Theoretical investigation of the complexation reaction of procaine-hydrochloride by β-cyclodextrin. Phys. Chem. Res. 2020, 8, 155–165. [Google Scholar] [CrossRef]
- Zaboub, A.; Madi, F.; Merdes, R.; Mohamedi, M.; Nouar, L. A combined DFT and experimental study of proline/β-cyclodextrin inclusion complex. J. Mol. Liq. 2016, 216, 716–723. [Google Scholar] [CrossRef]
- Papezhuk, M.V.; Volynkin, V.A.; Stroganova, T.A.; Krapivin, G.D.; Usacheva, T.R.; Thi, L.P. Theoretical and experimental study of inclusion complex formation of β-cyclodextrin with some 1,4-diazepine derivatives. Macroheterocycles 2020, 13, 64–73. [Google Scholar] [CrossRef]
- De Jesus, M.B.; de Matos Alves Pinto, L.; Fraceto, L.F.; Takahata, Y.; Lino, A.C.S.; Jaime, C.; de Paula, E. Theoretical and experimental study of a praziquantel and β-cyclodextrin inclusion complex using molecular mechanic calculations and 1H-nuclear magnetic resonance. J. Pharm. Biomed. Anal. 2006, 41, 1428–1432. [Google Scholar] [CrossRef] [PubMed]
- Abou-Okeil, A.; Rehan, M.; El-Sawy, S.M.; El-bisi, M.K.; Ahmed-Farid, O.A.; Abdel-Mohdy, F.A. Lidocaine/β-cyclodextrin inclusion complex as drug delivery system. Eur. Polym. J. 2018, 108, 304–310. [Google Scholar] [CrossRef]
- Cervelló, E.; Mazzucchi, F.; Jaime, C. Molecular mechanics and molecular dynamics calculations of the β-cyclodextrin inclusion complexes with m-, and p-nitrophenyl alkanoates. J. Mol. Struct. THEOCHEM 2000, 530, 155–163. [Google Scholar] [CrossRef]
- Rodrigues, S.G.; Chaves, I.D.S.; De Melo, N.F.S.; De Jesus, M.B.; Fraceto, L.F.; Fernandes, S.A.; De Paula, E.; De Freitas, M.P.; Pinto, L.D.M.A. Computational analysis and physico-chemical characterization of an inclusion compound between praziquantel and methyl-β-cyclodextrin for use as an alternative in the treatment of schistosomiasis. J. Incl. Phenom. Macrocycl. Chem. 2011, 70, 19–28. [Google Scholar] [CrossRef]
- Raffaini, G.; Ganazzoli, F. Understanding Surface Interaction and Inclusion Complexes between Piroxicam and Native or Crosslinked β-Cyclodextrins: The Role of Drug Concentration. Molecules 2020, 25, 2848. [Google Scholar] [CrossRef] [PubMed]
- Raffaini, G.; Ganazzoli, F. Molecular dynamics study of host-guest interactions in cyclodextrins: Methodology and data analysis for a comparison with solution data and the solid-state structure. J. Incl. Phenom. Macrocycl. Chem. 2007, 57, 683–688. [Google Scholar] [CrossRef]
- Fronza, G.; Mele, A.; Redenti, E.; Ventura, P. 1H NMR and molecular modeling study on the inclusion complex β-cyclodextrin-indomethacin. J. Org. Chem. 1996, 61, 909–914. [Google Scholar] [CrossRef]
- Zubiaur, M.; De Federico, M.; Burusco, K.K.; Beá, I.; Virgili, A.; Sánchez-Ferrando, F.; Jaime, C. Experimental (NMR) and theoretical (MD simulations) studies on the conformational preference of three cycloalkanols when included in β-cyclodextrin. J. Incl. Phenom. 2005, 51, 241–247. [Google Scholar] [CrossRef]
- Zubiaur, M.; Jaime, C. Complexation between tert-butyl ketones and β-cyclodextrin. Structural study by NMR and MD simulations. J. Org. Chem. 2000, 65, 8139–8145. [Google Scholar] [CrossRef] [PubMed]
- Raffaini, G.; Ganazzoli, F.; Malpezzi, L.; Fuganti, C.; Fronza, G.; Panzeri, W.; Mele, A. Validating a strategy for molecular dynamics simulations of cyclodextrin inclusion complexes through single-crystal x-ray and NMR experimental data: A case study. J. Phys. Chem. B 2009, 113, 9110–9122. [Google Scholar] [CrossRef]
