
Copper(II)-Mediated Iodination of 1-Nitroso-2-naphthol

 ,
,  , and
, and
Abstract
:1. Introduction
2. Results
2.1. Synthesis of 3-Iodo-1-nitrosonaphthalen-2-ol
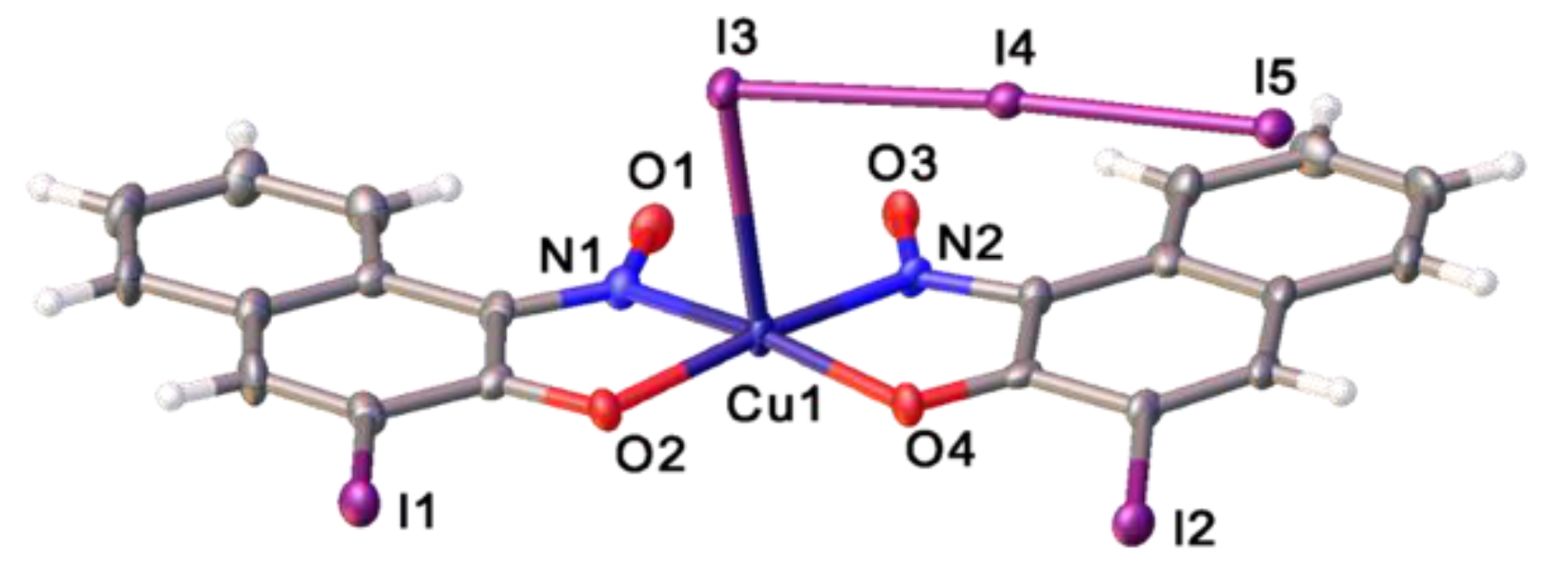
2.2. Copper(II) Complexes Featuring 3-Iodo-1-nitrosonaphthalen-2-ol
2.3. Halogen Bonding Interactions Involving Ligated I-NON
3. Discussion
4. Materials and Methods
4.1. Materials and Instrumentations
4.2. X-ray Structure Determinations
4.3. Synthetic Work
4.4. Computational Details
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pike, S.J.; Heliot, A.; Seaton, C.C. ortho-Substituent effect on the crystal packing and solid state speciation of aromatic C-nitroso compounds. CrystEngComm 2020, 22, 5040–5048. [Google Scholar] [CrossRef]
- Raić-Malić, S.; Tomašković, L.; Mrvoš-Sermek, D.; Prugovečki, B.; Cetina, M.; Grdiša, M.; Pavelić, K.; Mannschreck, A.; Balzarini, J.; De Clercq, E.; et al. Spirobipyridopyrans, spirobinaphthopyrans, indolinospiropyridopyrans, indolinospironaphthopyrans and indolinospironaphtho-1,4-oxazines: Synthesis, study of X-ray crystal structure, antitumoral and antiviral evaluation. Bioorg. Med. Chem. 2004, 12, 1037–1045. [Google Scholar] [CrossRef]
- Ambrose, J.A. Fluorometric measurement of tyrosine in serum and plasma. Clin. Chem. 1974, 20, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.A.; Robertson, G.; Wu, J.T. The chemical basis and specificity of the nitrosonaphthol reaction. Clin. Chem. 1983, 29, 1969–1971. [Google Scholar] [CrossRef]
- Fischer, V.; Mason, R.P. Formation of iminoxyl and nitroxide free radicals from nitrosonaphthols: An electron spin resonance study. Chem.-Biol. Interact. 1986, 57, 129–142. [Google Scholar] [CrossRef]
- Tôei, K.; Motomizu, S.; Korenaga, T. Nitrosophenol and nitrosonaphthol derivatives as reagents for the spectrophotometric determination of iron and determination of micro-amounts in waters with 2-nitroso-5-dimethylaminophenol. Analyst 1975, 100, 629–636. [Google Scholar] [CrossRef]
- Tôei, K.; Motomizu, S.; Yokosu, H. Extraction-spectrophotometric determination of nickel with 4-chloro-2-nitroso-1-naphthol and crystal violet. Anal. Chim. Acta 1979, 110, 329–334. [Google Scholar] [CrossRef]
- Sirén, H.; Riekkola, M.L. Separation and determination of metals as complexes of 1-nitroso-2-naphthol-6-sulphonic and 2-nitroso-1-naphthol-6-sulphonic acids-I: Ion-pair extraction and spectrophotometric method. Mikrochim. Acta 1986, 90, 159–173. [Google Scholar] [CrossRef]
