
Functional Analysis of the GPI Transamidase Complex by Screening for Amino Acid Mutations in Each Subunit

 ,
,  , , ,
, , , {kind=link}
{kind=link}
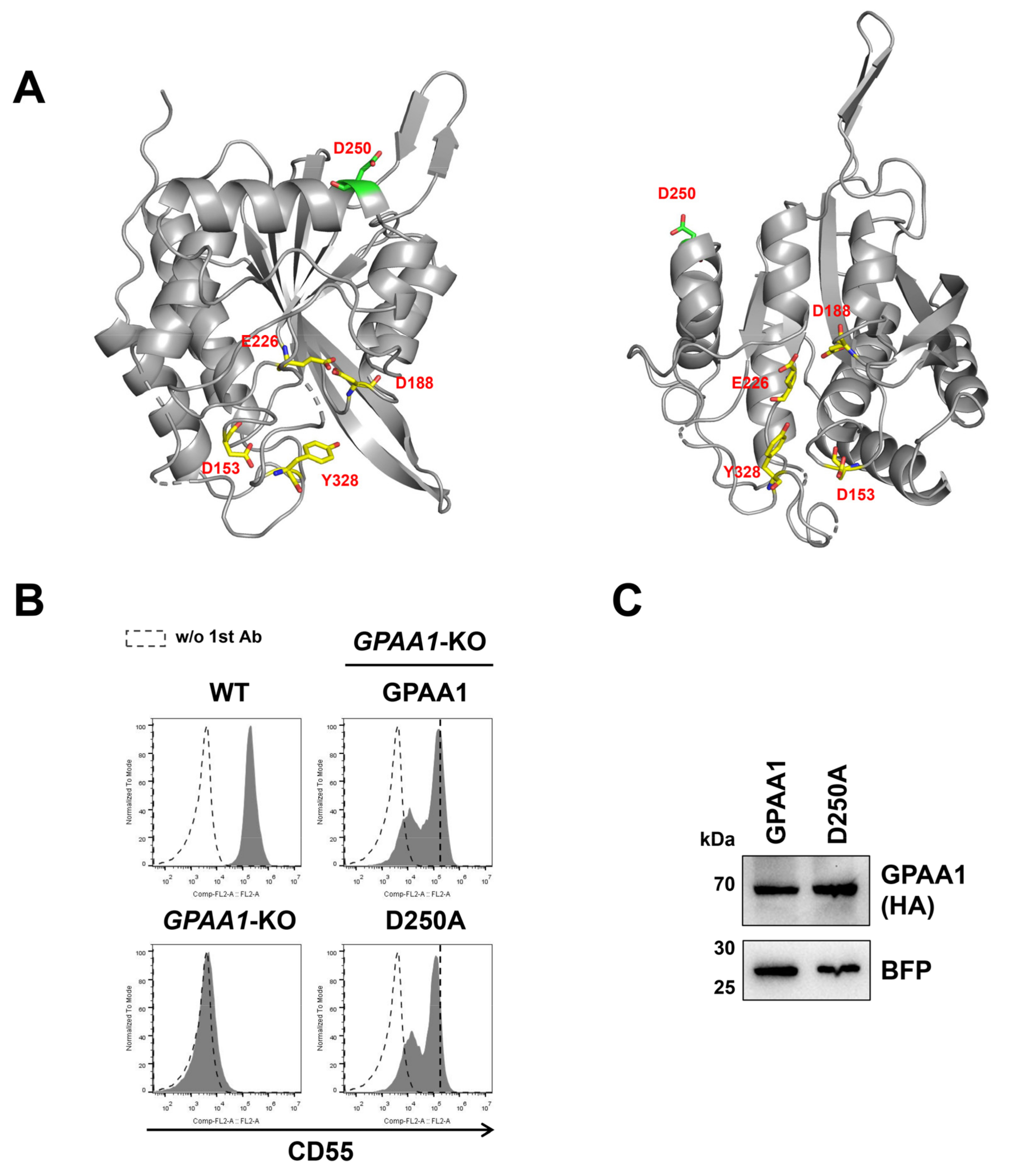{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Exploration of Functionally Important Residues of the GPI-TA Complex
2.2. Expression of PIGK Recombinant Protein
2.3. Optimization of the Purification Conditions of the GPI-TA Complex
2.4. Preliminary Cryo-EM Analysis of GPI-TA Complex
3. Conclusions
4. Materials and Methods
4.1. Cell Lines, Antibodies, and Reagents
4.2. Plasmids
4.3. Transient Transfection and Flow Cytometric Analysis
4.4. Western Blotting
4.5. Stable Transfection of hPIGK-StrepII or hPIGK-StrepII-GST
4.6. Purification of the GPI-TA Complex
4.7. Sliver Staining
4.8. EM Data Acquisition
4.9. Image Processing
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Liu, Y.S.; Fujita, M. Mammalian GPI-anchor modifications and the enzymes involved. Biochem. Soc. Trans. 2020, 48, 1129–1138. [Google Scholar] [CrossRef]
- Orlean, P.; Menon, A.K. Thematic review series: Lipid posttranslational modifications. GPI anchoring of protein in yeast and mammalian cells, or: How we learned to stop worrying and love glycophospholipids. J. Lipid Res. 2007, 48, 993–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pittet, M.; Conzelmann, A. Biosynthesis and function of GPI proteins in the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta 2007, 1771, 405–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borner, G.H.; Sherrier, D.J.; Stevens, T.J.; Arkin, I.T.; Dupree, P. Prediction of glycosylphosphatidylinositol-anchored proteins in Arabidopsis. A genomic analysis. Plant Physiol. 2002, 129, 486–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef]
- Paladino, S.; Sarnataro, D.; Pillich, R.; Tivodar, S.; Nitsch, L.; Zurzolo, C. Protein oligomerization modulates raft partitioning and apical sorting of GPI-anchored proteins. J. Cell Biol. 2004, 167, 699–709. [Google Scholar] [CrossRef]
- Brown, D.A.; Rose, J.K. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell 1992, 68, 533–544. [Google Scholar] [CrossRef]
- Sabharanjak, S.; Sharma, P.; Parton, R.G.; Mayor, S. GPI-anchored proteins are delivered to recycling endosomes via a distinct cdc42-regulated, clathrin-independent pinocytic pathway. Dev. Cell 2002, 2, 411–423. [Google Scholar] [CrossRef] [Green Version]
- Tansey, M.G.; Baloh, R.H.; Milbrandt, J.; Johnson, E.M., Jr. GFRalpha-mediated localization of RET to lipid rafts is required for effective downstream signaling, differentiation, and neuronal survival. Neuron 2000, 25, 611–623. [Google Scholar] [CrossRef] [Green Version]
- Eisenhaber, B.; Bork, P.; Eisenhaber, F. Sequence properties of GPI-anchored proteins near the omega-site: Constraints for the polypeptide binding site of the putative transamidase. Protein Eng. 1998, 11, 1155–1161. [Google Scholar] [CrossRef] [Green Version]