- Castiglione, F.; Ganazzoli, F.; Malpezzi, L.; Mele, A.; Panzeri, W.; Raffaini, G. Inclusion complexes of β-cyclodextrin with tricyclic drugs: An X-ray diffraction, NMR and molecular dynamics study. Beilstein J. Org. Chem. 2017, 13, 714–719. [Google Scholar] [CrossRef] [Green Version]
- Matei, I.; Nicolae, A.; Hillebrand, M. Fluorimetric and molecular mechanics study of the inclusion complex of 2-quinoxalinyl-phenoxathiin with β-cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 2007, 57, 597–601. [Google Scholar] [CrossRef]
- Pan, W.; Zhang, D.; Zhan, J. Theoretical investigation on the inclusion of TCDD with β-cyclodextrin by performing QM calculations and MD simulations. J. Hazard. Mater. 2011, 192, 1780–1786. [Google Scholar] [CrossRef]
- Benghodbane, S.; Khatmi, D. Quantum chemical calculations based on ONIOM and the DFT methods in the inclusion complex: Doxycycline/2-O-Me-β-cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 2013, 77, 231–240. [Google Scholar] [CrossRef]
- Bouchemela, H.; Madi, F.; Nouar, L. DFT investigation of host–guest interactions between α-Terpineol and β-cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 2019, 95, 247–258. [Google Scholar] [CrossRef]
- Li, W.; Lu, B.; Sheng, A.; Yang, F.; Wang, Z. Spectroscopic and theoretical study on inclusion complexation of beta-cyclodextrin with permethrin. J. Mol. Struct. 2010, 981, 194–203. [Google Scholar] [CrossRef]
- Christoforides, E.; Fourtaka, K.; Andreou, A.; Bethanis, K. X-ray crystallography and molecular dynamics studies of the inclusion complexes of geraniol in β-cyclodextrin, heptakis (2,6-di-O-methyl)-β-cyclodextrin and heptakis (2,3,6-tri-O-methyl)-β-cyclodextrin. J. Mol. Struct. 2020, 1202, 127350. [Google Scholar] [CrossRef]
- Khavani, M.; Kalantarinezhad, R.; Izadyar, M. A joint QM/MD study on -, β- and -cyclodextrins in selective complexation with cathinone. Supramol. Chem. 2018, 30, 687–696. [Google Scholar] [CrossRef]
- Bohle, F.; Grimme, S. Efficient structural and energetic screening of fullerene encapsulation in a large supramolecular double decker macrocycle. J. Serbian Chem. Soc. 2019, 84, 837–844. [Google Scholar] [CrossRef] [Green Version]
- Platts, J.A. Quantum chemical molecular dynamics and metadynamics simulation of aluminium binding to amyloid-β and related peptides. R. Soc. Open Sci. 2020, 7, 191562. [Google Scholar] [CrossRef] [Green Version]
- Boz, E.; Stein, M. Accurate receptor-ligand binding free energies from fast qm conformational chemical space sampling. Int. J. Mol. Sci. 2021, 22, 3078. [Google Scholar] [CrossRef]
- Fogueri, U.R.; Kozuch, S.; Karton, A.; Martin, J.M.L. The melatonin conformer space: Benchmark and assessment of wave function and DFT methods for a paradigmatic biological and pharmacological molecule. J. Phys. Chem. A 2013, 117, 2269–2277. [Google Scholar] [CrossRef]
- Stachowicz, A.; Styrcz, A.; Korchowiec, J.; Modaressi, A.; Rogalski, M. DFT studies of cation binding by β-cyclodextrin. Theor. Chem. Acc. 2011, 130, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Ugliengo, P.; Viterbo, D.; Chiari, G. MOLDRAW: Molecular graphics on a personal computer. Zeitschrift Kristallogr. Cryst. Struct. 1993, 207, 9–23. [Google Scholar] [CrossRef]
- Mostad, A.; Romming, C. The crystal and molecular structure of N-acetyl-5-methoxy-tryptamine (melatonin). Acta Chem. Scand. B. 1974, 28, 564–572. [Google Scholar] [CrossRef] [Green Version]