- Kitamori, T.; Suzuki, K.; Motojima, K.; Sawada, T.; Gohshi, Y. Determination of Sub-Part-per-Trillion Amounts of Cobalt by Extraction and Photoacoustic Spectroscopy. Anal. Chem. 1986, 58, 2275–2278. [Google Scholar] [CrossRef]
- Memon, S.Q.; Bhanger, M.I.; Hasany, S.M.; Khuhawar, M.Y. The efficacy of nitrosonaphthol functionalized XAD-16 resin for the preconcentration/sorption of Ni(II) and Cu(II) ions. Talanta 2007, 72, 1738–1745. [Google Scholar] [CrossRef] [PubMed]
- Tavana, J.; Edrisi, M. Synthesis of cobalt ferrite nanoparticles from thermolysis of prospective metal-nitrosonaphthol complexes and their photochemical application in removing methylene blue. Mater. Res. Express 2016, 3, 035009. [Google Scholar] [CrossRef]
- Malcik, N.; Oktar, O.; Ozser, M.E.; Caglar, P.; Bushby, L.; Vaughan, A.; Kuswandi, B.; Narayanaswamy, R. Immobilised reagents for optical heavy metal ions sensing. Sens. Actuators B Chem. 1998, 53, 211–221. [Google Scholar] [CrossRef]
- Basu, P.; Dey, T.K.; Ghosh, A.; Islam, S.M. Designing of a New Heterogeneous Polymer Supported Naphthyl-Azo Iron Catalyst for the Selective Oxidation of Substituted Methyl Benzenes. J. Inorg. Organomet. Polym. Mater. 2018, 28, 1158–1170. [Google Scholar] [CrossRef]
- Marschalk, C. Halogen derivatives of 2-hydroxynaphthalene not substituted in the 1-position. Kuhlmann Villers-St. Paul. Bull. Société Chim. France 1928, 43, 1361–1367. [Google Scholar]
- Issa, I.M.; Issa, R.M.; Amer, F.A.; Khattab, M.A. Spectrophotometric studies of some nitrosonaphthols. Indian J. Chem. Sect. A 1976, 14, 713–714. [Google Scholar]
- Baird, W.C.; Surridge, J.H. Halogenation with copper(II) halides. Synthesis of aryl iodides. J. Org. Chem. 1970, 35, 3436–3442. [Google Scholar] [CrossRef]
- Toshio, S.; Mitsushige, I.; Yoshiyuki, I.; Yoshinobu, T. Aromatic iodination with aluminum and copper(II) chlorides and iodine. Chem. Lett. 1982, 11, 1481–1484. [Google Scholar]
- Akira, H.C.; Yasuo, S.J. A Convenient Procedure for Iodination of Electron-rich Aromatic Compounds using Iodine–Copper(II) Acetate. Bull. Chem. Soc. Jpn. 1984, 57, 2691–2692. [Google Scholar]
- Saarinen, H.; Korvenranta, J. Crystal and molecular-structure of diacetone adduct of copper(II) complex of 1-Nitroso-2-Naphthol-CU(C10H6NO2)2.2(CH3)2CO. Acta Chem. Scand. Ser. A-Phys. Inorg. Chem. 1975, 29, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Carugo, O.; Djinović, K.; Rizzi, M.; Castellani, C.B. Studies on metal complexes of ortho-quinone monooximes. Part 9. Analysis of the charge distribution within the o-quinone monooxime ligands through crystallographic data. J. Chem. Soc. Dalton Trans. 