
Developing On-Site Trace Level Speciation of Lead, Cadmium and Zinc by Stripping Chronopotentiometry (SCP): Fast Screening and Quantification of Total Metal Concentrations


Abstract
:1. Introduction
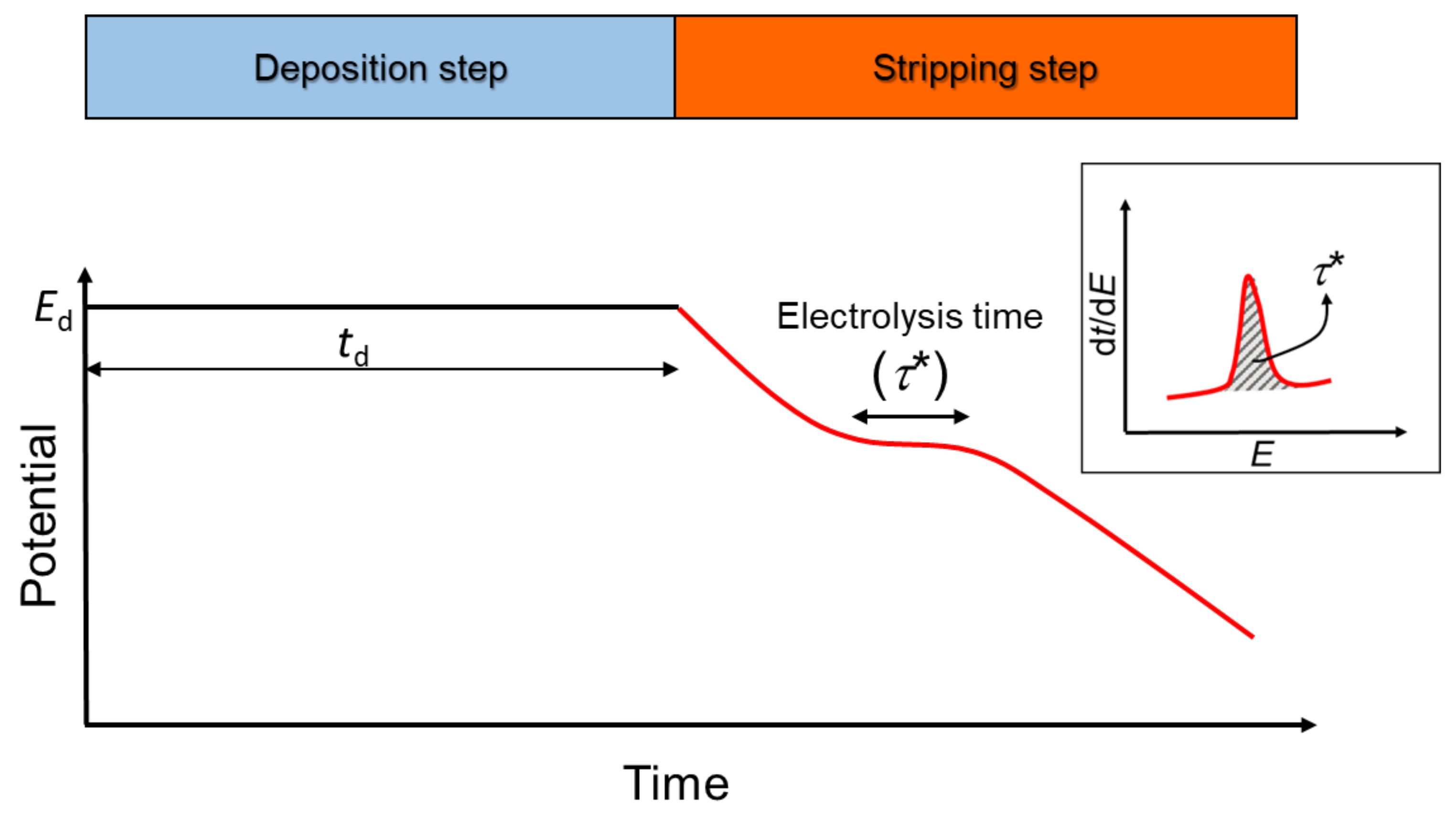
2. Theory
3. Results
3.1. Detection Limits for Cd(II), Pb(II) and Zn(II) for Calibration and Standard Addition Methods
3.2. Fast Screening INTERNAL Standard Method Using Cd(II)
3.3. Determination of Pb(II) and Zn(II) in Real Samples by Standard Addition and Cd(II) Internal Standard and Their Comparison with the ICP-MS Results
4. Discussion
5. Materials and Methods
5.1. Instruments
5.2. Reagents
5.3. Fabrication of the Carbon Screen-Printed Electrodes
5.4. Preparation of the Working Electrode
- (a)
- Electrode pretreatment consisted of a linear scan voltammetry sweep between −1.2 V and +1.5 V (vs. Ag/AgCl), using a scan rate of 0.1 V/s in H2SO4 (0.2 M) to remove any impurities from the electrode surface resulting from the fabrication process;
- (b)
- the conditioning consisted of applying 50 successive cyclic voltammetry cycles between −0.8 V and +0.8 V (vs. Ag/AgCl) at 0.1 V/s in NH4CH3COO (1 M)/HCl (0.5 M);
- (c)
- for the electrodeposition of the TMF, the electrode was immersed in a Hg(NO3)2 solution 0.18 mM (pH adjusted at 1.9), and a potential of −1.3 V (vs Ag/AgCl) was applied for 420 s. Following this, a square-wave voltammetry cycle was carried out (−1.3 V to −0.2 V, amplitude 25 mV, step 4 mV, frequency 25 Hz) to verify if the response corresponded to a good quality mercury film. If this voltammogram was not adequate, it was discarded, while if the voltammogram was satisfactory, the working electrode was deemed ready to be used;
- (d)
- at the end each day, the mercury must be recovered for proper disposal. This step also provides information on the amount of deposited mercury for purposes of quality control. Mercury reoxidation was carried out in a 5 mM ammonium thiocyanide (pH 3.4) using a linear scan voltammetric sweep from −0.1 V to +0.8 V at 0.005 V/s. The integral of the peak area multiplied by the scan rate provides the re-oxidized charge that was used to compute the amount of surface deposited mercury during the step (c). Scanning electron microscopy images of similar electrodes show that they are an assembly of nano-hemispheres of mercury that works as an array of microelectrodes with superposing diffusion layers [27].
5.5. Experimental Protocol