- Ramos, A.I.; Braga, T.M.; Silva, P.; Fernandes, J.A.; Ribeiro-Claro, P.; De Fátima Silva Lopes, M.; Paz, F.A.A.; Braga, S.S. Chloramphenicol·cyclodextrin inclusion compounds: Co-dissolution and mechanochemical preparations and antibacterial action. CrystEngComm 2013, 15, 2822–2834. [Google Scholar] [CrossRef]
- Bannwarth, C.; Caldeweyher, E.; Ehlert, S.; Hansen, A.; Pracht, P.; Seibert, J.; Spicher, S.; Grimme, S. Extended tight-binding quantum chemistry methods. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2021, 11, 1–49. [Google Scholar] [CrossRef]
- Grimme, S.; Bannwarth, C.; Shushkov, P. A Robust and Accurate Tight-Binding Quantum Chemical Method for Structures, Vibrational Frequencies, and Noncovalent Interactions of Large Molecular Systems Parametrized for All spd-Block Elements (Z = 1-86). J. Chem. Theory Comput. 2017, 13, 1989–2009. [Google Scholar] [CrossRef]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef] [Green Version]
- Spicher, S.; Grimme, S. Robust Atomistic Modeling of Materials, Organometallic, and Biochemical Systems. Angew. Chem. 2020, 132, 15795–15803. [Google Scholar] [CrossRef]
- Ehlert, S.; Stahn, M.; Spicher, S.; Grimme, S. Robust and Efficient Implicit Solvation Model for Fast Semiempirical Methods. J. Chem. Theory Comput. 2021, 17, 4250–4261. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 24103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Axilrod, B.M.; Teller, E. Interaction of the van der Waals type between three atoms. J. Chem. Phys. 1943, 11, 299–300. [Google Scholar] [CrossRef]
- Muto, Y. The Force Between Nonpolar Molecules. Proc. Phys. Math. Soc. Jpn. 1944, 17, 629–631. [Google Scholar]
- Brandenburg, J.G.; Alessio, M.; Civalleri, B.; Peintinger, M.F.; Bredow, T.; Grimme, S. Geometrical correction for the inter- and intramolecular basis set superposition error in periodic density functional theory calculations. J. Phys. Chem. A 2013, 117, 9282–9292. [Google Scholar] [CrossRef]
- Kruse, H.; Grimme, S. A geometrical correction for the inter- and intra-molecular basis set superposition error in Hartree-Fock and density functional theory calculations for large systems. J. Chem. Phys. 2012, 136, 154101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical condensed matter simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, 1360. [Google Scholar] [CrossRef]
- Dovesi, R.; Saunders, V.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.; Bush, I.; et al. CRYSTAL17 User’s Manual; University of Torino: Torino, Italy, 2017. [Google Scholar]
- Pascale, F.; Zicovich-Wilson, C.M.; López Gejo, F.; Civalleri, B.; Orlando, R.; Dovesi, R. The calculation of the vibrational frequencies of crystalline compounds and its implementation in the CRYSTAL code. J. Comput. Chem. 2004, 25, 888–897. [Google Scholar] [CrossRef]
- Zicovich-Wilson, C.M.; Pascale, F.; Roetti, C.; Saunders, V.R.; Orlando, R.; Dovesi, R. Calculation of the vibration frequencies of α-quartz: The effect of hamiltonian and basis set. J. Comput. Chem. 2004, 25, 1873–1881. [Google Scholar] [CrossRef]
- Pedrazzo, A.R.; Smarra, A.; Caldera, F.; Musso, G.; Dhakar, N.K.; Cecone, C.; Hamedi, A.; Corsi, I.; Trotta, F. Eco-Friendly β-cyclodextrin and Linecaps Polymers for the Removal of Heavy Metals. Polymers 2019, 11, 1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MT Structure N° | Relative Energy (Kcal/Mol) | ||||||
|---|---|---|---|---|---|---|---|
| B3LYP | PBEh-3c | GFN2 | GFNFF | B3LYP | PBEh-3c | GFN2 | GFNFF |
| 03 | 00 | 10 | 01 | 0.00 | 0.00 | 0.00 | 0.00 |
