
Chemistry of Peptide-Oligonucleotide Conjugates: A Review


Abstract
:1. Introduction
2. Nucleic Acid Therapeutics
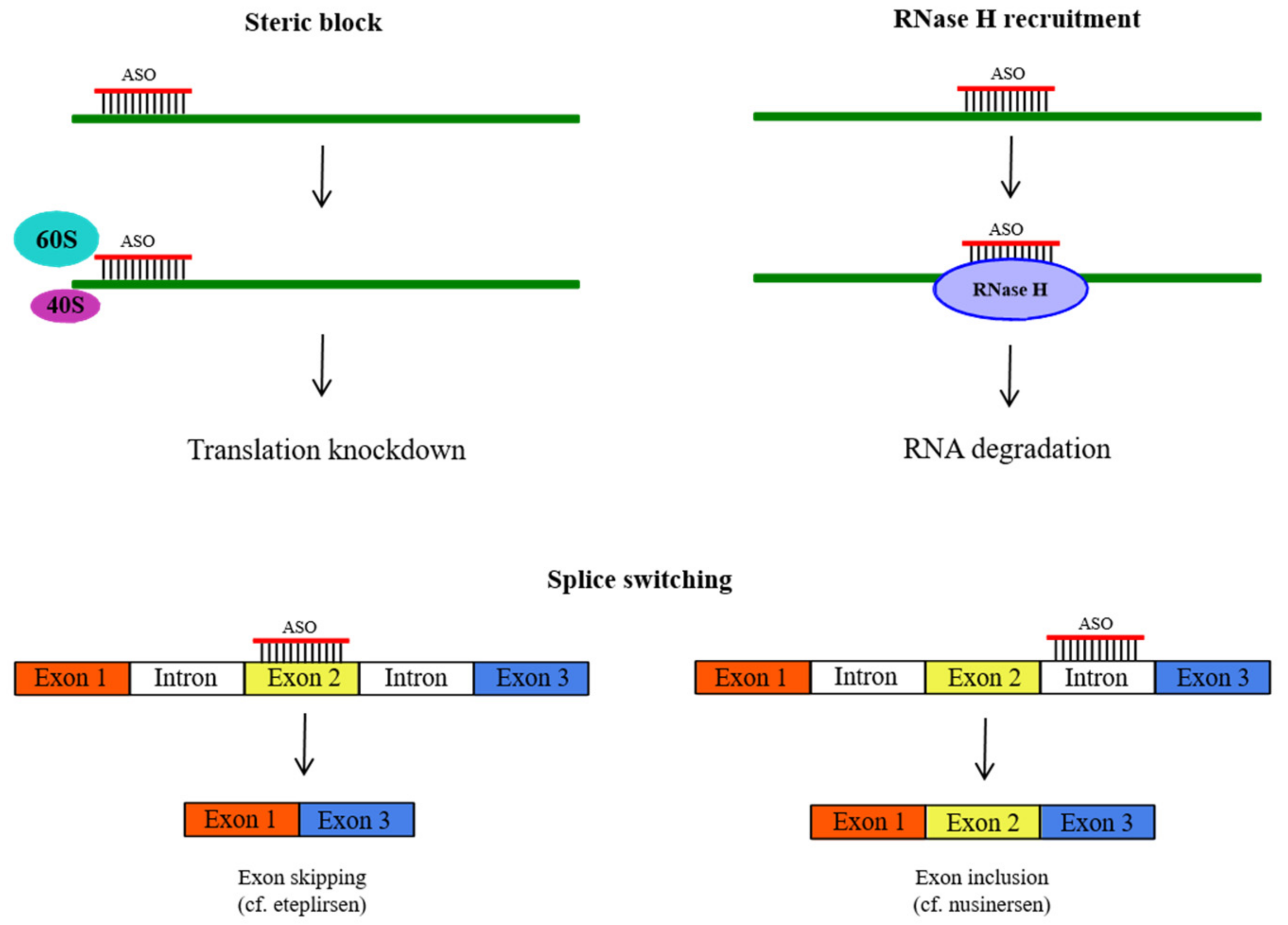
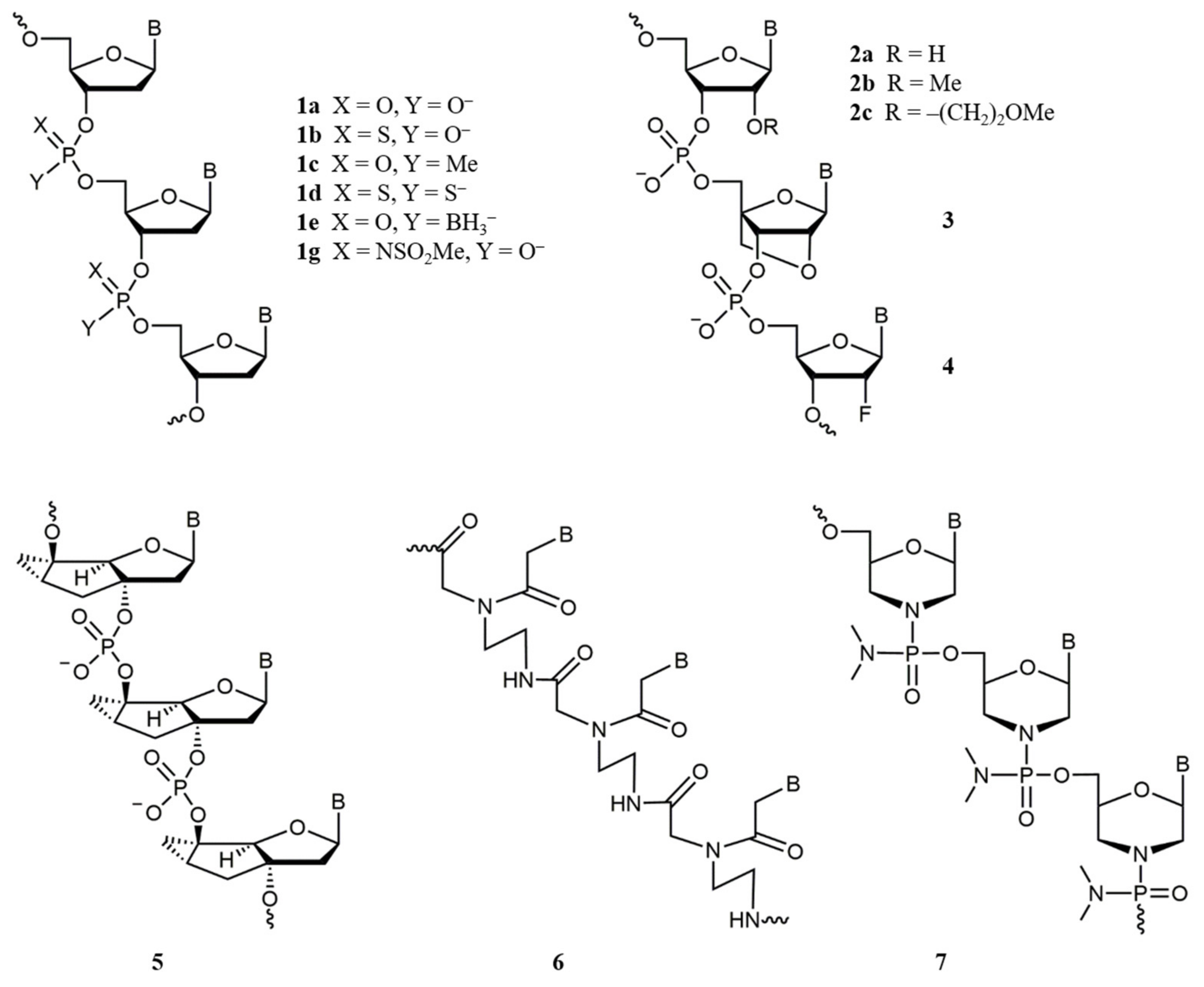
2.1. Antisense Oligonucleotides (ASOs)
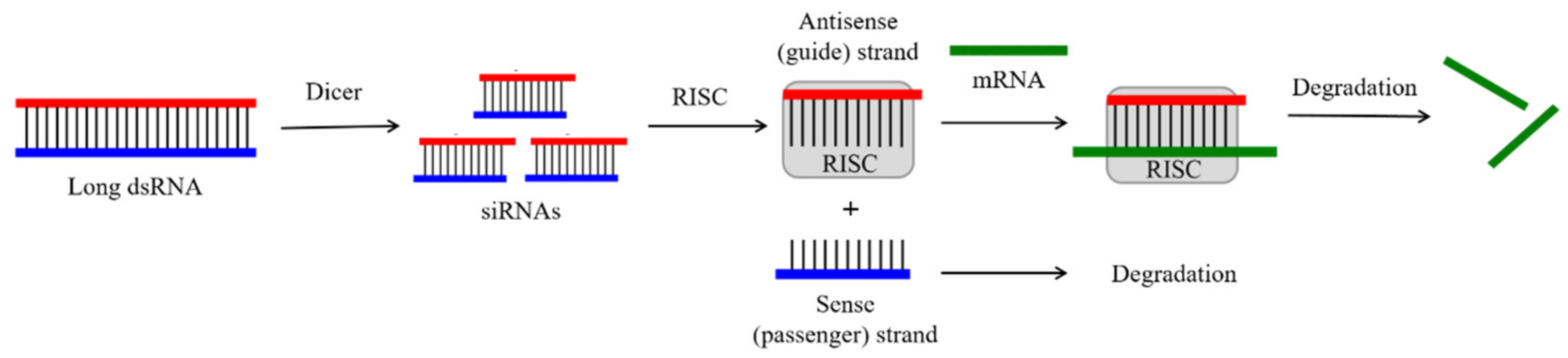
2.2. Small Interfering RNAs (siRNAs)
2.3. CRISPR/Cas9
