
Replacement of Volatile Acetic Acid by Solid SiO2@COOH Silica (Nano)Beads for (Ep)Oxidation Using Mn and Fe Complexes Containing BPMEN Ligand


 and
and
Abstract
:1. Introduction
2. Results and Discussion
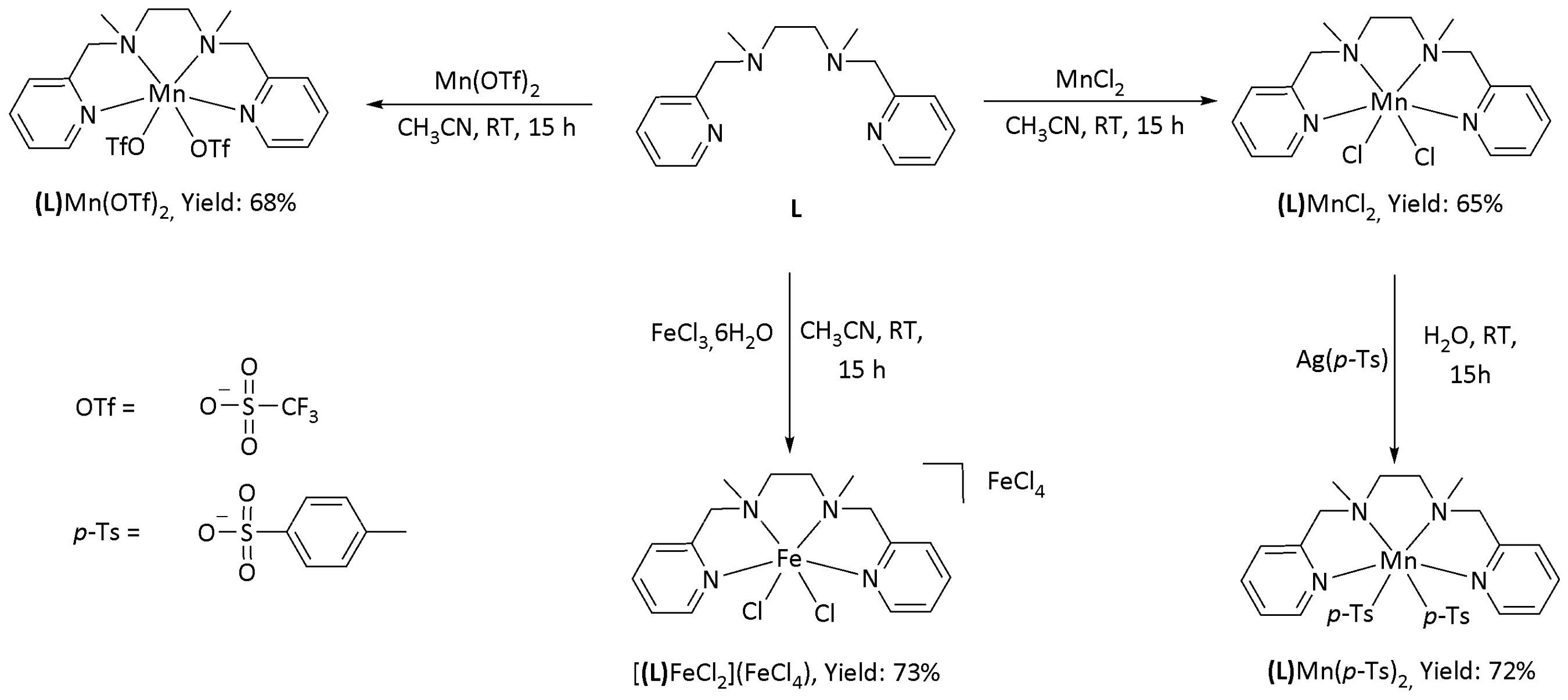
2.1. Metal Complexes
2.1.1. Synthesis
2.1.2. X-ray Characterization of the Complexes
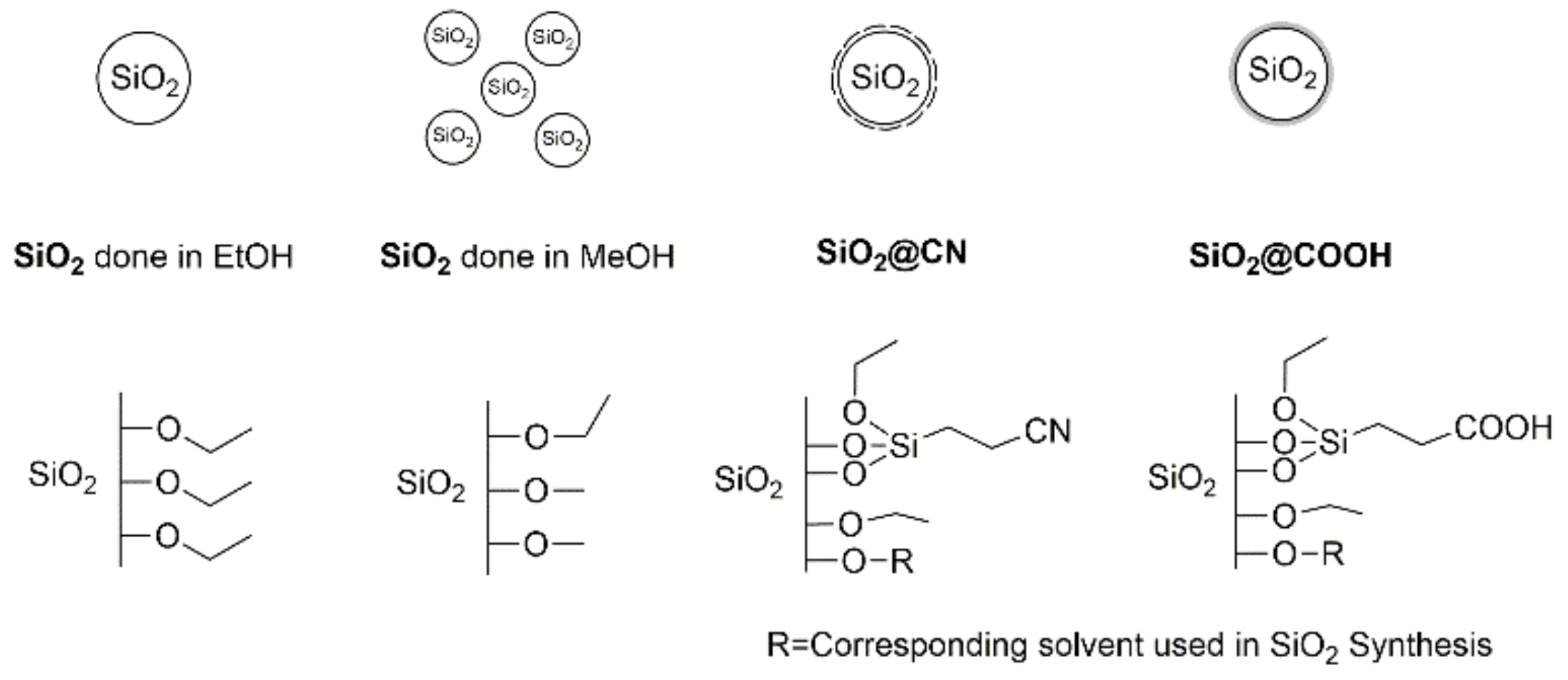
2.2. Silica Beads
2.2.1. Synthesis
2.2.2. Characterization
Morphological Study
- Transmission electron microscope (TEM) analysis
- Dynamic light scattering (DLS) measurements
Spectroscopic Characterization of the Grafting
- Infrared spectroscopy
- Solid state NMR
- Quantification by 1H NMR in solution
- Determination of function coverage of functionalized silica beads
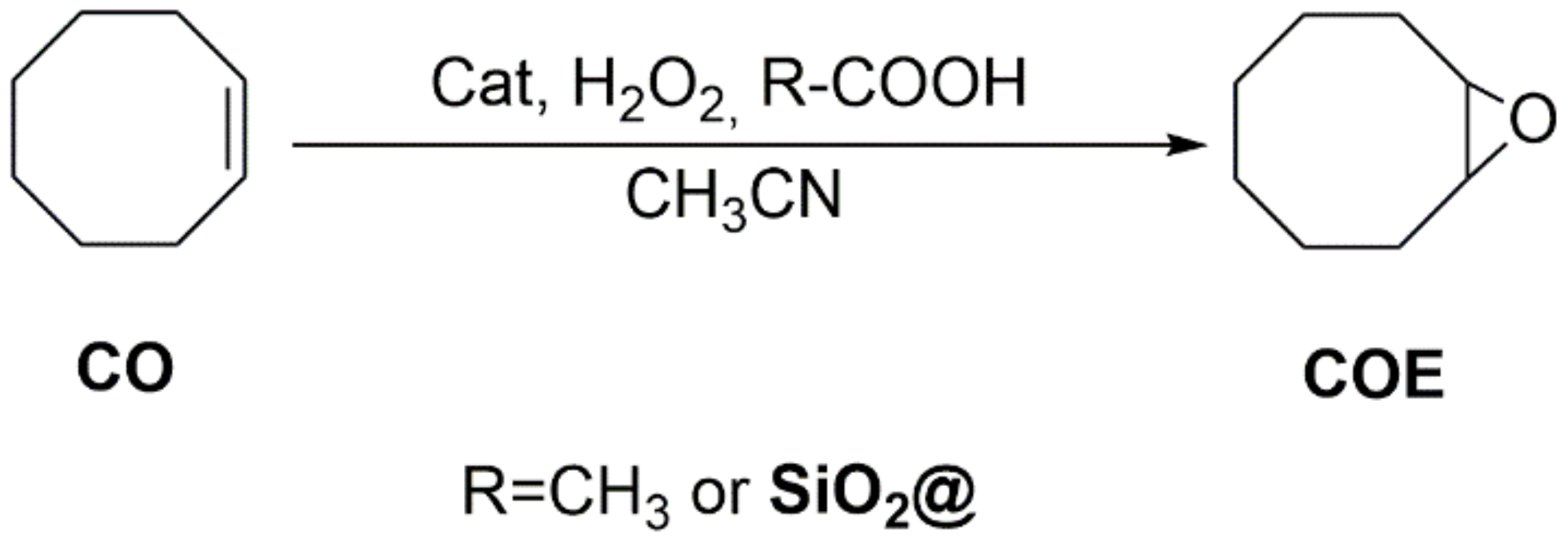
2.3. Catalysis
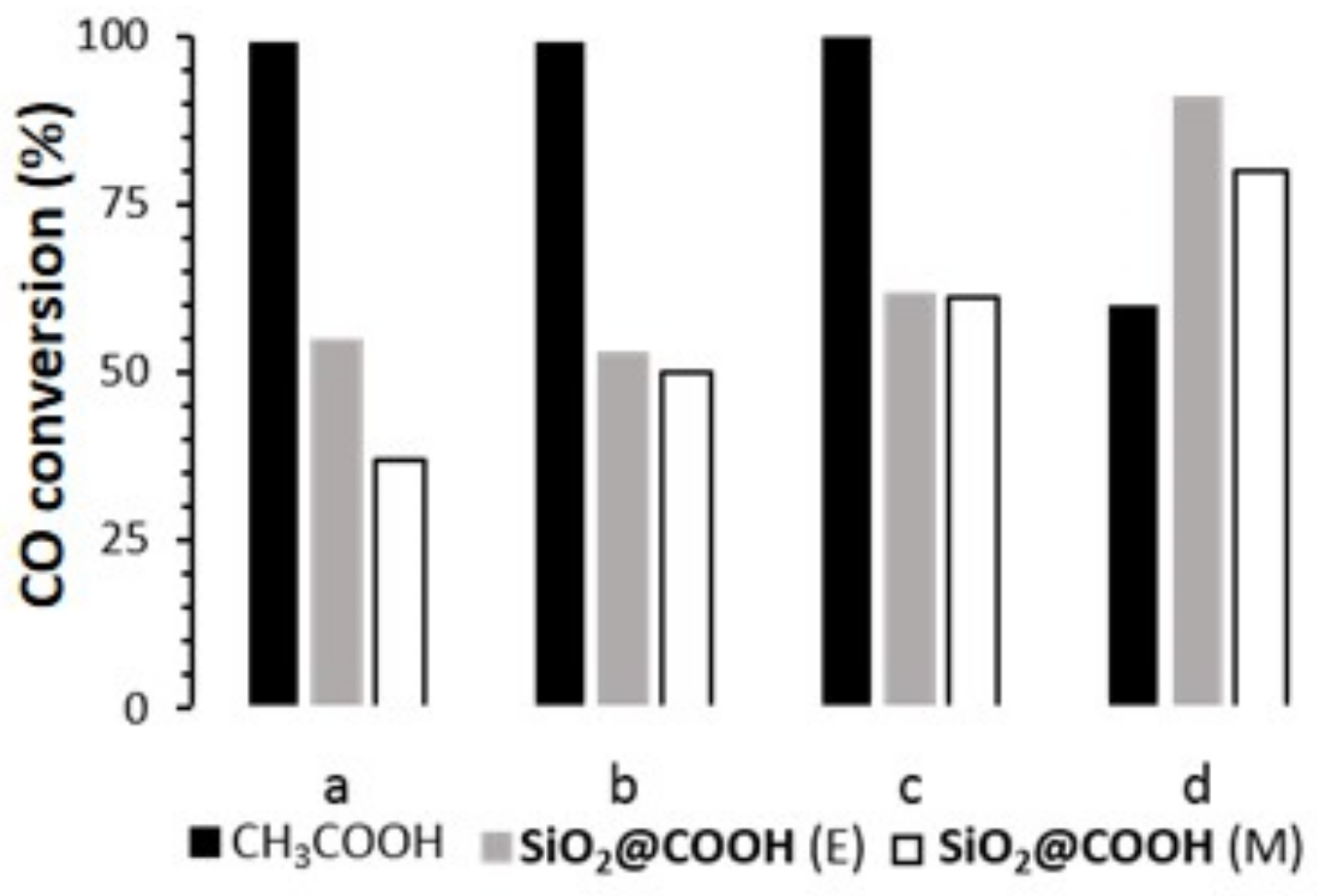
2.3.1. Oxidation of Cyclooctene
2.3.2. Oxidation of Cyclohexene
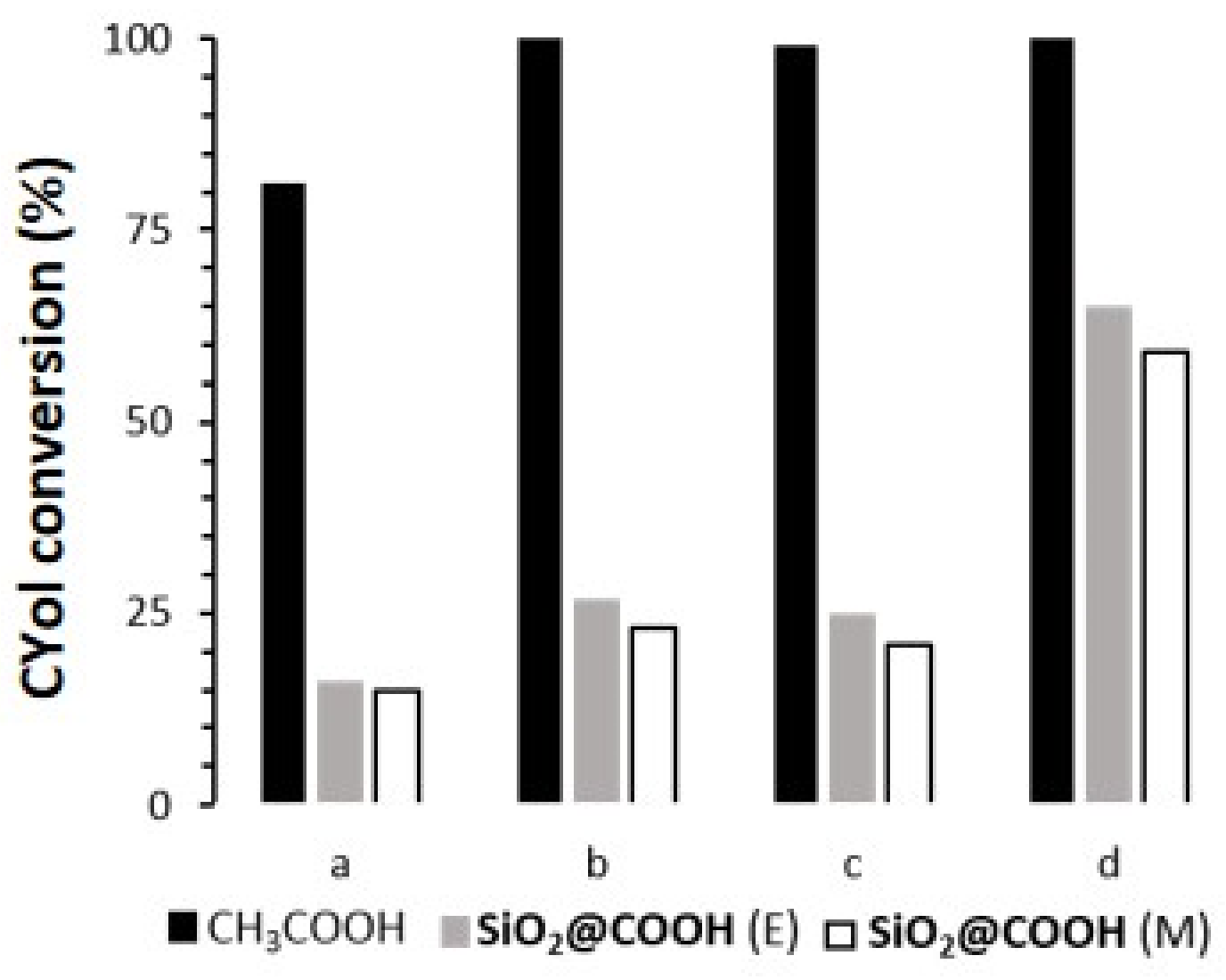
2.3.3. Oxidation of Cyclohexanol
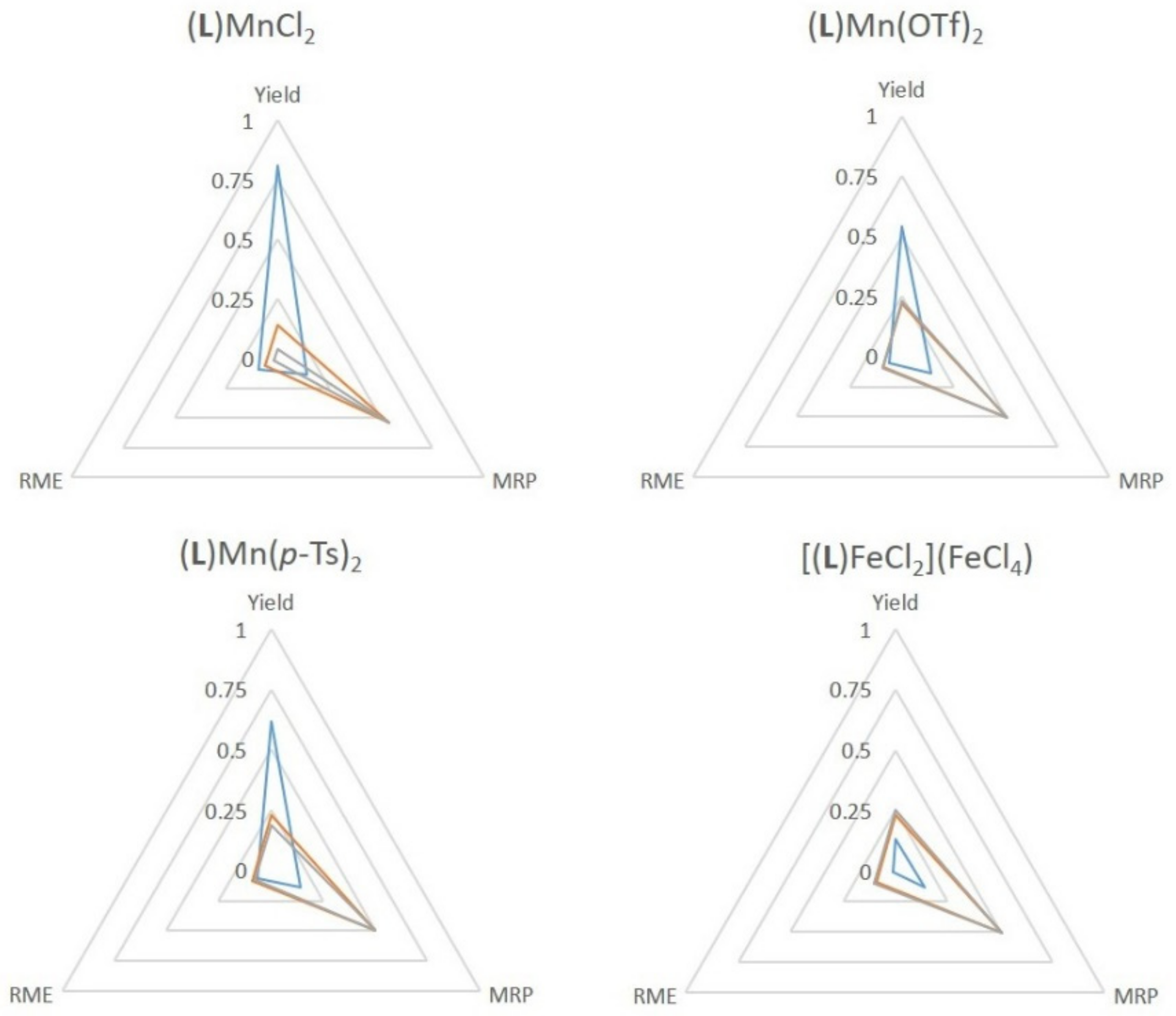
2.4. Green Metrics
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. X-ray Structural Analyses
3.2.2. Dynamic Light Scattering
3.2.3. TEM
3.2.4. Infrared Spectroscopy
3.2.5. Solid State NMR
3.2.6. Solution NMR
3.2.7. Elemental Analysis
3.2.8. Centrifugation
3.2.9. Gas Chromatography
3.2.10. Quantification of the Number of Functions per Gram of Grafted Silica through 1H NMR in Solution
3.3. Synthesis of Metal Complexes
3.3.1. (L)MnCl2
3.3.2. (L)Mn(OTf)2
3.3.3. (L)Mn(p-Ts)2
3.3.4. [(L)FeCl2](FeCl4)
3.4. Synthesis of Silica Particles
3.4.1. SiO2 Particles in EtOH (SiO2(E))
3.4.2. SiO2@CN(E) Particles
3.4.3. SiO2@COOH(E) Particles
3.4.4. SiO2 Nanoparticles in Methanol (SiO2(M))
3.4.5. SiO2@CN(M) Nanoparticles
3.4.6. SiO2@COOH(M) Nanoparticles
3.5. Catalytic Experiments
3.5.1. General Procedure of Catalysis with CH3COOH
3.5.2. General Procedure of Catalysis with SiO2@COOH
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Damico, R. Preparation, Characterization, and Reactions of Lithium and Sodium Tetraalkylboron Compounds. J. Org. Chem. 1964, 29, 1971–1976. [Google Scholar] [CrossRef]
- Organic Syntheses, Inc. m-Chloroperbenzoic Acid. Org. Synth. 1970, 50, 15. [Google Scholar] [CrossRef]
- Brulé, E.; De Miguel, Y.R. Supported manganese porphyrin catalysts as P450 enzyme mimics for alkene epoxidation. Tetrahedron Lett. 2002, 43, 8555–8558. [Google Scholar] [CrossRef]
- Burfield, D.R.; Eng, A.-H. Glass transition and crystallization phenomena in epoxidized trans-polyisoprene: A differential scanning calorimetry study. Polymer 1989, 30, 2019–2022. [Google Scholar] [CrossRef]
- Dryuk, V.G. Advances in the Development of Methods for the Epoxidation of Olefins. Russ. Chem. Rev. 1985, 54, 986–1005. [Google Scholar] [CrossRef]
- Swern, D. (Ed.) Organic Peroxides; Interscience: New York, NY, USA, 1971; Volume 2. [Google Scholar]
- Shen, Y.; Jiang, P.; Wai, P.T.; Gu, Q.; Zhang, W. Recent Progress in Application of Molybdenum-Based Catalysts for Epoxidation of Alkenes. Catalysts 2019, 9, 31. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, K.; Michaud, P.; Kochi, J.K. Epoxidation of olefins with cationic (salen)manganese(III) complexes. The modulation of catalytic activity by substituents. J. Am. Chem. Soc. 1986, 108, 2309–2320. [Google Scholar] [CrossRef]