- Benghezal, M.; Benachour, A.; Rusconi, S.; Aebi, M.; Conzelmann, A. Yeast Gpi8p is essential for GPI anchor attachment onto proteins. EMBO J. 1996, 15, 6575–6583. [Google Scholar] [CrossRef]
- Ikezawa, H. Glycosylphosphatidylinositol (GPI)-anchored proteins. Biol. Pharm. Bull. 2002, 25, 409–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenhaber, B.; Eisenhaber, S.; Kwang, T.Y.; Grüber, G.; Eisenhaber, F. Transamidase subunit GAA1/GPAA1 is a M28 family metallo-peptide-synthetase that catalyzes the peptide bond formation between the substrate protein’s omega-site and the GPI lipid anchor’s phosphoethanolamine. Cell Cycle 2014, 13, 1912–1917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohishi, K.; Nagamune, K.; Maeda, Y.; Kinoshita, T. Two subunits of glycosylphosphatidylinositol transamidase, GPI8 and PIG-T, form a functionally important intermolecular disulfide bridge. J. Biol. Chem. 2003, 278, 13959–13967. [Google Scholar] [CrossRef] [Green Version]
- Eisenhaber, B.; Maurer-Stroh, S.; Novatchkova, M.; Schneider, G.; Eisenhaber, F. Enzymes and auxiliary factors for GPI lipid anchor biosynthesis and post-translational transfer to proteins. BioEssays News Rev. Mol. Cell. Dev. Biol. 2003, 25, 367–385. [Google Scholar] [CrossRef] [PubMed]
- Su, C.T.; Sinha, S.; Eisenhaber, B.; Eisenhaber, F. Structural modelling of the lumenal domain of human GPAA1, the metallo-peptide synthetase subunit of the transamidase complex, reveals zinc-binding mode and two flaps surrounding the active site. Biol. Direct 2020, 15, 14. [Google Scholar] [CrossRef]
- Eisenhaber, B.; Sinha, S.; Wong, W.C.; Eisenhaber, F. Function of a membrane-embedded domain evolutionarily multiplied in the GPI lipid anchor pathway proteins PIG-B, PIG-M, PIG-U, PIG-W, PIG-V, and PIG-Z. Cell Cycle 2018, 17, 874–880. [Google Scholar] [CrossRef]
- Nguyen, T.T.M.; Murakami, Y.; Mobilio, S.; Niceta, M.; Zampino, G.; Philippe, C.; Moutton, S.; Zaki, M.S.; James, K.N.; Musaev, D.; et al. Bi-allelic Variants in the GPI Transamidase Subunit PIGK Cause a Neurodevelopmental Syndrome with Hypotonia, Cerebellar Atrophy, and Epilepsy. Am. J. Hum. Genet. 2020, 106, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.M.; Murakami, Y.; Sheridan, E.; Ehresmann, S.; Rousseau, J.; St-Denis, A.; Chai, G.; Ajeawung, N.F.; Fairbrother, L.; Reimschisel, T.; et al. Mutations in GPAA1, Encoding a GPI Transamidase Complex Protein, Cause Developmental Delay, Epilepsy, Cerebellar Atrophy, and Osteopenia. Am. J. Hum. Genet. 2017, 101, 856–865. [Google Scholar] [CrossRef] [Green Version]
- Kvarnung, M.; Nilsson, D.; Lindstrand, A.; Korenke, G.C.; Chiang, S.C.; Blennow, E.; Bergmann, M.; Stödberg, T.; Mäkitie, O.; Anderlid, B.M.; et al. A novel intellectual disability syndrome caused by GPI anchor deficiency due to homozygous mutations in PIGT. J. Med. Genet. 2013, 50, 521–528. [Google Scholar] [CrossRef]