2.4. The Problem of Oligonucleotide Delivery
3. Peptide-Mediated Cellular Delivery: A Brief Overview
4. Cell-Penetrating Peptides (CPPs): Types and Examples
4.1. Polycationic CPPs
4.2. Amphipathic CPPs
4.3. Hydrophobic CPP

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence | Reference |
|---|---|---|
| Polycationic | ||
| TAT | RKKRRQRRR | [182,183,184] |
| pAnt | RQIKIWFQNRRMKWKKGGC | [182,184] |
| Polyarginine | Rn (n = 8–12) | [164] |
| (RXR)4BR | RXRRXRRXRRXRXB | [185,186] |
| (KFF)3K | KFFKFFKFFK | [187] |
| Pip6a | RXRRBRRXRYQFLIRXRBRXRB | [188] |
| Pip7b | RXRRBRXYRFLIXRBRXRB | |
| Pip8b | RXRRBRXYQFLIRXRRBRB | |
| Pip9b | RXRRBRXFQILYRXRRBRB | |
| Pip9b2 | RXRRBRRFQILYRXRXRB | |
| Amphipathic | ||
| MPG | KETWWETWWTEWSQPKKRK | [167] |
| Pep-1 | GLAFLGFLGAAGSTMGAWSQPKKKRK | [168] |
| ARF (1–22) | MVRRFLVTLRIRRACGPPRVR | [169] |
| BPrPp (1–28) | MVKSKIGSWILVLFVAMWSDVGLCKKRPKP | [170] |
| MPrPp (1–30) | MANLGYWLLALFVTMWTDVGLCKKRPK | [171] |
| MAP | KLALKALKALKAALKLA | [172] |