- Rudolph, J.; Reddy, K.L.; Chiang, J.P.; Sharpless, K.B. Highly Efficient Epoxidation of Olefins Using Aqueous H2O2 and Catalytic Methyltrioxorhenium/Pyridine: Pyridine-Mediated Ligand Acceleration. J. Am. Chem. Soc. 1997, 119, 6189–6190. [Google Scholar] [CrossRef]
- Kobayashi, M.; Tawara, K. Method for Producing Epoxy Compound. Japan Patent JP 2007230908A, 13 September 2007. [Google Scholar]
- Bagherzadeh, M.; Tahsini, L.; Latifi, R.; Woo, L.K. cis-Dioxo-molybdenum(VI)-oxazoline complex catalyzed epoxidation of olefins by tert-butyl hydrogen peroxide. Inorg. Chim. Acta 2009, 362, 3698–3702. [Google Scholar] [CrossRef]
- Dallmann, K.; Buffon, R.; Loh, W. Catalyst recycling in the epoxidation of alkenes catalyzed by MoO2(acac)2 through precipitation with poly(ethylene oxide). J. Mol. Catal. A Chem. 2002, 178, 43–46. [Google Scholar] [CrossRef]
- Yamazaki, M.; Endo, H.; Tomoyama, M.; Kurusu, Y. Catalytic Epoxidation of Cyclohexene witht-Butyl Hydroperoxide in the Presence of Various Molybdenum Complexes. Bull. Chem. Soc. Jpn. 1983, 56, 3523–3524. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.C.; Smith, N.M.; Robertson, M.; Scott, M.S. An investigation into oxo analogues of molybdenum olefin metathesis complexes as epoxidation catalysts for alkenes. Tetrahedron Lett. 2009, 50, 5344–5346. [Google Scholar] [CrossRef]
- Sherwood, J. European Restrictions on 1,2-Dichloroethane: C−H Activation Research and Development Should Be Liberated and not Limited. Angew. Chem. Int. Ed. 2018, 57, 14286–14290. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Agustin, D.; Poli, R. Influence of ligand substitution on molybdenum catalysts with tridentate Schiff base ligands for the organic solvent-free oxidation of limonene using aqueous TBHP as oxidant. Mol. Catal. 2017, 443, 52–59. [Google Scholar] [CrossRef]
- Wang, W.; Daran, J.-C.; Poli, R.; Agustin, D. OH-substituted tridentate ONO Schiff base ligands and related molybdenum(VI) complexes for solvent-free (ep)oxidation catalysis with TBHP as oxidant. J. Mol. Catal. A Chem. 2016, 416, 117–126. [Google Scholar] [CrossRef]
- Cvijanović, D.; Pisk, J.; Pavlović, G.; Šišak-Jung, D.; Matković-Čalogović, D.; Cindric, M.; Agustin, D.; Vrdoljak, V. Discrete mononuclear and dinuclear compounds containing a MoO22+ core and 4-aminobenzhydrazone ligands: Synthesis, structure and organic-solvent-free epoxidation activity. New J. Chem. 2018, 43, 1791–1802. [Google Scholar] [CrossRef]
- Cordelle, C.; Agustin, D.; Daran, J.-C.; Poli, R. Oxo-bridged bis oxo-vanadium(V) complexes with tridentate Schiff base ligands (VOL)2O (L=SAE, SAMP, SAP): Synthesis, structure and epoxidation catalysis under solvent-free conditions. Inorg. Chim. Acta 2010, 364, 144–149. [Google Scholar] [CrossRef]
- Morlot, J.; Uyttebroeck, N.; Agustin, D.; Poli, R. Solvent-Free Epoxidation of Olefins Catalyzed by “[MoO2(SAP)]”: A New Mode of tert -Butylhydroperoxide Activation. ChemCatChem 2012, 5, 601–611. [Google Scholar] [CrossRef]
- Guérin, B.; Fernandes, D.M.; Daran, J.-C.; Agustin, D.; Poli, R. Investigation of induction times, activity, selectivity, interface and mass transport in solvent-free epoxidation by H2O2 and TBHP: A study with organic salts of the [PMo12O40]3− anion. New J. Chem. 2013, 37, 3466–3475. [Google Scholar] [CrossRef]