- Nguyen, T.T.M.; Murakami, Y.; Wigby, K.M.; Baratang, N.V.; Rousseau, J.; St-Denis, A.; Rosenfeld, J.A.; Laniewski, S.C.; Jones, J.; Iglesias, A.D.; et al. Mutations in PIGS, Encoding a GPI Transamidase, Cause a Neurological Syndrome Ranging from Fetal Akinesia to Epileptic Encephalopathy. Am. J. Hum. Genet. 2018, 103, 602–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knaus, A.; Kortüm, F.; Kleefstra, T.; Stray-Pedersen, A.; Đukić, D.; Murakami, Y.; Gerstner, T.; van Bokhoven, H.; Iqbal, Z.; Horn, D.; et al. Mutations in PIGU Impair the Function of the GPI Transamidase Complex, Causing Severe Intellectual Disability, Epilepsy, and Brain Anomalies. Am. J. Hum. Genet. 2019, 105, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellai-Dussault, K.; Nguyen, T.T.M.; Baratang, N.V.; Jimenez-Cruz, D.A.; Campeau, P.M. Clinical variability in inherited glycosylphosphatidylinositol deficiency disorders. Clin. Genet. 2019, 95, 112–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krawitz, P.M.; Höchsmann, B.; Murakami, Y.; Teubner, B.; Krüger, U.; Klopocki, E.; Neitzel, H.; Hoellein, A.; Schneider, C.; Parkhomchuk, D.; et al. A case of paroxysmal nocturnal hemoglobinuria caused by a germline mutation and a somatic mutation in PIGT. Blood 2013, 122, 1312–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höchsmann, B.; Murakami, Y.; Osato, M.; Knaus, A.; Kawamoto, M.; Inoue, N.; Hirata, T.; Murata, S.; Anliker, M.; Eggermann, T.; et al. Complement and inflammasome overactivation mediates paroxysmal nocturnal hemoglobinuria with autoinflammation. J. Clin. Investig. 2019, 129, 5123–5136. [Google Scholar] [CrossRef]
- Gamage, D.G.; Hendrickson, T.L. GPI transamidase and GPI anchored proteins: Oncogenes and biomarkers for cancer. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 446–464. [Google Scholar] [CrossRef]
- Toh, Y.K.; Kamariah, N.; Maurer-Stroh, S.; Roessle, M.; Eisenhaber, F.; Adhikari, S.; Eisenhaber, B.; Grüber, G. Structural insight into the glycosylphosphatidylinositol transamidase subunits PIG-K and PIG-S from yeast. J. Struct. Biol. 2011, 173, 271–281. [Google Scholar] [CrossRef]
- Saw, W.G.; Eisenhaber, B.; Eisenhaber, F.; Grüber, G. Low-resolution structure of the soluble domain GPAA1 (yGPAA170-247) of the glycosylphosphatidylinositol transamidase subunit GPAA1 from Saccharomyces cerevisiae. Biosci. Rep. 2013, 33, e00033. [Google Scholar] [CrossRef]
- Liu, S.S.; Liu, Y.S.; Guo, X.Y.; Murakami, Y.; Yang, G.; Gao, X.D.; Kinoshita, T.; Fujita, M. A knockout cell library of GPI biosynthetic genes for functional studies of GPI-anchored proteins. Commun. Biol. 2021, 4, 777. [Google Scholar] [CrossRef] [PubMed]