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Companys, E.; Galceran, J.; Pinheiro, J.P.; Puy, J.; Salaün, P. A Review on Electrochemical Methods for Trace Metal Speciation in Environmental Media. Curr. Opin. Electrochem. 2017, 3, 144–162. [Google Scholar] [CrossRef] [Green Version]
- Van Leeuwen, H.P.; Buffle, J. Chemodynamics of Aquatic Metal Complexes: From Small Ligands to Colloids. Environ. Sci. Technol. 2009, 43, 7175–7183. [Google Scholar] [CrossRef]
- Mebane, C.A.; Chowdhury, M.J.; De Schamphelaere, K.A.C.; Lofts, S.; Paquin, P.R.; Santore, R.C.; Wood, C.M. Metal Bioavailability Models: Current Status, Lessons Learned, Considerations for Regulatory Use, and the Path Forward. Env. Toxicol Chem 2020, 39, 60–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omanović, D.; Garnier, C.; Gibbon–Walsh, K.; Pižeta, I. Electroanalysis in Environmental Monitoring: Tracking Trace Metals—A Mini Review. Electrochem. Commun. 2015, 61, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Mota, A.M.; Pinheiro, J.P.; Simões Gonçalves, M.L. Electrochemical Methods for Speciation of Trace Elements in Marine Waters. Dynamic Aspects. J. Phys. Chem. A 2012, 116, 6433–6442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mota, A.M.; Correia dos Santos, M.M. Trace Metal Speciation of Labile Chemical Species in Natural Water: Electrochemical Methods. In Metal Speciation and Bioavailability; Tessier, A., Turner, D., Eds.; John Wiley & Sons: New York, NY, USA, 1995; Volume 5. [Google Scholar]
- Pinheiro, J.; Mota, A.; Goncalves, M.; van Leeuwen, H. Kinetics of Adsorption of Humic Matter on Mercury. Environ. Sci. Technol. 1994, 28, 2112–2119. [Google Scholar] [CrossRef] [PubMed]
- Bugarin, M.; Mota, A.; Pinheiro, J.; Goncalves, M. Influence of Metal Concentration at the Electrode Surface in Differential-Pulse Anodic-Stripping Voltammetry in the Presence of Humic Matter. Anal. Chim. Acta 1994, 294, 271–281. [Google Scholar] [CrossRef]
- Tercier-Waeber, M.-L.; Confalonieri, F.; Abdou, M.; Dutruch, L.; Bossy, C.; Fighera, M.; Bakker, E.; Graziottin, F.; van der Wal, P.; Schäfer, J. Advanced Multichannel Submersible Probe for Autonomous High-Resolution in Situ Monitoring of the Cycling of the Potentially Bioavailable Fraction of a Range of Trace Metals. Chemosphere 2021, 282, 131014. [Google Scholar] [CrossRef]
- Town, R.M.; van Leeuwen, H.P. Fundamental Features of Metal Ion Determination by Stripping Chronopotentiometry. J. Electroanal. Chem. 2001, 509, 58–65. [Google Scholar] [CrossRef]