1991, 1255–1258. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavallo, G.; Abate, A.; Rosati, M.; Paolo Venuti, G.; Pilati, T.; Terraneo, G.; Resnati, G.; Metrangolo, P. Tuning of Ionic Liquid Crystal Properties by Combining Halogen Bonding and Fluorous Effect. ChemPlusChem 2021, 86, 469–474. [Google Scholar] [CrossRef]
- Taylor, M.S. Anion recognition based on halogen, chalcogen, pnictogen and tetrel bonding. Coord. Chem. Rev. 2020, 413, 213270. [Google Scholar] [CrossRef]
- Scheiner, S.; Michalczyk, M.; Zierkiewicz, W. Coordination of anions by noncovalently bonded σ-hole ligands. Coord. Chem. Rev. 2020, 405, 213136. [Google Scholar] [CrossRef]
- Riel, A.M.S.; Rowe, R.K.; Ho, E.N.; Carlsson, A.-C.C.; Rappé, A.K.; Berryman, O.B.; Ho, P.S. Hydrogen Bond Enhanced Halogen Bonds: A Synergistic Interaction in Chemistry and Biochemistry. Acc. Chem. Res. 2019, 52, 2870–2880. [Google Scholar] [CrossRef]
- Wirth, S.; Rohbogner, C.J.; Cieslak, M.; Kazmierczak-Baranska, J.; Donevski, S.; Nawrot, B.; Lorenz, I.P. Rhodium(III) and iridium(III) complexes with 1,2-naphthoquinone-1-oximate as a bidentate ligand: Synthesis, structure, and biological activity. J. Biol. Inorg. Chem. 2010, 15, 429–440. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711. [Google Scholar] [CrossRef]
- Osman, A.M.; Khalaf, A.A.; Amer, F.A. Synthesis of some naphthoxazoles from nitrosonaphthols. Indian J. Chem. 1974, 12, 120–123. [Google Scholar]
- Zincke, T.; Schmunk, L. Ueber die Einwirkung von Chlor auf Chinon0xime (Nitrosophenole). Justus Liebigs Ann. Chem. 1890, 257, 133–155. [Google Scholar] [CrossRef] [Green Version]
- Bolotin, D.S.; Bokach, N.A.; Demakova, M.Y.; Kukushkin, V.Y. Metal-Involving Synthesis and Reactions of Oximes. Chem. Rev. 2017, 117, 13039–13122. [Google Scholar] [CrossRef] [PubMed]
- Orpen, A.G.; Brammer, L.; Allen, F.H.; Kennard, O.; Watson, D.G.; Taylor, R. Supplement. Tables of bond lengths determined by X-ray and neutron diffraction. Part 2. Organometallic compounds and co-ordination complexes of the d- and f-block metals. J. Chem. Soc. Dalton Trans. 1989, S1–S83. [Google Scholar] [CrossRef]
- Bikbaeva, Z.M.; Ivanov, D.M.; Novikov, A.S.; Ananyev, I.V.; Bokach, N.A.; Kukushkin, V.Y. Electrophilic–Nucleophilic Dualism of Nickel(II) toward Ni···I Noncovalent Interactions: Semicoordination of Iodine Centers via Electron Belt and Halogen Bonding via σ-Hole. Inorg. Chem. 2017, 56, 13562–13578. [Google Scholar] [CrossRef] [PubMed]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii of Metals in Covalent Compounds. J. Phys. Chem. 1966, 70, 3006–3007. [Google Scholar] [CrossRef]