- Ohishi, K.; Inoue, N.; Maeda, Y.; Takeda, J.; Riezman, H.; Kinoshita, T. Gaa1p and gpi8p are components of a glycosylphosphatidylinositol (GPI) transamidase that mediates attachment of GPI to proteins. Mol. Biol. Cell 2000, 11, 1523–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dall, E.; Brandstetter, H. Mechanistic and structural studies on legumain explain its zymogenicity, distinct activation pathways, and regulation. Proc. Natl. Acad. Sci. USA 2013, 110, 10940–10945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemu, X.; El Sahili, A.; Hu, S.; Wong, K.; Chen, Y.; Wong, Y.H.; Zhang, X.; Serra, A.; Goh, B.C.; Darwis, D.A.; et al. Structural determinants for peptide-bond formation by asparaginyl ligases. Proc. Natl. Acad. Sci. USA 2019, 116, 11737–11746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dall, E.; Zauner, F.B.; Soh, W.T.; Demir, F.; Dahms, S.O.; Cabrele, C.; Huesgen, P.F.; Brandstetter, H. Structural and functional studies of Arabidopsis thaliana legumain beta reveal isoform specific mechanisms of activation and substrate recognition. J. Biol. Chem. 2020, 295, 13047–13064. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, B.; Kolenko, P.; Buchholz, M.; Carrillo, D.R.; Parthier, C.; Wermann, M.; Rahfeld, J.U.; Reuter, G.; Schilling, S.; Stubbs, M.T.; et al. Crystal structures of glutaminyl cyclases (QCs) from Drosophila melanogaster reveal active site conservation between insect and mammalian QCs. Biochemistry 2012, 51, 7383–7392. [Google Scholar] [CrossRef]
- Ruiz-Carrillo, D.; Koch, B.; Parthier, C.; Wermann, M.; Dambe, T.; Buchholz, M.; Ludwig, H.H.; Heiser, U.; Rahfeld, J.U.; Stubbs, M.T.; et al. Structures of glycosylated mammalian glutaminyl cyclases reveal conformational variability near the active center. Biochemistry 2011, 50, 6280–6288. [Google Scholar] [CrossRef]
- Huang, K.F.; Hsu, H.L.; Karim, S.; Wang, A.H. Structural and functional analyses of a glutaminyl cyclase from Ixodes scapularis reveal metal-independent catalysis and inhibitor binding. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014, 70, 789–801. [Google Scholar] [CrossRef]
- Schilling, S.; Niestroj, A.J.; Rahfeld, J.U.; Hoffmann, T.; Wermann, M.; Zunkel, K.; Wasternack, C.; Demuth, H.U. Identification of human glutaminyl cyclase as a metalloenzyme. Potent inhibition by imidazole derivatives and heterocyclic chelators. J. Biol. Chem. 2003, 278, 49773–49779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.F.; Liu, Y.L.; Cheng, W.J.; Ko, T.P.; Wang, A.H. Crystal structures of human glutaminyl cyclase, an enzyme responsible for protein N-terminal pyroglutamate formation. Proc. Natl. Acad. Sci. USA 2005, 102, 13117–13122. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.F.; Liaw, S.S.; Huang, W.L.; Chia, C.Y.; Lo, Y.C.; Chen, Y.L.; Wang, A.H. Structures of human Golgi-resident glutaminyl cyclase and its complexes with inhibitors reveal a large loop movement upon inhibitor binding. J. Biol. Chem. 2011, 286, 12439–12449. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.F.; Liu, Y.L.; Wang, A.H. Cloning, expression, characterization, and crystallization of a glutaminyl cyclase from human bone marrow: A single zinc metalloenzyme. Protein Expr. Purif. 2005, 43, 65–72. [Google Scholar] [CrossRef]