- Town, R.M.; van Leeuwen, H.P. Effects of Adsorption in Stripping Chronopotentiometric Metal Speciation Analysis. J. Electroanal. Chem. 2002, 523, 1–15. [Google Scholar] [CrossRef]
- Town, R.M.; van Leeuwen, H.P. Stripping Chronopotentiometry at Scanned Deposition Potential (SSCP). Part 5. Features of Multi-Metal Systems. J. Electroanal. Chem. 2004, 573, 147–157. [Google Scholar] [CrossRef]
- Galceran, J.; Companys, E.; Puy, J.; Cecilia, J.; Garces, J.L. AGNES: A New Electroanalytical Technique for Measuring Free Metal Ion Concentration. J. Electroanal. Chem. 2004, 566, 95–109. [Google Scholar] [CrossRef] [Green Version]
- Domingos, R.F.; Huidobro, C.; Companys, E.; Galceran, J.; Puy, J.; Pinheiro, J.P. Comparison of AGNES (Absence of Gradients and Nernstian Equilibrium Stripping) and SSCP (Scanned Stripping Chronopotentiometry) for Trace Metal Speciation Analysis. J. Electroanal. Chem. 2008, 617, 141–148. [Google Scholar] [CrossRef]
- Costa Monteiro, A.S.; Parat, C.; Rosa, A.H.; Pinheiro, J.P. Towards Field Trace Metal Speciation Using Electroanalytical Techniques and Tangential Ultrafiltration. Talanta 2016, 152, 112–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, C.E. Biogeochemistry in Mineral Exploration. In Biogeochemistry in Mineral Exploration; Elsevier Science Bv: Amsterdam, The Netherlands, 2007; Volume 9, pp. 1–462. ISBN 978-0-08-054649-0. [Google Scholar]
- Vydra, F.; Štulík, K.; Juláková, E. Electrochemical Stripping Analysis; Prentice Hall Europe: Chichester, UK, 1976. [Google Scholar]
- Domingos, R.F.; Benedetti, M.F.; Croue, J.P.; Pinheiro, J.P. Electrochemical Methodology to Study Labile Trace Metal/Natural Organic Matter Complexation at Low Concentration Levels in Natural Waters. Anal. Chim. Acta 2004, 521, 77–86. [Google Scholar] [CrossRef]
- Mocak, J.; Bond, A.M.; Mitchell, S.; Scollary, G. A Statistical Overview of Standard (IUPAC and ACS) and New Procedures for Determining the Limits of Detection and Quantification: Application to Voltammetric and Stripping Techniques (Technical Report). Pure Appl. Chem. 1997, 69, 297–328. [Google Scholar] [CrossRef]
- Ziegel, E.R. Statistics and Chemometrics for Analytical Chemistry. Technometrics 2004, 46, 498–499. [Google Scholar] [CrossRef]
- Goncalves, D.A.; Jones, B.T.; Donati, G.L. The Reversed-Axis Method to Estimate Precision in Standard Additions Analysis. Microchem. J. 2016, 124, 155–158. [Google Scholar] [CrossRef]