- Pisk, J.; Agustin, D.; Poli, R. Organic Salts and Merrifield Resin Supported [PM12O40]3− (M = Mo or W) as Catalysts for Adipic Acid Synthesis. Molecules 2019, 24, 783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Gayet, F.; Guillo, P.; Agustin, D. Organic Solvent-Free Olefins and Alcohols (ep)oxidation Using Recoverable Catalysts Based on [PM12O40]3- (M = Mo or W) Ionically Grafted on Amino Functionalized Silica Nanobeads. Materials 2019, 12, 3278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, C.; Wang, B.; Wang, Y.; Xia, C.; Lee, Y.-M.; Nam, W.; Sun, W. Proton-Promoted and Anion-Enhanced Epoxidation of Olefins by Hydrogen Peroxide in the Presence of Nonheme Manganese Catalysts. J. Am. Chem. Soc. 2016, 138, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Ottenbacher, R.V.; Samsonenko, D.G.; Talsi, E.P.; Bryliakov, K.P. Enantioselective Epoxidations of Olefins with Various Oxidants on Bioinspired Mn Complexes: Evidence for Different Mechanisms and Chiral Additive Amplification. ACS Catal. 2016, 6, 979–988. [Google Scholar] [CrossRef]
- Du, J.; Miao, C.; Xia, C.; Lee, Y.-M.; Nam, W.; Sun, W. Mechanistic Insights into the Enantioselective Epoxidation of Olefins by Bioinspired Manganese Complexes: Role of Carboxylic Acid and Nature of Active Oxidant. ACS Catal. 2018, 8, 4528–4538. [Google Scholar] [CrossRef]
- Balleste, R.M.; Que, L. Iron-Catalyzed Olefin Epoxidation in the Presence of Acetic Acid: Insights into the Nature of the Metal-Based Oxidant. J. Am. Chem. Soc. 2007, 129, 15964–15972. [Google Scholar] [CrossRef] [PubMed]
- White, M.C.; Doyle, A.G.; Jacobsen, E.N. A synthetically useful, self-assembling MMO mimic system for catalytic alkene epoxidation with aqueous H2O2. J. Am. Chem. Soc. 2001, 123, 7194–7195. [Google Scholar] [CrossRef]
- Lorenz, S.; Plietker, B. Selectivity Trends in Olefin Epoxidations Catalyzed by (NNNN)Manganese(+II) Complexes using Trifluoroethanol as the Solvent. ChemCatChem 2016, 8, 3203–3206. [Google Scholar] [CrossRef]
- Duban, E.A.; Bryliakov, K.P.; Talsi, E.P. The Active Intermediates of Non-Heme-Iron-Based Systems for Catalytic Alkene Epoxidation with H2O2/CH3COOH. Eur. J. Inorg. Chem. 2007, 2007, 852–857. [Google Scholar] [CrossRef]
- Clemente-Tejeda, D.; Bermejo, F.A. Oxidation of alkenes with non-heme iron complexes: Suitability as an organic synthetic method. Tetrahedron 2014, 70, 9381–9386. [Google Scholar] [CrossRef]
- Clemente-Tejeda, D.; López-Moreno, A.; Bermejo, F.A. Non-heme iron catalysis in CC, C–H, and CH2 oxidation reactions. Oxidative transformations on terpenoids catalyzed by Fe(bpmen)(OTf)2. Tetrahedron 2013, 69, 2977–2986. [Google Scholar] [CrossRef]
- Clemente-Tejeda, D.; López-Moreno, A.; Bermejo, F.A. Oxidation of unsaturated steroid ketones with hydrogen peroxide catalyzed by Fe(bpmen)(OTf)2. New methodology to access biologically active steroids by chemo-, and stereoselective processes. Tetrahedron 2012, 68, 9249–9255. [Google Scholar] [CrossRef]
- Duban, E.A.; Brylyakov, K.P.; Talsi, E.P. The nature of active species in catalytic systems based on non-heme iron complexes, hydrogen peroxide, and acetic acid for selective olefin epoxidation. Kinet. Catal. 2008, 49, 379–385. [Google Scholar] [CrossRef]
- Taktak, S.; Kryatov, S.V.; Haas, T.E.; Rybak-Akimova, E.V. Diiron(III) oxo-bridged complexes with BPMEN and additional monodentate or bidentate ligands: Synthesis and reactivity in olefin epoxidation with H2O2. J. Mol. Catal. A Chem. 2006, 259, 24–34. [Google Scholar] [CrossRef]
- Chen, K.; Que, L., Jr. cis-Dihydroxylation of Olefins by a Non-Heme Iron Catalyst: A Functional Model for Rieske Dioxygenases. Angew. Chem. Int. Ed. 1999, 38, 2227–2229. [Google Scholar] [CrossRef]