| Transportan | GWTLNSAGYLLGKINLKALAALAKKIL | [189] |
| TP-10 | AGYLLGKINLKALAALAKKIL | [173] |
| CADY | GLWRALWRLLRSLWRLLWRA | [174,190] |
| RICK | KWLLRWLSRLLRWLARWLG | [191] |
| 599 | GLFEAIEGFIENGWEGMIDGWYGGGGRRRRRRRRRK | [192,193] |
| p28 | LSTAADMQGVVTDGMASGLDKDYLKPD | [175,176] |
| Bac7 | RRIRPRPPRLPRPRPRPLPFP | [177,178] |
| Proline-rich peptides | (PPR)n or (PRR)n (n = 3–6) | [179] |
| Hydrophobic | ||
| C105Y | PFVYLI | [180] |
| Pep-7 | SDLWEMMMVSLACQ | [181] |
| P4 | LGAQSNF | [194] |
| Pept1 | PLILLRLLRGQF | [195] |
5. Mechanisms of Peptide-Mediated Delivery
5.1. Direct Translocation
5.2. Endocytosis
6. Peptide Additives (Non-Covalent) and Peptide Conjugates (Covalent)
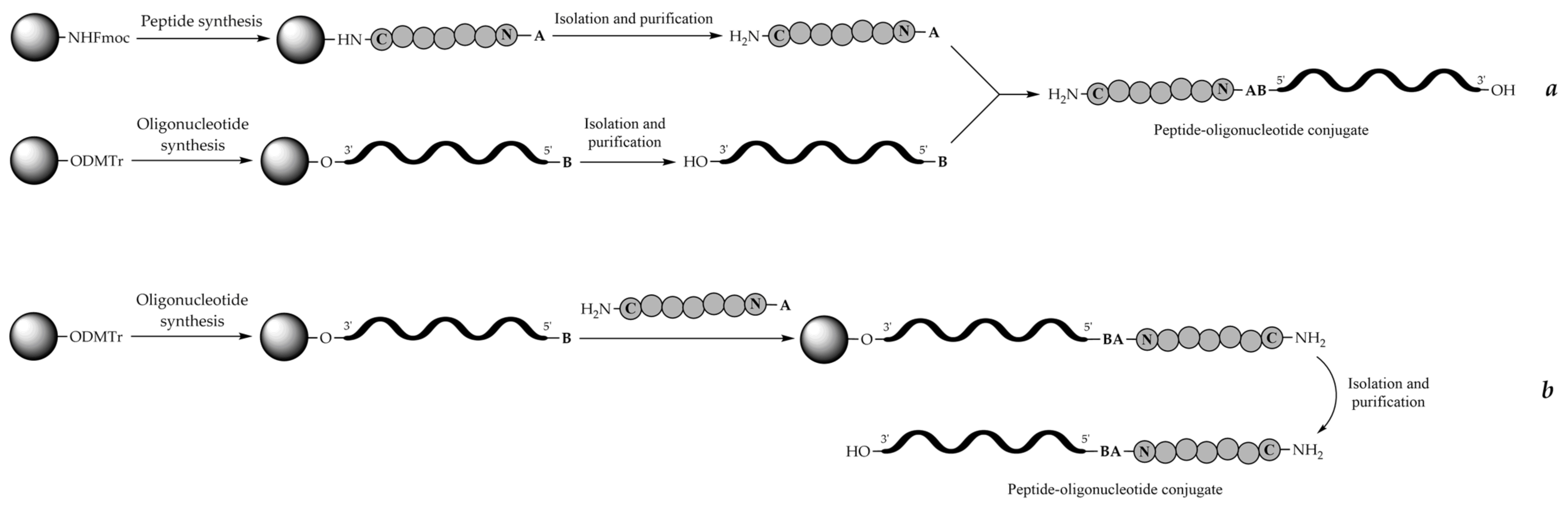
7. Synthetic Approaches
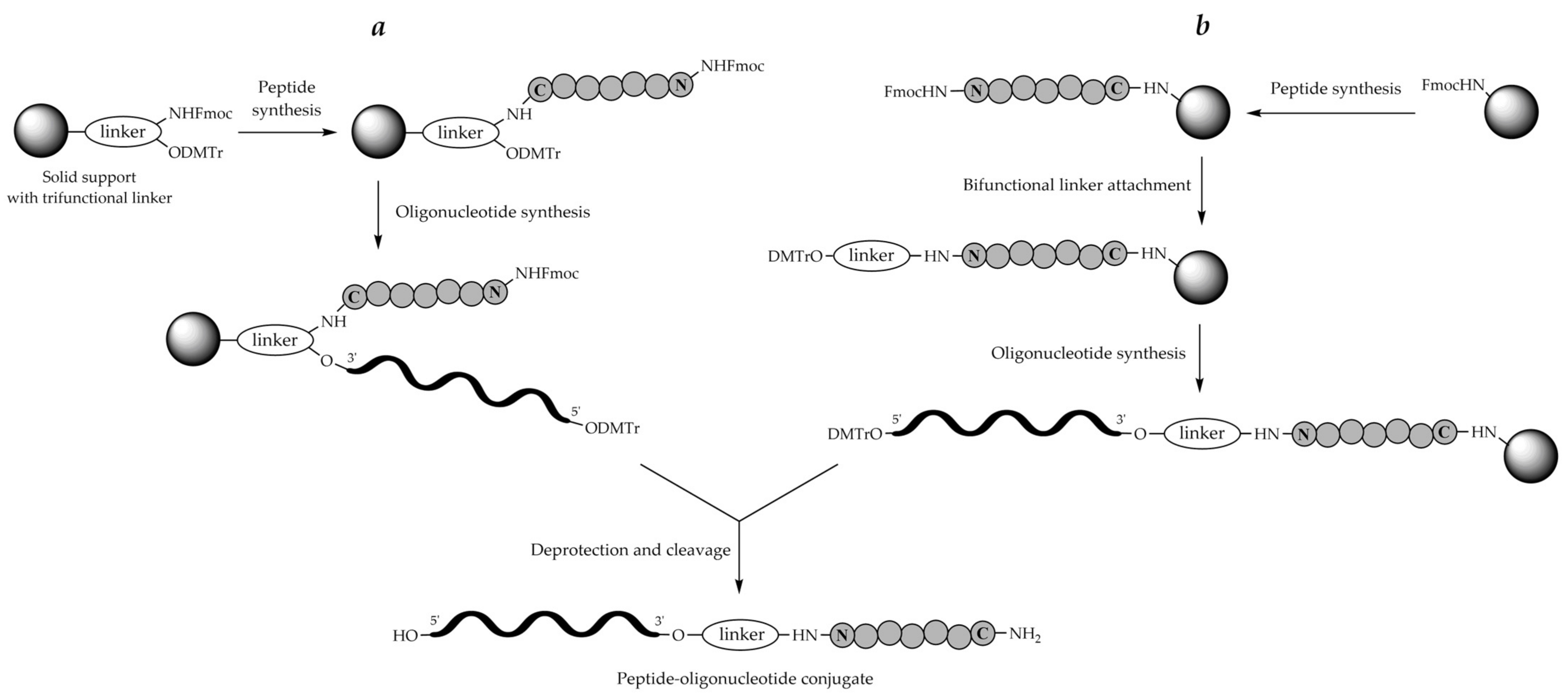
8. Stepwise Solid-Phase Synthesis Approach (On-Line or In-Line Synthesis)
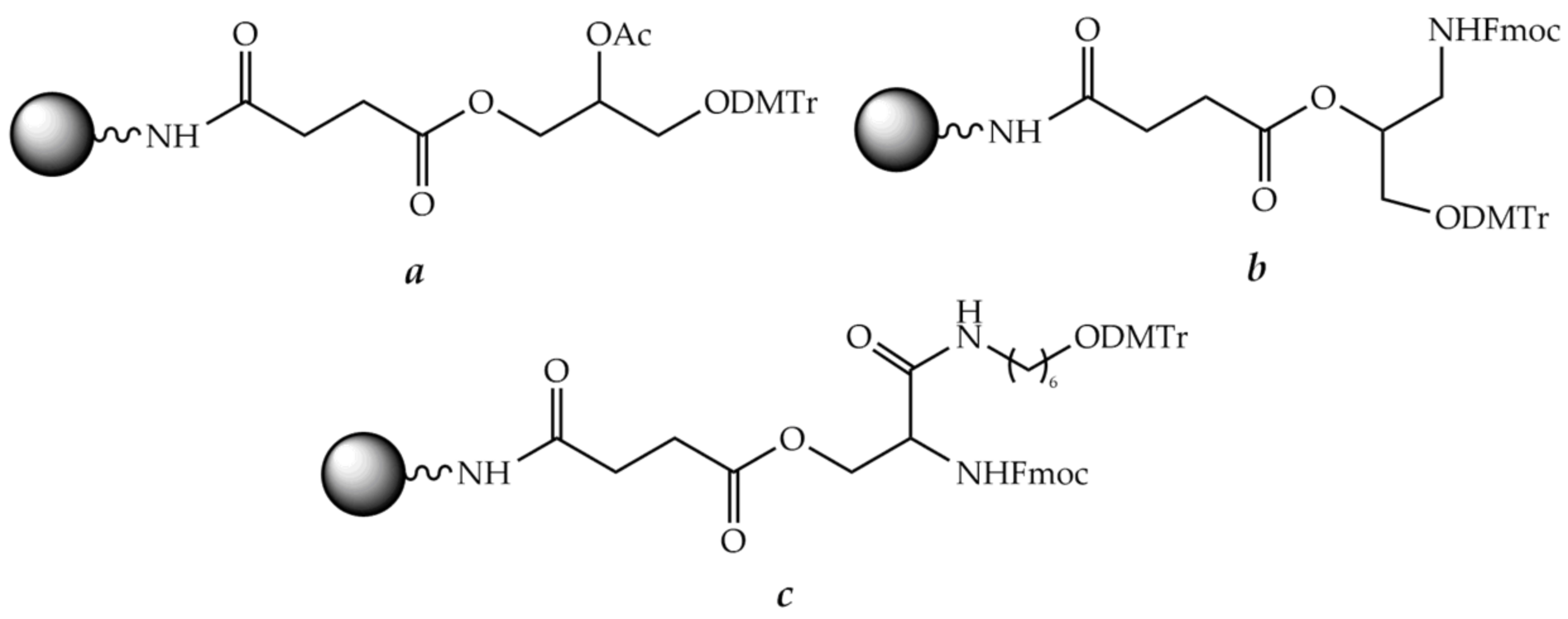
8.1. Solid Support
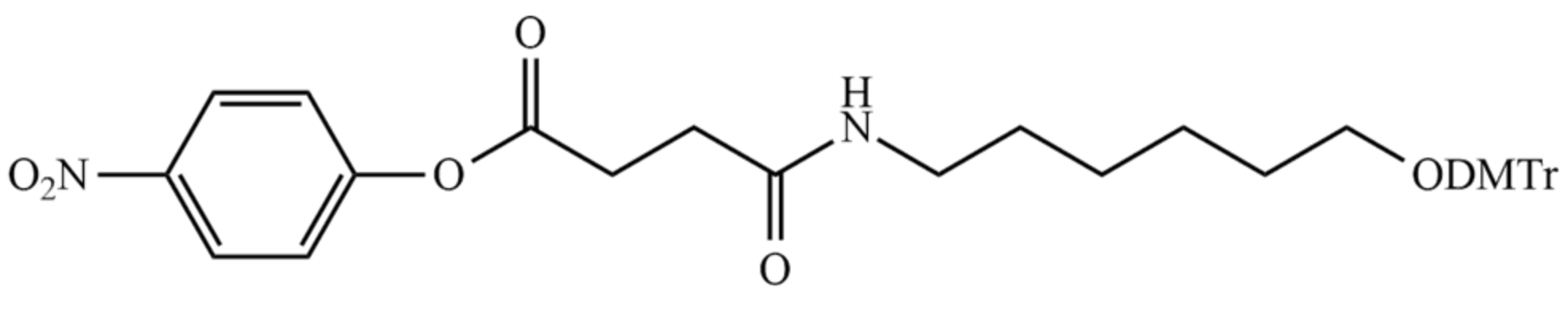
8.2. Linkers