- Kariuki, S.; Dewald, H.D. Evaluation of Diffusion Coefficients of Metallic Ions in Aqueous Solutions. Electroanalysis 1996, 8, 307–313. [Google Scholar] [CrossRef]
- Companys, E.; Cecilia, J.; Codina, G.; Puy, J.; Galceran, J. Determination of Zn2+ Concentration with AGNES Using Different Strategies to Reduce the Deposition Time. J. Electroanal. Chem. 2005, 576, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Chito, D.; Galceran, J.; Companys, E. The Impact of Intermetallic Compounds CuxZn in the Determination of Free Zn2+ Concentration with AGNES. Electroanalysis 2010, 22, 2024–2033. [Google Scholar] [CrossRef]
- Rotureau, E.; Gajdar, J.; Herzog, G.; Waldvogel, Y.; Pinheiro, J.-P.; Etienne, M. Electroanalytical Metal Sensor with Built-in Oxygen Filter. Anal. Chim. Acta 2021, 1167, 338544. [Google Scholar] [CrossRef] [PubMed]
- Morrin, A.; Killard, A.J.; Smyth, M.R. Electrochemical Characterization of Commercial and Home-Made Screen-Printed Carbon Electrodes. Anal. Lett. 2003, 36, 2021–2039. [Google Scholar] [CrossRef] [Green Version]
- Monterroso, S.C.C.; Carapuca, H.M.; Simao, J.E.J.; Duarte, A.C. Optimisation of Mercury Film Deposition on Glassy Carbon Electrodes: Evaluation of the Combined Effects of PH, Thiocyanate Ion and Deposition Potential. Anal. Chim. Acta 2004, 503, 203–212. [Google Scholar] [CrossRef]
- Pinheiro, J.P.; Bosker, W. Polystyrene Film-Coated Glassware: A New Means of Reducing Metal Losses in Trace Metal Speciation. Anal. Bioanal. Chem. 2004, 380, 964–968. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Deposition Time | Zn(II) nM | Cd(II) nM | Pb(II) nM | |||
|---|---|---|---|---|---|---|
| (s) | Range | LODsy | Range | LODsy | Range | LODsy |
| 45 | 7.5–75 | 1.7 | 7.5–75 | 2.2 | 7.5–75 | 0.9 |
| 45 | 7.5–75 | 1.8 | 7.5–75 | 2.0 | 7.5–75 | 1.0 |
| 45 | 7.5–75 | 2.4 | 7.5–75 | 1.9 | 7.5–75 | 1.2 |
| 45 | 7.5–75 | 2.6 | 7.5–75 | 1.8 | 7.5–75 | 1.5 |
| 45 | 7.5–75 | 2.5 | 7.5–75 | 1.4 | 7.5–75 | 2.3 |
| 45 | 7.5–75 | 1.8 | 7.5–75 | 2.9 | 7.5–75 | 2.2 |
| 45 | 7.5–75 | 1.5 | 7.5–75 | 1.1 | 7.5–75 | 1.7 |
| 45 | 7.5–75 | 1.7 | 7.5–75 | 1.5 | 7.5–75 | 1.8 |
| 45 | 5.0–50 | 0.9 | 5.0–50 | 1.3 | 5.0–50 | 1.0 |
| 45 | 5.0–50 | 1.5 | 5.0–50 | 1.5 | 5.0–50 | 1.2 |
| 45 | 5.0–50 | 2.0 | 5.0–50 | 1.5 | 5.0–50 | 1.0 |
| 90 | 10–50 | 2.0 | 10–50 | 1.1 | 3.0–15 | 0.4 |
| 90 | 10–50 | 1.2 | 10–50 | 1.0 | 3.0–15 | 0.4 |
| 180 | - | - | 5.0–25 | 0.4 | 0.5–4.0 | 0.08 |