- Cussó, O.; Garcia-Bosch, I.; Ribas, X.; Fillol, J.L.; Costas, M. Asymmetric Epoxidation with H2O2 by Manipulating the Electronic Properties of Non-heme Iron Catalysts. J. Am. Chem. Soc. 2013, 135, 14871–14878. [Google Scholar] [CrossRef] [PubMed]
- Bautz, J.; Comba, P.; De Laorden, C.L.; Menzel, M.; Rajaraman, G. Biomimetic High-Valent Non-Heme Iron Oxidants for thecis-Dihydroxylation and Epoxidation of Olefins. Angew. Chem. Int. Ed. 2007, 46, 8067–8070. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.-Y.; Huang, Y.-B.; Niu, X.; Pan, H. Highly Efficient Silica-Supported Peroxycarboxylic Acid for the Epoxidation of Unsaturated Fatty Acid Methyl Esters and Vegetable Oils. ACS Sustain. Chem. Eng. 2016, 4, 3840–3849. [Google Scholar] [CrossRef]
- Crucho, C.I.C.; Baleizão, C.; Farinha, J.P.S. Functional Group Coverage and Conversion Quantification in Nanostructured Silica by 1H NMR. Anal. Chem. 2017, 89, 681–687. [Google Scholar] [CrossRef]
- Cohen, R.; Sukenik, C.N. Highly loaded COOH functionalized silica particles. Colloids Surf. A Physicochem. Eng. Asp. 2016, 504, 242–251. [Google Scholar] [CrossRef]
- Feinle, A.; Leichtfried, F.; Straßer, S.; Hüsing, N. Carboxylic acid-functionalized porous silica particles by a co-condensation approach. J. Sol-Gel Sci. Technol. 2017, 81, 138–146. [Google Scholar] [CrossRef] [Green Version]
- Boullanger, A.; Gracy, G.; Bibent, N.; Devautour-Vinot, S.; Clément, S.; Mehdi, A. From an Octakis(3-cyanopropyl)silsesquioxane Building Block to a Highly COOH-Functionalized Hybrid Organic-Inorganic Material. Eur. J. Inorg. Chem. 2011, 2012, 143–150. [Google Scholar] [CrossRef]
- Ghaida, F.A.; Clément, S.; Mehdi, A. Heterogenized Catalysis on Metals Impregnated Mesoporous Silica. In Novel Nanoscale Hybrid Materials; Wiley: Hoboken, NJ, USA, 2018; pp. 323–349. [Google Scholar]
- Touisni, N.; Kanfar, N.; Ulrich, S.; Dumy, P.; Supuran, C.T.; Mehdi, A.; Winum, J.-Y. Fluorescent Silica Nanoparticles with Multivalent Inhibitory Effects towards Carbonic Anhydrases. Chem.- Eur. J. 2015, 21, 10306–10309. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, Y.; Pi, H.; Zeng, G. Controlling the shear thickening behavior of suspensions by changing the surface properties of dispersed microspheres. RSC Adv. 2019, 9, 3469–3478. [Google Scholar] [CrossRef] [Green Version]
- Atta, S.; Fatima, M.; Islam, A.; Gull, N.; Sultan, M. Grafting of Silica Particles with Linoleic Acid via Modified Stober’s Method for Preconcenrtration of Pesticides in Drinking Water. Key Eng. Mater. 2018, 778, 316–324. [Google Scholar] [CrossRef] [Green Version]
- Yadav, M.; Akita, T.; Tsumori, N.; Xu, Q. Strong metal–molecular support interaction (SMMSI): Amine-functionalized gold nanoparticles encapsulated in silica nanospheres highly active for catalytic decomposition of formic acid. J. Mater. Chem. 2012, 22, 12582–12586. [Google Scholar] [CrossRef]
- Berg, R.V.D.; Parmentier, T.E.; Elkjær, C.F.; Gommes, C.; Sehested, J.; Helveg, S.; De Jongh, P.E.; De Jong, K.P. Support Functionalization To Retard Ostwald Ripening in Copper Methanol Synthesis Catalysts. ACS Catal. 2015, 5, 4439–4448. [Google Scholar] [CrossRef]
- Yantasee, W.; Rutledge, R.D.; Chouyyok, W.; Sukwarotwat, V.; Orr, G.; Warner, C.L.; Warner, M.G.; Fryxell, G.E.; Wiacek, R.J.; Timchalk, C.; et al. Functionalized Nanoporous Silica for the Removal of Heavy Metals from Biological Systems: Adsorption and Application. ACS Appl. Mater. Interfaces 2010, 2, 2749–2758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-S.; Chah, S.; Yi, J. Preparation of modified silica for heavy metal removal. Korean J. Chem. Eng. 2000, 17, 118–121. [Google Scholar] [CrossRef]