- Batsanov, S.S. Van der Waals Radii of Elements. Inorg. Mater. 2001, 37, 871–885. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Lu, Y.-X.; Zou, J.-W.; Yu, Q.-S. Theoretical investigation on charge-assisted halogen bonding interactions in the complexes of bromocarbons with some anions. Int. J. Quantum Chem. 2008, 108, 90–99. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Bilewicz, E. Cooperativity halogen bonding effect–Ab initio calculations on H2CO⋯(ClF)n complexes. Chem. Phys. Lett. 2006, 427, 51–55. [Google Scholar] [CrossRef]
- Lu, Y.-X.; Zou, J.-W.; Wang, Y.-H.; Jiang, Y.-J.; Yu, Q.-S. Ab Initio Investigation of the Complexes between Bromobenzene and Several Electron Donors: Some Insights into the Magnitude and Nature of Halogen Bonding Interactions. J. Phys. Chem. A 2007, 111, 10781–10788. [Google Scholar] [CrossRef]
- Forni, A. Experimental and Theoretical Study of the Br···N Halogen Bond in Complexes of 1,4-Dibromotetrafluorobenzene with Dipyridyl Derivatives. J. Phys. Chem. A 2009, 113, 3403–3412. [Google Scholar] [CrossRef]
- Sheldrick, G. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Palatinus, L.; Chapuis, G. SUPERFLIP-a computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Crystallogr. 2007, 40, 786–790. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian: Wallingford, CT, USA, 2013. [Google Scholar]
- Jorge, F.E.; Neto, A.C.; Camiletti, G.G.; Machado, S.F. Contracted Gaussian basis sets for Douglas–Kroll–Hess calculations: Estimating scalar relativistic effects of some atomic and molecular properties. J. Chem. Phys. 2009, 130, 064108. [Google Scholar] [CrossRef]
- Canal Neto, A.; Muniz, E.P.; Centoducatte, R.; Jorge, F.E. Gaussian basis sets for correlated wave functions. Hydrogen, helium, first- and second-row atoms. J. Mol. Struct. THEOCHEM 2005, 718, 219–224. [Google Scholar] [CrossRef]
- Camiletti, G.G.; Machado, S.F.; Jorge, F.E. Gaussian basis set of double zeta quality for atoms K through Kr: Application in DFT calculations of molecular properties. J. Comput. Chem. 2008, 29, 2434–2444. [Google Scholar] [CrossRef]