| 180 | - | - | 0.5–2.5 | 0.03 | 0.5–4.0 | 0.07 |
| 180 | - | - | 0.5–3.0 | 0.04 | - | - |
| 180 | - | - | 0.5–3.0 | 0.04 | - | - |
| 180 | - | - | 0.5–3.0 | 0.05 | - | - |
| 240 | - | - | 5.0–25 | 0.3 | 0.5–4.0 | 0.06 |
| 240 | - | - | 0.5–3.0 | 0.05 | - | - |
| 240 | - | - | 0.5–3.0 | 0.04 | - | - |
| 240 | - | - | 0.5–3.0 | 0.06 | - | - |
| Sample | pH | Cd(II) nM | Pb(II) nM | ||||
|---|---|---|---|---|---|---|---|
| Addition | LODsb | Addition | LODsb | ||||
| Bethdwn 20.02.03 | 3.5 | 1.0–2.5 | - | 0.5–3.0 | 0.02 | ||
| <2 | 0.5–2.0 | 0.03 | 1.0–4.0 | 0.03 | |||
| Bethdwn 20.02.04 | 3.5 | 0.5–2.0 | 0.04 | 1.0–4.0 | 0.04 | ||
| <2 | - | - | 1.0–4.0 | 0.04 | |||
| Bethdwn 20.02.06 | 3.5 | 0.5–2.0 | 0.02 | 1.0–4.0 | 0.04 | ||
| <2 | 0.5–2.0 | 0.04 | 1.0–4.0 | 0.04 | |||
| Bethdwn 20.02.07 | 3.5 | 0.5–2.0 | 0.02 | 1.0–4.0 | 0.06 | ||
| <2 | - | - | 1.0–4.0 | 0.04 | |||
| Aub 20.02.04 | 3.5 | 0.5–2.0 | 0.03 | 1.0–4.0 | 0.05 | ||
| <2 | 0.5–2.0 | 0.07 | 1.0–4.0 | 0.06 | |||
| Rich 20.02.04 | 3.5 | 0.5–2.0 | 0.04 | 1.0–4.0 | 0.07 | ||
| <2 | - | - | 1.0–4.0 | 0.07 | |||
| Deposition Time (s) | Pb(II) Slope ×105 | Zn(II) Slope ×105 | Cd(II) Slope ×105 | Pb/Cd Ratio | Zn/Cd Ratio |
|---|---|---|---|---|---|
| 45 | 15.43 ± 0.09 | 5.22 ± 0.03 | 10.90 ± 0.11 | 1.41 | 0.48 |
| 45 | 16.95 ± 0.07 | 8.45 ± 0.05 | 12.16 ± 0.08 | 1.39 | 0.69 |
| 45 | 11.89 ± 0.05 | 6.77 ± 0.05 | 9.58 ± 0.06 | 1.24 | 0.71 |
| 45 | 14.15 ± 0.07 | 8.47 ± 0.07 | 11.9 ± 0.08 | 1.19 | 0.71 |
| 45 | 12.37 ± 0.09 | 6.77 ± 0.05 | 9.86 ± 0.05 | 1.26 | 0.69 |
| 45 | 10.84 ± 0.08 | 5.58 ± 0.03 | 8.79 ± 0.09 | 1.23 | 0.63 |
| 45 | 13.01 ± 0.09 | 7.08 ± 0.03 | 10.5 ± 0.04 | 1.24 | 0.67 |
| 45 | 13.76 ± 0.08 | 8.28 ± 0.04 | 10.4 ± 0.06 | 1.33 | 0.80 |
| 45 | 11.92 ± 0.08 | 6.77 ± 0.03 | 9.23 ± 0.08 | 1.29 | 0.73 |
| 45 | 9.56 ± 0.06 | 5.39 ± 0.04 | 8.39 ± 0.08 | 1.14 | 0.64 |
| 45 | 19.52 ± 0.13 | 7.60 ± 0.08 | 12.51 ± 0.10 | 1.56 | 0.61 |
| 90 | 24.51 ± 0.18 | 15.50 ± 0.12 | 18.49 ± 0.12 | 1.32 | 0.84 |
| 90 | 16.10± 0.10 | 9.82 ± 0.10 | 14.61 ± 0.09 | 1.10 | 0.67 |
| Sample | Pb(II) Slope ×105 | Zn(II) Slope ×105 | Cd(II) Slope ×105 | Pb/Cd Ratio | Zn/Cd Ratio |
|---|---|---|---|---|---|
| Bethdwn 19.11.29 | 15.5 ± 0.2 | 4.14 ± 0.09 | 13.1 ± 0.2 | 1.18 | 0.32 |
| Bethdwn 19.11.30.a | 18.3 ± 0.2 | 4.08 ± 0.08 | 18.2 ± 0.1 | 1.01 | 0.22 |
| Bethdwn 19.11.30.b | 20.3 ± 0.2 | 4.12 ± 0.06 | 20.2 ± 0.1 | 1.00 | 0.20 |
| Bethdwn 19.12.01.a | 16.5 ± 0.1 | 6.36 ± 0.06 | 16.1 ± 0.2 | 1.02 | 0.39 |