- Leon, P.A.A.I.-D.; Contreras, C.A.; Thornburg, N.E.; Thompson, A.B.; Notestein, J.M. Catalyst structure and substituent effects on epoxidation of styrenics with immobilized Mn(tmtacn) complexes. Appl. Catal. A Gen. 2016, 511, 78–86. [Google Scholar] [CrossRef]
- Schoenfeldt, N.J.; Ni, Z.; Korinda, A.W.; Meyer, R.J.; Notestein, J.M. Manganese Triazacyclononane Oxidation Catalysts Grafted under Reaction Conditions on Solid Cocatalytic Supports. J. Am. Chem. Soc. 2011, 133, 18684–18695. [Google Scholar] [CrossRef]
- Ottenbacher, R.V.; Talsi, E.P.; Bryliakov, K.P. Bioinspired Mn-aminopyridine catalyzed epoxidations of olefins with various oxidants: Enantioselectivity and mechanism. Catal. Today 2016, 278, 30–39. [Google Scholar] [CrossRef]
- Cussó, O.; Serrano-Plana, J.; Costas, M. Evidence of a Sole Oxygen Atom Transfer Agent in Asymmetric Epoxidations with Fe-pdp Catalysts. ACS Catal. 2017, 7, 5046–5053. [Google Scholar] [CrossRef]
- Hureau, C.; Blondin, G.; Charlot, M.-F.; Philouze, C.; Nierlich, M.; Cesario, M.; Anxolabéhère-Mallart, E. Synthesis, Structure, and Characterization of New Mononuclear Mn(II) Complexes. Electrochemical Conversion into New Oxo-Bridged Mn2(III,IV) Complexes. Role of Chloride Ions. Inorg. Chem. 2005, 44, 3669–3683. [Google Scholar] [CrossRef]
- Chow, T.W.-S.; Wong, E.L.-M.; Guo, Z.; Liu, Y.; Huang, J.-S.; Che, C.M. cis-Dihydroxylation of Alkenes with Oxone Catalyzed by Iron Complexes of a Macrocyclic Tetraaza Ligand and Reaction Mechanism by ESI-MS Spectrometry and DFT Calculations. J. Am. Chem. Soc. 2010, 132, 13229–13239. [Google Scholar] [CrossRef]
- To, W.-P.; Chow, T.W.-S.; Tse, C.-W.; Guan, X.; Huang, J.-S.; Che, C.-M. Water oxidation catalysed by iron complex of N,N′-dimethyl-2,11-diaza[3,3](2,6)pyridinophane. Spectroscopy of iron–oxo intermediates and density functional theory calculations. Chem. Sci. 2015, 6, 5891–5903. [Google Scholar] [CrossRef] [Green Version]
- Murphy, A.; Dubois, G.; Stack, T.D.P. Efficient Epoxidation of Electron-Deficient Olefins with a Cationic Manganese Complex. J. Am. Chem. Soc. 2003, 125, 5250–5251. [Google Scholar] [CrossRef] [PubMed]
- Dexuan, W.; Guian, L.; Qingyan, H.; Ziqiang, W.; Liping, P.; Zhongyue, Z.; Hairong, Z. Synthesis of Au-SiO2 Composite Nanospheres and Their Catalytic Activity. J. Nanosci. Nanotechnol. 2016, 16, 3821–3826. [Google Scholar] [CrossRef]
- Bourebrab, M.A.; Oben, D.T.; Durand, G.G.; Taylor, P.G.; Bruce, J.I.; Bassindale, A.R.; Taylor, A. Influence of the initial chemical conditions on the rational design of silica particles. J. Sol-Gel Sci. Technol. 2018, 88, 430–441. [Google Scholar] [CrossRef] [Green Version]
- Green, D.; Jayasundara, S.; Lam, Y.-F.; Harris, M. Chemical reaction kinetics leading to the first Stober silica nanoparticles—NMR and SAXS investigation. J. Non-Cryst. Solids 2003, 315, 166–179. [Google Scholar] [CrossRef]
- Suratwala, T.; Hanna, M.; Whitman, P. Effect of humidity during the coating of Stöber silica sols. J. Non- Cryst. Solids 2004, 349, 368–376. [Google Scholar] [CrossRef]
- Stöber, W.; Fink, A.; Bohn, E. Controlled growth of monodisperse silica spheres in the micron size range. J. Colloid Interface Sci. 1968, 26, 62–69. [Google Scholar] [CrossRef]