- Barros, C.L.; de Oliveira, P.J.P.; Jorge, F.E.; Canal Neto, A.; Campos, M. Gaussian basis set of double zeta quality for atoms Rb through Xe: Application in non-relativistic and relativistic calculations of atomic and molecular properties. Mol. Phys. 2010, 108, 1965–1972. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Douglas, M.; Kroll, N.M. Quantum electrodynamical corrections to the fine structure of helium. Ann. Phys. 1974, 82, 89–155. [Google Scholar] [CrossRef]
- Hess, B.A. Applicability of the no-pair equation with free-particle projection operators to atomic and molecular structure calculations. Phys. Rev. A 1985, 32, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Keith, T.A.; Gristmill, T.K. AIMAll; Gristmill Software: Overland Park, KS, USA, 2014. [Google Scholar]
- Lefebvre, C.; Rubez, G.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Hénon, E. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 2017, 19, 17928–17936. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, C.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Piquemal, J.-P.; Hénon, E. The Independent Gradient Model: A New Approach for Probing Strong and Weak Interactions in Molecules from Wave Function Calculations. ChemPhysChem 2018, 19, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Simon, S.; Duran, M.; Dannenberg, J.J. How does basis set superposition error change the potential surfaces for hydrogen-bonded dimers? J. Chem. Phys. 1996, 105, 11024–11031. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Conditions | Product Identification | |||
|---|---|---|---|---|---|
| M Salt | M/NON Ratio | I2/NON Ratio | I-NON/NON Ratio | Yield of I-NON (%) (1H NMR) | |
| 1 | CuCl2·2H2O | 1/2 | 1/1 | 1.15/1 | 38 |
| 2 | Cu(SO4)2·5H2O | 1/2 | 1/1 | 1/1 | 32 |
| 3 | Cu(acac)2 | 1/2 | 1/1 | 1.7/1 | 6 |
| 4 | Сu(NO3)2·2H2O | 1/2 | 1/1 | 1.14 | 21 |
| 5 | Cu(OAc)2·H2O | 1/2 | 1/1 | 2/1 | 18 |
| 6 | Cu(OAc)2·H2O | 1/2 | 2/1 | 3.5/1 | 30 |
| 7 | Cu(OAc)2·H2O | 1/2 | 3/1 | 16/1 | 81 |
| 8 | Cu(OAc)2·H2O | 1/2 | 4/1 | 13.5/1 | 94 |
| 9 | Cu(OAc)2·H2O | 1/4 | 1/1 | 0.04/1 | 2.6 |
| 10 | Cu(OAc)2·H2O | 1/4 | 3/1 | 2.7/1 | 53 |
| 11 | CuI | 1/2 | 4/1 | –/1 | – |
| 12 | Ni(OAc)2·4H2O | 1/2 | 4/1 | 0.31/1 | <1 |
| 13 | Co(OAc)2·4H2O | 1/2 | 4/1 | –/1 | – |
| 14 | FeCl3·6H2O | 1/2 | 4/1 | 2/1 | 23 |
| 15 | – 2 | – | 2/1 | 1/1.5 | 24 |
| Bond | Bond Length, Å | ||
|---|---|---|---|
| 1 | 3 | 4 | |
| Cu–N | 1.963(5) 1.955(5) | 1.964(5) | 2.018(4) |
| 1.967(5) | |||
| 1.969(5) | |||
| 1.977(5) | |||
| Cu–O | 1.954(4) 1.976(4) | 1.953(4) | 2.302(3) |
| 1.955(4) | |||
| 1.954(4) | |||
| 1.957(4) | |||
| N–O | 1.299(7) 1.330(7) | 1.260(5) | 1.282(5) |