| Bethdwn 19.12.01.b | 22.5 ± 0.1 | 9.23 ± 0.09 | 19.3 ± 0.1 | 1.16 | 0.48 * |
| Bethdwn 19.12.02.a | 16.9 ± 0.1 | 6.25 ± 0.06 | 18.5 ± 0.2 | 0.91 | 0.34 |
| Bethdwn 19.12.02.b | 19.3 ± 0.1 | 6.46 ± 0.10 | 18.6 ± 0.1 | 1.04 | 0.35 |
| Bethdwn 19.12.03 | 22.9 ± 0.2 | 5.51 ± 0.08 | 21.6 ± 0.2 | 1.06 | 0.25 |
| Bethdwn 19.12.04 | 22.0 ± 0.1 | 4.79 ± 0.06 | 19.6 ± 0.2 | 1.12 | 0.24 |
| Bethdwn 20.02.03 | 29.6 ± 0.3 | 6.01 ± 0.10 | 27.4 ± 0.2 | 1.08 | 0.22 |
| Bethdwn 20.02.04 | 27.8 ± 0.3 | 7.23 ± 0.11 | 25.5 ± 0.3 | 1.09 | 0.28 |
| Bethdwn 20.02.06 | 38.5 ± 0.3 | 7.91 ± 0.07 | 31.8 ± 0.2 | 1.21 | 0.25 |
| Bethdwn 20.02.07 | 27.7 ± 0.2 | 7.48 ± 0.08 | 24.9 ± 0.1 | 1.11 | 0.31 |
| Aub 19.11.30 | 17.8 ± 0.1 | 5.02 ± 0.03 | 17.6 ± 0.1 | 1.01 | 0.29 |
| Aub 19.12.01 | 11.7 ± 0.1 | 3.12 ± 0.06 | 12.7 ± 0.7 | 0.92 | 0.25 |
| Aub 19.12.02 | 32.7 ± 0.3 | 7.05 ± 0.11 | 24.2 ± 0.3 | 1.35 | 0.29 |
| Aub 19.12.03 | 18.9 ± 0.1 | 1.58 ± 0.05 | 16.9 ± 0.2 | 1.12 | 0.09 * |
| Aub 19.12.04 | 24.5 ± 0.1 | 4.66 ± 0.05 | 21.8 ± 0.2 | 1.12 | 0.21 |
| Aub 20.02.04 | 26.9 ± 0.2 | 8.03 ± 0.07 | 26.3 ± 0.3 | 1.02 | 0.31 |
| Rich 20.02.04 | 41.2 ± 0.4 | 9.07 ± 0.12 | 32.7 ± 0.3 | 1.26 | 0.28 |
| Sample | Filtered | Zn(II) nM | Zn(II) LODsb nM | Zn(II) Int. Std Zn/Cd; 0.28 | ICP-MS (SARM) nM | ICP-MS (LIEC) nM |
|---|---|---|---|---|---|---|
| Bethdwn 19.11.29 | No | 51 ± 3 | 4 | 51 ± 6; 57 ± 11 | 35 ± 5 | - |
| Bethdwn 19.11.30.a | No | 70 ± 2 | 2 | 66 ± 5; 54 ± 8 | 35 ± 5 | - |
| Bethdwn 19.11.30.b | No | 76 ± 3 | 3 | 68 ± 10; 50 ± 12 | 35 ± 5 | - |
| Bethdwn 19.12.01.a | No | 18 ± 2 | 2 | 16 ± 4; 23 ± 7 | 29 ± 4 | - |
| Bethdwn 19.12.01.b | No | 43 ± 2 | 4 | 36 ± 4; 62 ± 12 | 29 ± 4 | - |
| Bethdwn 19.12.02.a | No | 34 ± 1 | 2 | 25 ± 3; 30 ± 6 | 51 ± 8 | - |
| Bethdwn 19.12.02.b | No | 26 ± 1 | 2 | 25 ± 3; 31 ± 6 | 51 ± 8 | - |
| Bethdwn 19.12.03 | No | 54 ± 2 | 2 | 48 ± 7; 43 ± 9 | 32 ± 5 | - |
| Bethdwn 19.12.04 | No | 52 ± 3 | 2 | 34 ± 5; 29 ± 7 | 35 ± 5 | - |
| Aub 19.11.30 | No | 25 ± 1 | 2 | 26 ± 3; 27 ± 5 | 32 ± 5 | - |
| Aub 19.12.01 | No | 88 ± 3 | 3 | 82 ± 6; 73 ± 10 | 39 ± 5 | - |
| Aub 19.12.02 | No | 18 ± 1 | 3 | 19 ± 3; 19 ± 5 | 27 ± 4 | - |
| Aub 19.12.03 | No | 116 ± 11 | 7 | 102 ± 21; 34 ± 8 | 26 ± 4 | - |
| Aub 19.12.04 | No | 28 ± 1 | 2 | 31 ± 5; 24 ± 5 | 16 ± 2 | - |
| Bethdwn 20.02.03 | Yes | 18 ± 2 | 3 | 16 ± 2; 13 ± 3 | 12 ± 2 | 27 ± 4 |
| Bethdwn 20.02.04 | Yes | 16 ± 2 | 3 | 15 ± 2; 16 ± 3 | 43 ± 6 | 38 ± 6 |