- Wang, X.-D.; Shen, Z.-X.; Sang, T.; Cheng, X.-B.; Li, M.-F.; Chen, L.-Y.; Wang, Z.-S. Preparation of spherical silica particles by Stöber process with high concentration of tetra-ethyl-orthosilicate. J. Colloid Interface Sci. 2010, 341, 23–29. [Google Scholar] [CrossRef]
- Green, D.; Lin, J.; Lam, Y.-F.; Hu, M.; Schaefer, D.W.; Harris, M. Size, volume fraction, and nucleation of Stober silica nanoparticles. J. Colloid Interface Sci. 2003, 266, 346–358. [Google Scholar] [CrossRef]
- Malay, O.; Yilgor, I.; Menceloglu, Y.Z. Effects of solvent on TEOS hydrolysis kinetics and silica particle size under basic conditions. J. Sol-Gel Sci. Technol. 2013, 67, 351–361. [Google Scholar] [CrossRef]
- Bu, J.; Li, R.; Quah, C.W.; Carpenter, K.J. Propagation of PAMAM Dendrons on Silica Gel: A Study on the Reaction Kinetics. Macromolecules 2004, 37, 6687–6694. [Google Scholar] [CrossRef]
- Aneja, K.S.; Bohm, S.; Khanna, A.S.; Bohm, H.L.M. Graphene based anticorrosive coatings for Cr(vi) replacement. Nanoscale 2015, 7, 17879–17888. [Google Scholar] [CrossRef]
- Das, D.; Yang, Y.; O’Brien, J.S.; Breznan, D.; Nimesh, S.; Bernatchez, S.; Hill, M.; Sayari, A.; Vincent, R.; Kumarathasan, P. Synthesis and Physicochemical Characterization of MesoporousSiO2Nanoparticles. J. Nanomater. 2014, 2014, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Feifel, S.C.; Lisdat, F. Silica nanoparticles for the layer-by-layer assembly of fully electro-active cytochrome c multilayers. J. Nanobiotechnology 2011, 9, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trébosc, J.; Wiench, J.W.; Huh, S.; Lin, V.S.-Y.; Pruski, M. Solid-State NMR Study of MCM-41-type Mesoporous Silica Nanoparticles. J. Am. Chem. Soc. 2005, 127, 3057–3068. [Google Scholar] [CrossRef] [PubMed]
- Mouawia, R.; Mehdi, A.; Reyé, C.; Corriu, R. Direct synthesis of ordered and highly functionalized organosilicas containing carboxylic acid groups. J. Mater. Chem. 2007, 17, 616–618. [Google Scholar] [CrossRef]
- Sharma, R.K.; Sharma, S.; Gulati, S.; Pandey, A. Fabrication of a novel nano-composite carbon paste sensor based on silica-nanospheres functionalized with isatin thiosemicarbazone for potentiometric monitoring of Cu2+ ions in real samples. Anal. Methods 2013, 5, 1414–1426. [Google Scholar] [CrossRef]
- Ribeiro, S.O.; Granadeiro, C.; de Almeida, P.M.; Pires, J.; Sánchez, M.D.C.C.; Campos-Martin, J.M.; Gago, S.; de Castro, B.; Balula, S.S. Oxidative desulfurization strategies using Keggin-type polyoxometalate catalysts: Biphasic versus solvent-free systems. Catal. Today 2019, 333, 226–236. [Google Scholar] [CrossRef]
- Park, J.S.; Hah, J.; Koo, S.M.; Lee, Y.S. Effect of Alcohol Chain Length on Particle Growth in a Mixed Solvent System. J. Ceram. Process. Res. 2006, 7, 83–89. [Google Scholar]
- Beganskienė, A.; Sirutkaitis, V.; Kurtinaitienė, M.; Juškėnas, R.; Kareiva, A. FTIR, TEM and NMR Iinvestigations of Stöber Silica Nanoparticles. Mater. Sci. (Medžiagotyra) 2004, 10, 287–290. [Google Scholar] [CrossRef]
- Van De Vyver, S.; Roman-Leshkov, Y. Emerging catalytic processes for the production of adipic acid. Catal. Sci. Technol. 2013, 3, 1465–1479. [Google Scholar] [CrossRef] [Green Version]
- Cavani, F.; Alini, S. Synthesis of Adipic Acid: On the Way to More Sustainable Production. In Sustainable Industrial Processes; Cavani, F., Centi, G., Perathoner, S., Trifiro, F., Eds.; Wiley: Weinheim, Germany, 2009; pp. 367–426. [Google Scholar]