| 1.268(5) | |||
| 1.240(6) | |||
| 1.240(6) | |||
| N–C | 1.313(8) 1.313(8) | 1.339(7) | 1.341(5) |
| 1.343(6) | |||
| 1.354(6) | |||
| 1.348(6) | |||
| O–C | 1.280(8) 1.249(8) | 1.263(6) | 1.236(5) |
| 1.269(6) | |||
| 1.271(6) | |||
| 1.270(6) | |||
| Cu–OTHF | 2.535(5) | ||
| 2.448(4) | |||
| 2.473(6) | |||
| 2.514(6) | |||
| Cu–NPy | 2.028(4) | ||
| Cu–I | 2.9417(8) | ||
| Contacting Atoms | Distance, Å (Nc) 1 | Angle, ° | Contact Type/Name |
|---|---|---|---|
| 1 | |||
| Cu1···I4 | 3.5935(8) (1.06) | Cu1–I4–I5 88.225(17) | semicoordination |
| I1···I3 | 3.9656(6) (1.00) | C3–I1–I3 178.76(17) I1–I3–Cu1 97.06(2) | HaB (I···I)1 |
| I2···I4 | 3.7872(6) (0.95) | C–I2–I4 150.6(1) I2–I4–I5 101.229(17) | HaB (I···I)2 |
| 3 | |||
| I2···O1 | 3.159(4) (0.90) | C13–I2–O1 152.33 (15) I2–O1–N1 156.7(3) | (I···O)1 |
| I1···O3 | 3.146(4) (0.90) | C3–I1–O3 150.64(16) I1–O3–N2 152.2(3) | (I···O)2 |
| I2A···O3A | 3.187(4) (0.91) | C13A–I2A–O3A 152.97(12) I2A–O3A–N2A 160.9(3) | (I···O)3 |
| I1A···O1A | 3.566(4) (1.02) 2 | C3A–I1A–O1A 138.07(16) I1A–O1A–N1A 142.7(3) | (I···O)4 |
| Cluster | HaB | d(I…I/O) | ρb | ∇2ρb | Vb | Gb | λ2,b | Eint(BSSE) | Eint(S) |
|---|---|---|---|---|---|---|---|---|---|
| BM11 | (I···I)1 | 3.966 | 0.0058 | 0.0191 | −0.0022 | 0.0035 | −0.0026 | −5.1 | −9.0 |
| BM12 | (I···I)2 | 3.787 | 0.0089 | 0.0290 | −0.0039 | 0.0056 | −0.0046 | −9.2 | −10.4 |
| BM31 | (I···O)1 | 3.159 | 0.0095 | 0.0458 | −0.0063 | 0.0089 | −0.0059 | −4.0 | −7.2 |
| BM32 | (I···O)2 | 3.146 | 0.0100 | 0.0471 | −0.0065 | 0.0091 | −0.0065 | −4.0 | −6.9 |
| BM33 | (I···O)3 | 3.187 | 0.0089 | 0.0430 | −0.0057 | 0.0082 | −0.0058 | −7.23 1 | −7.7 |
| BM33 | (I···O)4 | 3.566 | 0.0046 | 0.0191 | −0.0023 | 0.0035 | −0.0026 | −5.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Efimenko, Z.M.; Rozhkov, A.V.; Suslonov, V.V.; Kuznetsov, M.L.; Kukushkin, V.Y.; Bokach, N.A. Copper(II)-Mediated Iodination of 1-Nitroso-2-naphthol. Molecules 2021, 26, 5708. https://doi.org/10.3390/molecules26185708
Efimenko ZM, Rozhkov AV, Suslonov VV, Kuznetsov ML, Kukushkin VY, Bokach NA. Copper(II)-Mediated Iodination of 1-Nitroso-2-naphthol. Molecules. 2021; 26(18):5708. https://doi.org/10.3390/molecules26185708
Chicago/Turabian StyleEfimenko, Zarina M., Anton V. Rozhkov, Vitalii V. Suslonov, Maxim L. Kuznetsov, Vadim Yu. Kukushkin, and Nadezhda A. Bokach. 2021. "Copper(II)-Mediated Iodination of 1-Nitroso-2-naphthol" Molecules 26, no. 18: 5708. https://doi.org/10.3390/molecules26185708