| Bethdwn 20.02.06 | Yes | 17 ± 2 | 2 | 16 ± 2; 14 ± 3 | 32 ± 5 | 27 ± 4 |
| Bethdwn 20.02.07 | Yes | 12 ± 1 | 2 | 11 ± 1; 12 ± 3 | 35 ± 5 | 31 ± 5 |
| Aub 20.02.04 | Yes | 9 ± 1 | 2 | 9 ± 2; 11 ± 3 | 26 ± 4 | 33 ± 5 |
| Rich 20.02.04 | Yes | 17 ± 1 | 2 | 18 ± 4; 18 ± 5 | 31 ± 5 | 30 ± 5 |
| Sample | Pb(II) nM | Pb(II) LODsb nM | Pb(II) Int. Std Pb/Cd; 1.09 | ICP-MS (SARM) nM |
|---|---|---|---|---|
| Bethdwn 19.11.29 | 2.5 ± 0.5 | 0.6 | - | 0.4 ± 0.1 |
| Bethdwn 19.11.30.a | 1.8 ± 0.2 | 0.3 | 1.7 ± 0.3; 1.6 ± 0.4 | 0.4 ± 0.1 |
| Bethdwn 19.11.30.b | 3.2 ± 0.3 | 0.4 | 2.6 ± 0.6; 2.4 ± 0.7 | 0.4 ± 0.1 |
| Bethdwn 19.12.01.a | 0.3 ± 0.1 | 0.2 | 0.3 ± 0.1; 0.3 ± 0.1 | 0.3 ± 0.1 |
| Bethdwn 19.12.01.b | 2.1 ± 0.2 | 0.3 | 1.8 ± 0.2; 1.9 ± 0.4 | 0.3 ± 0.1 |
| Bethdwn 19.12.02.a | 0.7 ± 0.2 | 0.3 | 0.5 ± 0.2; 0.6 ± 0.2 | 0.3 ± 0.1 |
| Bethdwn 19.12.02.b | 0.2 ± 0.2 | 0.3 | 0.2 ± 0.1; 0.2 ± 0.1 | 0.3 ± 0.1 |
| Bethdwn 19.12.03 | 1.3 ± 0.2 | 0.3 | 1.1 ± 0.3; 1.0 ± 0.4 | 0.6 ± 0.2 |
| Bethdwn 19.12.04 | 1.1 ± 0.1 | 0.2 | 0.7 ± 0.3; 0.7 ± 0.3 | 0.5 ± 0.2 |
| Aub 19.11.30 | 1.4 ± 0.2 | 0.3 | 1.4 ± 0.2; 1.3 ± 0.3 | 0.4 ± 0.1 |
| Aub 19.12.01 | 2.1 ± 0.2 | 0.3 | 1.8 ± 0.2; 1.5 ± 0.3 | 0.6 ± 0.2 |
| Aub 19.12.02 | 0.5 ± 0.2 | 0.3 | 0.8 ± 0.2; 0.7 ± 0.1 | 0.3 ± 0.1 |
| Aub 19.12.03 | 1.0 ± 0.2 | 0.3 | 0.9 ± 0.2; 0.9 ± 0.2 | 0.3 ± 0.1 |
| Aub 19.12.04 | 0.3 ± 0.1 | 0.2 | 0.4 ± 0.2; 0.4 ± 0.3 | 0.2 ± 0.1 |
| Sample | Dep. Time/pH | Pb(II) nM | Pb(II) LODsb nM | Pb(II) Int. Std Pb/Cd; 1.09 | ICP-MS (SARM) nM | ICP-MS (LIEC) nM |
|---|---|---|---|---|---|---|
| Bethdwn 20.02.03 | 90 s/3.5 | 0.35 < LOD | 0.37 | - | 0.51 ± 0.17 | 0.29 ± 0.09 |
| 180 s/3.5 | 0.11 ± 0.02 | 0.02 | - | |||
| 180 s/<2 | 0.21 ± 0.02 | 0.03 | 0.20 ± 0.05; 0.40 ± 0.13 | |||
| Bethdwn 20.02.04 | 90 s/3.5 | 1.16 ± 0.22 | 0.36 | 1.05 ± 0.15; 1.05 ± 0.10 | 0.45 ± 0.15 | 0.53 ± 0.16 |
| 180 s/3.5 | 0.17 ± 0.02 | 0.04 | 0.16 ± 0.01; 0.19 ± 0.02 | |||
| 180 s/<2 | 0.28 ± 0.03 | 0.04 | - | |||
| Bethdwn 20.02.06 | 90 s/3.5 | 2.00 ± 0.21 | 0.29 | 1.74 ± 0.40; 1.94 ± 0.55 | 0.33 ± 0.11 | 0.16 ± 0.05 |
| 180 s/3.5 | 0.20 ± 0.03 | 0.04 | 0.20 ± 0.05; 0.29 ± 0.09 | |||
| 180 s/<2 | 0.10 ± 0.03 | 0.04 | 0.09 ± 0.05; 0.18 ± 0.09 | |||
| Bethdwn 20.02.07 | 90 s/3.5 | 0.29 < LOD | 0.31 | - | 0.28 ± 0.09 | 0.15 ± 0.05 |
| 180 s/3.5 | 0.42 ± 0.04 | 0.06 | 0.43 ± 0.09; 0.69 ± 0.19 | |||
| 180 s/<2 | 0.27 ± 0.03 | 0.04 | - | |||
| Aub 20.02.04 | 90 s/3.5 | 0.68 < LOQ | 0.24 | 0.79 ± 0.19; 0.84 ± 0.22 | 0.32 ± 0.11 | 0.43 ± 0.13 |