- Booth, R.E.; Lovell, S.C.; Misquitta, S.A.; Bateman, R.C. Human glutaminyl cyclase and bacterial zinc aminopeptidase share a common fold and active site. BMC Biol. 2004, 2, 2. [Google Scholar] [CrossRef] [Green Version]
- Schilling, S.; Hoffmann, T.; Rosche, F.; Manhart, S.; Wasternack, C.; Demuth, H.U. Heterologous expression and characterization of human glutaminyl cyclase: Evidence for a disulfide bond with importance for catalytic activity. Biochemistry 2002, 41, 10849–10857. [Google Scholar] [CrossRef] [PubMed]
- Schilling, S.; Manhart, S.; Hoffmann, T.; Ludwig, H.H.; Wasternack, C.; Demuth, H.U. Substrate specificity of glutaminyl cyclases from plants and animals. Biol. Chem. 2003, 384, 1583–1592. [Google Scholar] [CrossRef]
- Schilling, S.; Cynis, H.; von Bohlen, A.; Hoffmann, T.; Wermann, M.; Heiser, U.; Buchholz, M.; Zunkel, K.; Demuth, H.U. Isolation, catalytic properties, and competitive inhibitors of the zinc-dependent murine glutaminyl cyclase. Biochemistry 2005, 44, 13415–13424. [Google Scholar] [CrossRef]
- Yu, J.; Nagarajan, S.; Knez, J.J.; Udenfriend, S.; Chen, R.; Medof, M.E. The affected gene underlying the class K glycosylphosphatidylinositol (GPI) surface protein defect codes for the GPI transamidase. Proc. Natl. Acad. Sci. USA 1997, 94, 12580–12585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherezov, V.; Rosenbaum, D.M.; Hanson, M.A.; Rasmussen, S.G.; Thian, F.S.; Kobilka, T.S.; Choi, H.J.; Kuhn, P.; Weis, W.I.; Kobilka, B.K.; et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 2007, 318, 1258–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, S.G.; Choi, H.J.; Fung, J.J.; Pardon, E.; Casarosa, P.; Chae, P.S.; Devree, B.T.; Rosenbaum, D.M.; Thian, F.S.; Kobilka, T.S.; et al. Structure of a nanobody-stabilized active state of the β(2) adrenoceptor. Nature 2011, 469, 175–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenbaum, D.M.; Zhang, C.; Lyons, J.A.; Holl, R.; Aragao, D.; Arlow, D.H.; Rasmussen, S.G.; Choi, H.J.; Devree, B.T.; Sunahara, R.K.; et al. Structure and function of an irreversible agonist-β(2) adrenoceptor complex. Nature 2011, 469, 236–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, M.; Hibbs, R.E.; Gouaux, E. A fluorescence-detection size-exclusion chromatography-based thermostability assay for membrane protein precrystallization screening. Structure 2012, 20, 1293–1299. [Google Scholar] [CrossRef] [Green Version]