- Chen, K.; Costas, M.; Kim, J.; Tipton, A.A.K.; Que, J.L. Olefin Cis-Dihydroxylation versus Epoxidation by Non-Heme Iron Catalysts: Two Faces of an FeIII−OOH Coin. J. Am. Chem. Soc. 2002, 124, 3026–3035. [Google Scholar] [CrossRef]
- Gelasco, A.; Askenas, A.; Pecoraro, V.L. Catalytic Disproportionation of Hydrogen Peroxide by the Tetranuclear Manganese Complex [Mnll(2-OHpicpn)]4. Inorg. Chem. 1996, 35, 1419–1420. [Google Scholar] [CrossRef]
- Fenton, H.J.H. LXXIII.—Oxidation of tartaric acid in presence of iron. J. Chem. Soc. Trans. 1894, 65, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Jaouen, F.; Dodelet, J.-P. O2 Reduction Mechanism on Non-Noble Metal Catalysts for PEM Fuel Cells. Part I: Experimental Rates of O2 Electroreduction, H2O2 Electroreduction, and H2O2 Disproportionation. J. Phys. Chem. C 2009, 113, 15422–15432. [Google Scholar] [CrossRef]
- Sengupta, K.; Chatterjee, S.; Dey, A. Catalytic H2O2 Disproportionation and Electrocatalytic O2 Reduction by a Functional Mimic of Heme Catalase: Direct Observation of Compound 0 and Compound I in Situ. ACS Catal. 2016, 6, 1382–1388. [Google Scholar] [CrossRef] [Green Version]
- Nourian, M.; Zadehahmadi, F.; Kardanpour, R.; Tangestaninejad, S.; Moghadam, M.; Mirkhani, V.; Mohammadpoor-Baltork, I. Highly efficient oxidative cleavage of alkenes and cyanosilylation of aldehydes catalysed by magnetically recoverable MIL-101. Appl. Organomet. Chem. 2018, 32, e3957. [Google Scholar] [CrossRef]
- Wang, J.Y.; Zhou, M.D.; Yuan, Y.G.; Fu, N.H.; Zang, S.L. Oxidation of cyclooctene to suberic acid using perrhenate-containing composite ionic liquids as green catalysts. Russ. J. Gen. Chem. 2015, 85, 2378–2385. [Google Scholar] [CrossRef]
- Chen, J.; Chen, M.; Zhang, B.; Nie, R.; Huang, A.; Goh, T.W.; Volkov, A.; Zhang, Z.; Ren, Q.; Huang, W. Allylic oxidation of olefins with a manganese-based metal–organic framework. Green Chem. 2019, 21, 3629–3636. [Google Scholar] [CrossRef]
- Chavan, S.; Srinivas, D.; Ratnasamy, P. Oxidation of Cyclohexane, Cyclohexanone, and Cyclohexanol to Adipic Acid by a Non-HNO3 Route over Co/Mn Cluster Complexes. J. Catal. 2002, 212, 39–45. [Google Scholar] [CrossRef]
- Schuchardt, U.; Cardoso, D.; Sercheli, R.; Pereira, R.; da Cruz, R.S.; Guerreiro, M.C.; Mandelli, D.; Spinacé, E.V.; Pires, E.L. Cyclohexane oxidation continues to be a challenge. Appl. Catal. A Gen. 2001, 211, 1–17. [Google Scholar] [CrossRef]
- Shen, D.; Miao, C.; Xu, D.; Xia, C.; Sun, W. Highly Efficient Oxidation of Secondary Alcohols to Ketones Catalyzed by Manganese Complexes of N4 Ligands with H2O2. Org. Lett. 2014, 17, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Nehru, K.; Kim, S.J.; Kim, I.Y.; Seo, M.S.; Kim, Y.; Kim, S.-J.; Kim, J.; Nam, W. A highly efficient non-heme manganese complex in oxygenation reactions. Chem. Commun. 2007, 44, 4623–4625. [Google Scholar] [CrossRef]
- Andraos, J.; Sayed, M. On the Use of “Green” Metrics in the Undergraduate Organic Chemistry Lecture and Lab To Assess the Mass Efficiency of Organic Reactions. J. Chem. Educ. 2007, 84, 1004. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Farrugia, L.J. ORTEP-3 for Windows—A version ofORTEP-III with a Graphical User Interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar] [CrossRef]
- Burnett, M.N.; Johnson, C.K. ORTEPIII. Report ORNL-6895; Oak Ridge National Laboratory: Oak Ridge, TN, USA, 1996.