| 180 s/3.5 | 0.80 ± 0.03 | 0.05 | 0.74 ± 0.07; 0.98 ± 0.14 | |||
| 180 s/<2 | 0.35 ± 0.04 | 0.06 | 0.28 ± 0.09; 0.80 ± 0.27 | |||
| Rich 20.02.04 | 90 s/3.5 | 0.63 < LOQ | 0.36 | 0.83 ± 0.20; 0.96 ± 0.28 | 0.83 ± 0.28 | 0.25 ± 0.08 |
| 180 s/3.5 | 0.31 ± 0.05 | 0.07 | 0.33 ± 0.15; 0.52 ± 0.27 | |||
| 180 s/<2 | 0.40 ± 0.05 | 0.07 | - | |||
| Rich 21.02.23 | 180 s/<2 | 0.33 ± 0.08 | 0.10 | - | 0.21 ± 0.06 | - |
| Aub 21.02.22 | 180 s/<2 | 0.29 ± 0.04 | 0.05 | - | 0.21 ± 0.06 | - |
| Bethdwn 21.01.26 | 180 s/<2 | 0.38 ± 0.06 | 0.08 | - | 0.32 ± 0.11 | - |
| Bethdwn 21.01.29 | 180 s/<2 | 0.54 ± 0.08 | 0.10 | - | 0.16 ± 0.05 | - |
| Bethdwn 21.02.03 | 180 s/<2 | 0.41 ± 0.05 | 0.07 | - | 0.25 ± 0.08 | - |
| Bethdwn 21.02.05 | 180 s/<2 | 0.30 ± 0.04 | 0.06 | - | 0.23 ± 0.07 | - |
| Bethdwn 21.02.12 | 180 s/<2 | 0.32 ± 0.04 | 0.05 | - | 0.14 ± 0.04 | - |
| Bethdwn 21.02.22 | 180 s/<2 | 0.34 ± 0.03 | 0.04 | - | 0.27 ± 0.08 | - |
| Sample | pH | Cd(II) nM | Cd(II) LODsb nM | ICP_MS (SARM) nM |
|---|---|---|---|---|
| Bethdwn 20.02.03 | <2 | 0.22 ± 0.03 | 0.03 | 0.20 ± 0.06 |
| Bethdwn 20.02.06 | 3.5 | 0.10 ± 0.02 | 0.02 | 0.19 ± 0.06 |
| Bethdwn 20.02.06 | <2 | 0.18 ± 0.03 | 0.04 | 0.19 ± 0.06 |
| Bethdwn 20.02.07 | 3.5 | 0.14 ± 0.02 | 0.02 | 0.06 ± 0.02 |
| Aub 20.02.04 | 3.5 | 0.18 ± 0.02 | 0.03 | 0.25 ± 0.06 |
| Aub 20.02.04 | <2 | 0.32 ± 0.05 | 0.07 | 0.25 ± 0.06 |
| Rich 20.02.04 | 3.5 | 0.09 ± 0.03 | 0.04 | 0.30 ± 0.09 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hackel, L.; Rotureau, E.; Morrin, A.; Pinheiro, J.P. Developing On-Site Trace Level Speciation of Lead, Cadmium and Zinc by Stripping Chronopotentiometry (SCP): Fast Screening and Quantification of Total Metal Concentrations. Molecules 2021, 26, 5502. https://doi.org/10.3390/molecules26185502
Hackel L, Rotureau E, Morrin A, Pinheiro JP. Developing On-Site Trace Level Speciation of Lead, Cadmium and Zinc by Stripping Chronopotentiometry (SCP): Fast Screening and Quantification of Total Metal Concentrations. Molecules. 2021; 26(18):5502. https://doi.org/10.3390/molecules26185502
Chicago/Turabian StyleHackel, Laetitia, Elise Rotureau, Aoife Morrin, and José Paulo Pinheiro. 2021. "Developing On-Site Trace Level Speciation of Lead, Cadmium and Zinc by Stripping Chronopotentiometry (SCP): Fast Screening and Quantification of Total Metal Concentrations" Molecules 26, no. 18: 5502. https://doi.org/10.3390/molecules26185502