- She, J.; Guo, J.; Chen, Q.; Zeng, W.; Jiang, Y.; Bai, X.C. Structural insights into the voltage and phospholipid activation of the mammalian TPC1 channel. Nature 2018, 556, 130–134. [Google Scholar] [CrossRef]
- Singh, A.K.; McGoldrick, L.L.; Sobolevsky, A.I. Structure and gating mechanism of the transient receptor potential channel TRPV3. Nat. Struct. Mol. Biol. 2018, 25, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Suzuki, T.; Rubinstein, J.L. Structure of a bacterial ATP synthase. eLife 2019, 8, e43128. [Google Scholar] [CrossRef]
- Pan, X.; Li, Z.; Zhou, Q.; Shen, H.; Wu, K.; Huang, X.; Chen, J.; Zhang, J.; Zhu, X.; Lei, J.; et al. Structure of the human voltage-gated sodium channel Na(v)1.4 in complex with β1. Science 2018, 362, eaau2486–eaau2596. [Google Scholar] [CrossRef] [PubMed]
- Mannervik, B.; Danielson, U.H. Glutathione transferases—Structure and catalytic activity. CRC Crit. Rev. Biochem. 1988, 23, 283–337. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Medof, M.E.; Silber, R.; Nussenzweig, V. Distribution of decay-accelerating factor in the peripheral blood of normal individuals and patients with paroxysmal nocturnal hemoglobinuria. J. Exp. Med. 1985, 162, 75–92. [Google Scholar] [CrossRef]
- Maeda, Y.; Tashima, Y.; Houjou, T.; Fujita, M.; Yoko-o, T.; Jigami, Y.; Taguchi, R.; Kinoshita, T. Fatty acid remodeling of GPI-anchored proteins is required for their raft association. Mol. Biol. Cell 2007, 18, 1497–1506. [Google Scholar] [CrossRef]
- Zheng, S.Q.; Palovcak, E.; Armache, J.P.; Verba, K.A.; Cheng, Y.; Agard, D.A. MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 2017, 14, 331–332. [Google Scholar] [CrossRef] [Green Version]
- Rohou, A.; Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 2015, 192, 216–221. [Google Scholar] [CrossRef]
- Zivanov, J.; Nakane, T.; Forsberg, B.O.; Kimanius, D.; Hagen, W.J.; Lindahl, E.; Scheres, S.H. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 2018, 7, e42166. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.-S.; Jin, F.; Liu, Y.-S.; Murakami, Y.; Sugita, Y.; Kato, T.; Gao, X.-D.; Kinoshita, T.; Hattori, M.; Fujita, M. Functional Analysis of the GPI Transamidase Complex by Screening for Amino Acid Mutations in Each Subunit. Molecules 2021, 26, 5462. https://doi.org/10.3390/molecules26185462
Liu S-S, Jin F, Liu Y-S, Murakami Y, Sugita Y, Kato T, Gao X-D, Kinoshita T, Hattori M, Fujita M. Functional Analysis of the GPI Transamidase Complex by Screening for Amino Acid Mutations in Each Subunit. Molecules. 2021; 26(18):5462. https://doi.org/10.3390/molecules26185462
Chicago/Turabian StyleLiu, Si-Si, Fei Jin, Yi-Shi Liu, Yoshiko Murakami, Yukihiko Sugita, Takayuki Kato, Xiao-Dong Gao, Taroh Kinoshita, Motoyuki Hattori, and Morihisa Fujita. 2021. "Functional Analysis of the GPI Transamidase Complex by Screening for Amino Acid Mutations in Each Subunit" Molecules 26, no. 18: 5462. https://doi.org/10.3390/molecules26185462