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (L)Mn(p-Ts)2 | [(L)FeCl2](FeCl4) | |
|---|---|---|
| Bonds (Å) | ||
| M-Npy | 2.308(5)–2.352(2) | 2.1408(12), 2.1556(12) |
| M-Namine | 2.249(2)–2.283(2) | 2.2233(11), 2.2264(12) |
| Angles (°) | ||
| Namine-M-Namine | 75.46(9)–76.06(8) | 79.70(4) |
| NPy-M-NPy | 168.55(8)–168.87(7) | 166.17(5) |
| S | ρ(f) (mmol F/g S) | |||
|---|---|---|---|---|
| OCH2CH3 | OCH3 | CN | COOH | |
| SiO2 (E) | 0.43 | |||
| SiO2@CN (E) | 0.64 | 0.29 | ||
| SiO2@COOH (E) | 0.45 | 0.04 | ||
| SiO2 (M) | 1.18 | 0.05 | ||
| SiO2@CN (M) | 1.85 | 0.04 | 1.40 | |
| SiO2@COOH (M) | 0.08 | 0.05 | 0.31 | |
| Solvent Used for SiO2 Synthesis | SiO2@CN | SiO2@COOH |
|---|---|---|
| Ethanol | 20.6 | 2.8 |
| Methanol | 16.6 | 3.2 |
| Catalyst | RCOOH | CO | COE | TON (e) | |
|---|---|---|---|---|---|
| Conv (b) | Sel (c) | Yield (d) | |||
| (L)MnCl2 | no | 1 | - | - | - |
| CH3COOH | 99 | 81 | 81 | 100 | |
| CH3COOH (f) | 1 | - | - | - | |
| SiO2@COOH(M) | 37 | 9 | 4 | 38 | |
| SiO2@COOH(E) | 55 | 26 | 14 | 55 | |
| (L)Mn(OTf)2 | no | 5 | 7 | <1 | 3 |
| CH3COOH | 99 | 54 | 54 | 99 | |
| SiO2@COOH(M) | 50 | 45 | 23 | 50 | |
| SiO2@COOH(E) | 53 | 43 | 23 | 52 | |
| (L)Mn(p-Ts)2 | no | 5 | 50 | 2.7 | 6 |
| CH3COOH | 100 | 62 | 62 | 100 | |
| SiO2@COOH(M) | 61 | 30 | 19 | 61 | |
| SiO2@COOH(E) | 62 | 28 | 23 | 62 | |
| [(L)FeCl2](FeCl4) | no | 0 | - | - | - |
| CH3COOH | 60 | 21 | 13 | 60 | |
| SiO2@COOH(M) | 80 | 31 | 25 | 80 | |
| SiO2@COOH(E) | 91 | 25 | 23 | 91 | |
| Catalyst | RCOOH | Conv (b) | Selectivity (c) | TON (d) | |||
|---|---|---|---|---|---|---|---|
| CH | CHO | CHD | CHol | CHone | |||
| (L)MnCl2 | CH3COOH | 100 | 89 | 0 | 0 | 0 | 100 |
| SiO2@COOH(M) | 63 | 3.3 | 0 | 2 | 2 | 63 | |
| SiO2@COOH(E) | 74 | 14 | 23 | 0 | 0 | 74 | |
| (L)Mn(OTf)2 | CH3COOH | 98 | 57 | 3 | 0 | 1 | 98 |
| SiO2@COOH(M) | 57 | 13 | 0 | 0 | 0 | 56 | |
| SiO2@COOH(E) | 83 | 27 | 0 | 0 | 0 | 83 | |
| (L)Mn(p-Ts)2 | CH3COOH | 96 | 68 | 2 | 0 | 2 | 96 |
| SiO2@COOH(M) | 53 | 16 | 0 | 0 | 0 | 53 | |
| SiO2@COOH(E) | 80 | 28 | 0 | 0 | 0 | 80 | |
| [(L)FeCl2](FeCl4) | CH3COOH | 11 | 0 | 0 | 0 | 0 | 11 |
| SiO2@COOH(M) | 87 | 9 | 23 | 6 | 17 | 86 | |
| SiO2@COOH(E) | 96 | 4 | 5 | 0 | 9 | 96 | |
| Catalyst | RCOOH | CYol | CYone | TON (e) | |
|---|---|---|---|---|---|
| Conv (b) | Sel (c) | Yield (d) | |||
| (L)MnCl2 | CH3COOH | 81 | 91 | 74 | 81 |
| SiO2@COOH(M) | 15 | 46 | 7 | 15 | |
| SiO2@COOH(E) | 16 | 90 | 14 | 16 | |
| (L)Mn(OTf)2 | CH3COOH | 100 | 79 | 79 | 100 |
| SiO2@COOH(M) | 23 | 90 | 21 | 23 | |
| SiO2@COOH(E) | 27 | 87 | 24 | 27 | |
| (L)Mn(p-Ts)2 | CH3COOH | 99 | 85 | 85 | 99 |
| SiO2@COOH(M) | 21 | 97 | 21 | 21 | |
| SiO2@COOH(E) | 25 | 87 | 22 | 25 | |
| [(L)FeCl2](FeCl4) | CH3COOH | 100 | 79 | 79 | 99 |
| SiO2@COOH(M) | 59 | 45 | 27 | 59 | |
| SiO2@COOH(E) | 65 | 36 | 23 | 65 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Gayet, F.; Daran, J.-C.; Guillo, P.; Agustin, D. Replacement of Volatile Acetic Acid by Solid SiO2@COOH Silica (Nano)Beads for (Ep)Oxidation Using Mn and Fe Complexes Containing BPMEN Ligand. Molecules 2021, 26, 5435. https://doi.org/10.3390/molecules26185435
Wang Y, Gayet F, Daran J-C, Guillo P, Agustin D. Replacement of Volatile Acetic Acid by Solid SiO2@COOH Silica (Nano)Beads for (Ep)Oxidation Using Mn and Fe Complexes Containing BPMEN Ligand. Molecules. 2021; 26(18):5435. https://doi.org/10.3390/molecules26185435
Chicago/Turabian StyleWang, Yun, Florence Gayet, Jean-Claude Daran, Pascal Guillo, and Dominique Agustin. 2021. "Replacement of Volatile Acetic Acid by Solid SiO2@COOH Silica (Nano)Beads for (Ep)Oxidation Using Mn and Fe Complexes Containing BPMEN Ligand" Molecules 26, no. 18: 5435. https://doi.org/10.3390/molecules26185435