| 00 | 01 | 11 | 00 | 0.28 | 1.32 | 0.00 | 0.59 |
| 02 | 03 | 07 | 02 | 0.30 | 1.36 | 0.01 | 2.02 |
| 15 | 02 | 04 | 15 | 0.86 | 1.77 | 0.06 | 2.73 |
| 05 | 04 | 15 | 05 | 1.07 | 2.47 | 0.07 | 2.73 |
| 09 | 15 | 05 | 04 | 1.20 | 2.47 | 0.08 | 2.77 |
| 04 | 05 | 00 | 11 | 1.53 | 2.48 | 0.12 | 2.93 |
| 11 | 09 | 06 | 10 | 1.74 | 3.02 | 0.17 | 2.93 |
| 01 | 07 | 09 | 07 | 1.80 | 3.12 | 0.65 | 2.95 |
| 07 | 10 | 02 | 03 | 1.83 | 3.12 | 0.93 | 3.10 |
| 10 | 11 | 18 | 06 | 1.89 | 3.12 | 0.98 | 3.29 |
| 22 | 06 | 21 | 09 | 2.73 | 3.12 | 0.98 | 5.84 |
| 21 | 22 | 22 | 18 | 4.75 | 3.86 | 1.01 | 5.92 |
| 18 | 18 | 01 | 21 | 5.40 | 3.87 | 1.15 | 5.93 |
| 06 | 21 | 03 | 22 | 5.85 | 3.88 | 1.53 | 5.95 |
| GFN2 | GFNFF | PBEh-3c | B3LYP | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| System | ΔE | ΔH | ΔG | ΔE | ΔH | ΔG | ΔE | ΔH | ΔG | ΔE |
| DIM-HH | −4.0 | −2.7 | 9.2 | −7.5 | −5.3 | 7.2 | −2.0 | −9.6 | 5.2 | −5.4 |
| DIM-HT | −6.0 | −4.6 | 6.7 | −5.0 | −3.3 | 10.4 | −3.9 | −11.2 | 3.2 | −8.6 |
| DIM-TT | −9.3 | −8.0 | 3.9 | −11.3 | −9.0 | 3.1 | −9.0 | −16.3 | −1.4 | -- |
| DIM-II | −15.0 | −13.6 | 0.8 | −16.6 | −14.1 | 0.3 | −11.9 | −19.1 | −2.4 | −18.8 |
| Method | Solvent | Energy Difference | CD2–CD1 |
|---|---|---|---|
| PBEh-3c | Gas phase | ΔE | −4.4 |
| B3LYP | Gas phase | ΔE | 7.4 |
| GFN2 | Gas phase | ΔE | 7.0 |
| ΔH | 7.1 | ||
| ΔG | 3.7 | ||
| Water | ΔE | −5.0 | |
| ΔH | −5.0 | ||
| ΔG | −10.1 | ||
| Acetonitrile | ΔE | −4.4 | |
| ΔH | −4.4 | ||
| ΔG | −8.7 | ||
| GFNFF | Gas phase | ΔE | 5.3 |
| ΔH | 4.6 | ||
| ΔG | 0.7 | ||
| Water | ΔE | −5.0 | |
| ΔH | −5.0 | ||
| ΔG | −10.1 | ||
| Acetonitrile | ΔE | −4.4 | |
| ΔH | −4.4 | ||
| ΔG | −8.7 |
| Method | ΔEdisp | ΔEnoD |
|---|---|---|
| GFN2 | −30.3 | −13.3 |
| GFNFF | −41.3 | −18.1 |
| PBEh-3c | −34.5 | −19.9 |
| B3LYP | −35.3 | −6.4 |
| Method | Structure | ΔE | ΔH | ΔG |
|---|---|---|---|---|
| PBEh-3c | EXT | −39.4 | −38.5 | −18.2 |
| GFN2—GP | EXT | −30.3 | −28.7 | −11.0 |
| MT-βCD1 | −25.6 | −23.7 | −6.3 | |
| MT-βCD2 | −39.5 | −37.2 | −16.9 | |
| MT-βCD3 | −25.3 | −23.4 | −4.9 | |
| GFN2—W | EXT | −12.5 | −10.2 | 7.2 |
| MT-βCD1 | −14.2 | −12.5 | 6.4 | |
| MT-βCD2 | −15.4 | −12.7 | 4.7 | |
| MT-βCD3 | −13.5 | −11.1 | 7.2 | |
| GFN2—ACN | EXT | −7.4 | −5.3 | 10.4 |
| MT-βCD1 | −7.0 | −4.9 | 11.9 | |
| MT-βCD2 | −7.5 | −5.2 | 12.4 | |
| MT-βCD3 | −5.5 | −4.3 | 11.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrero, R.; Pantaleone, S.; Delle Piane, M.; Caldera, F.; Corno, M.; Trotta, F.; Brunella, V. On the Interactions of Melatonin/β-Cyclodextrin Inclusion Complex: A Novel Approach Combining Efficient Semiempirical Extended Tight-Binding (xTB) Results with Ab Initio Methods. Molecules 2021, 26, 5881. https://doi.org/10.3390/molecules26195881
Ferrero R, Pantaleone S, Delle Piane M, Caldera F, Corno M, Trotta F, Brunella V. On the Interactions of Melatonin/β-Cyclodextrin Inclusion Complex: A Novel Approach Combining Efficient Semiempirical Extended Tight-Binding (xTB) Results with Ab Initio Methods. Molecules. 2021; 26(19):5881. https://doi.org/10.3390/molecules26195881
Chicago/Turabian StyleFerrero, Riccardo, Stefano Pantaleone, Massimo Delle Piane, Fabrizio Caldera, Marta Corno, Francesco Trotta, and Valentina Brunella. 2021. "On the Interactions of Melatonin/β-Cyclodextrin Inclusion Complex: A Novel Approach Combining Efficient Semiempirical Extended Tight-Binding (xTB) Results with Ab Initio Methods" Molecules 26, no. 19: 5881. https://doi.org/10.3390/molecules26195881