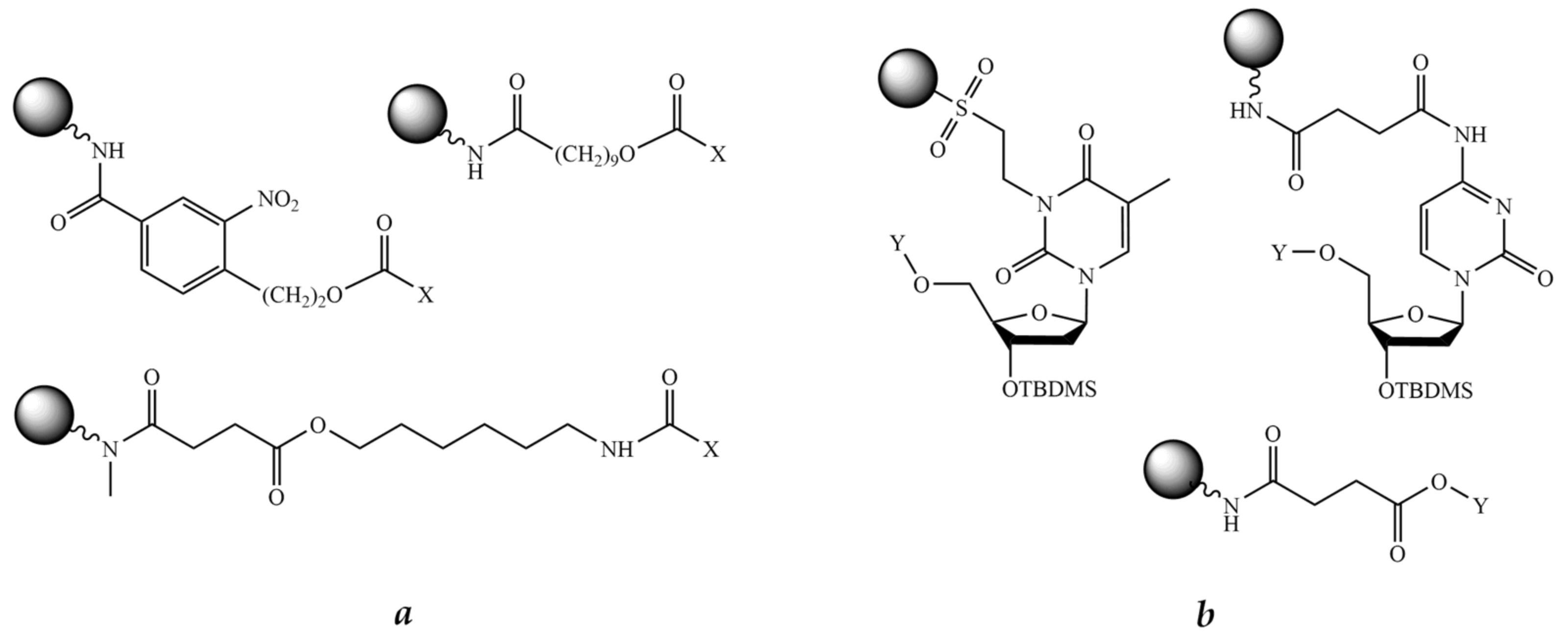
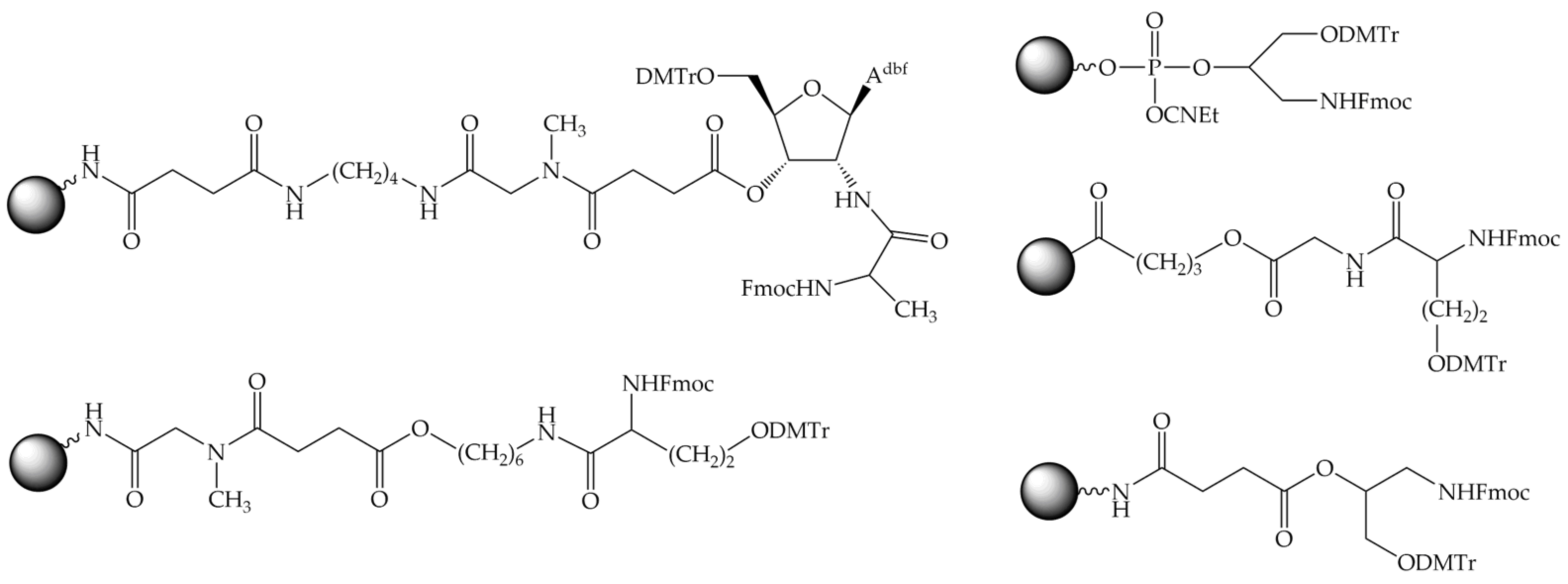
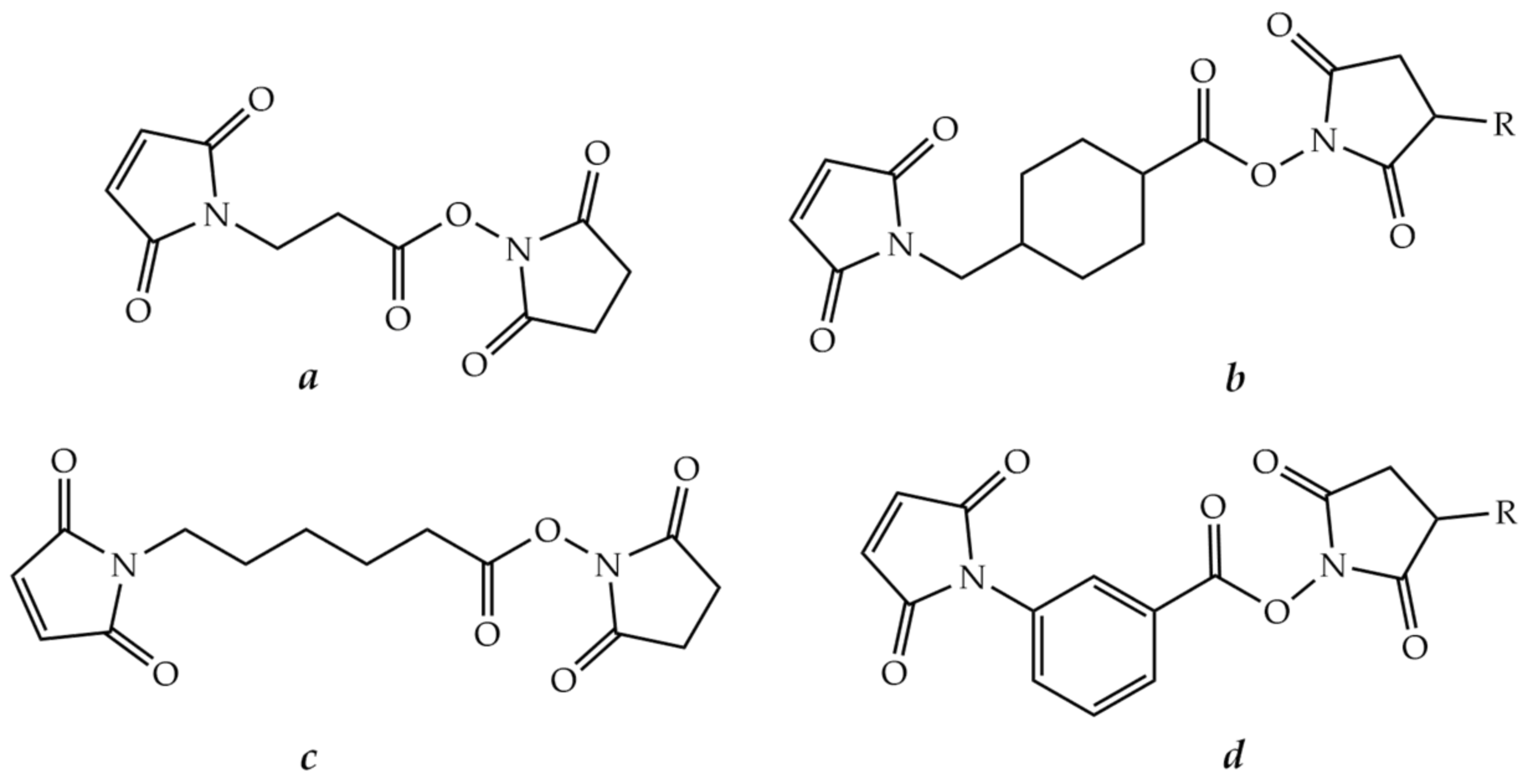
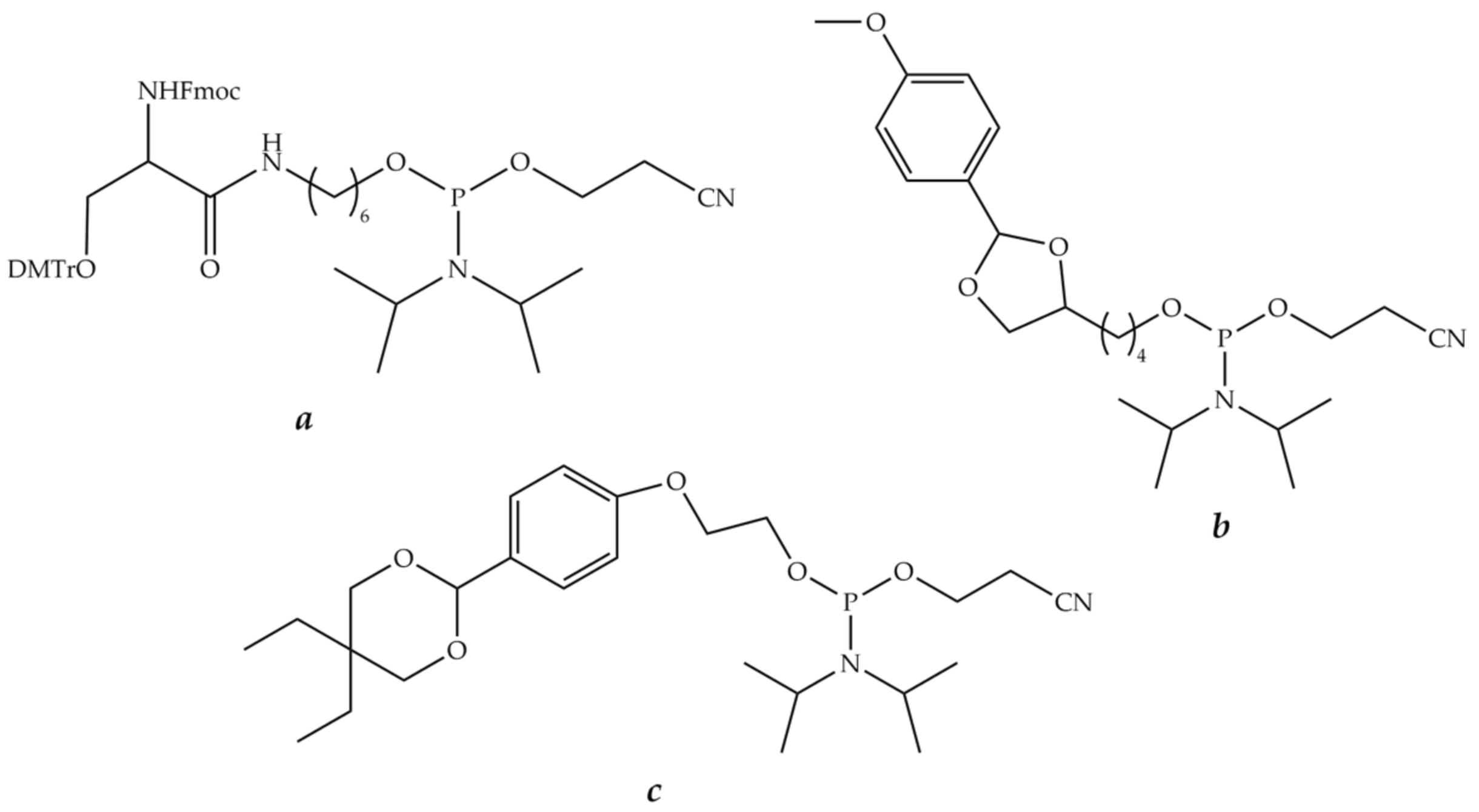
8.2.1. Bifunctional Linkers
8.2.2. Trifunctional Linkers
9. Post-Synthetic Conjugation Approaches
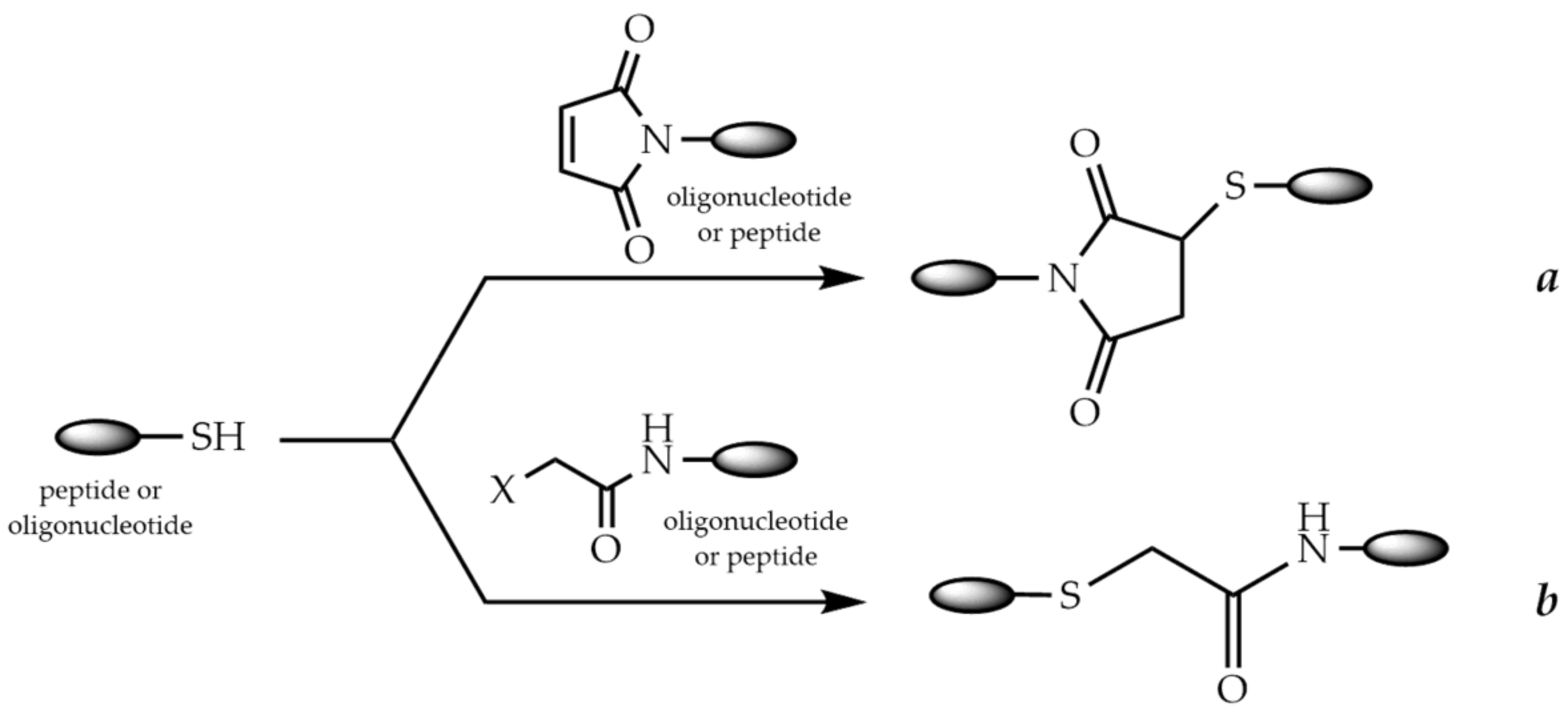
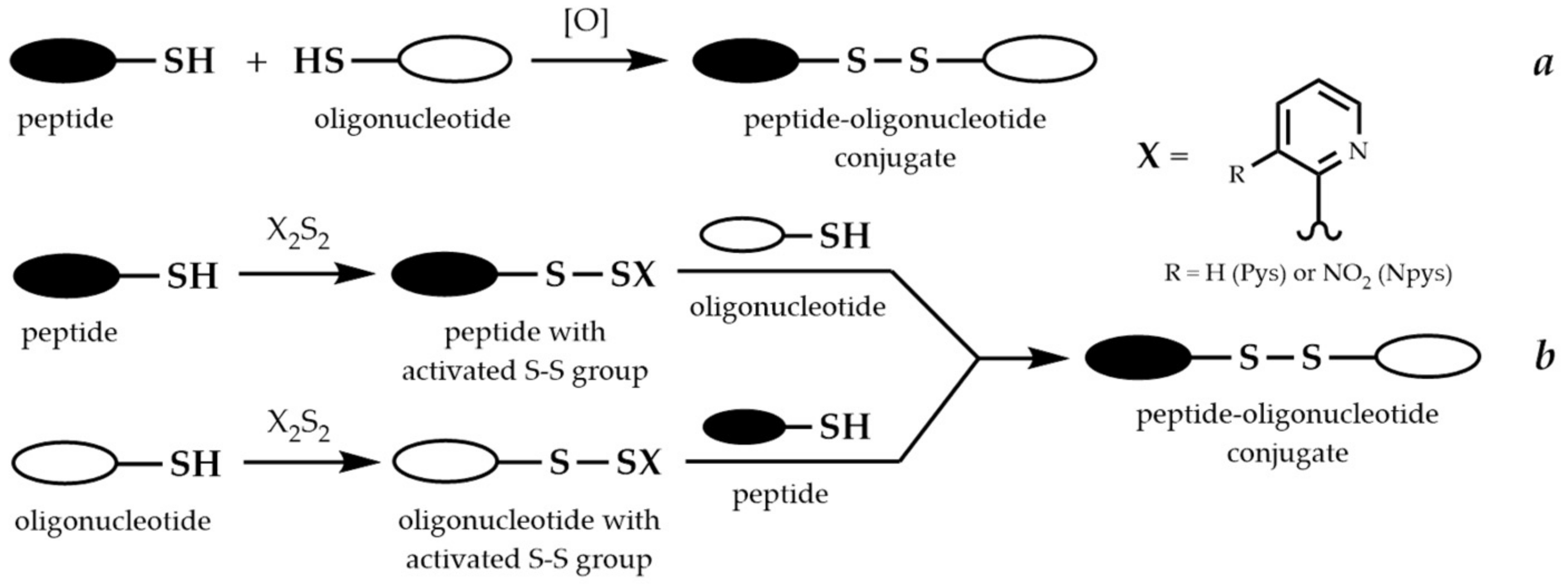
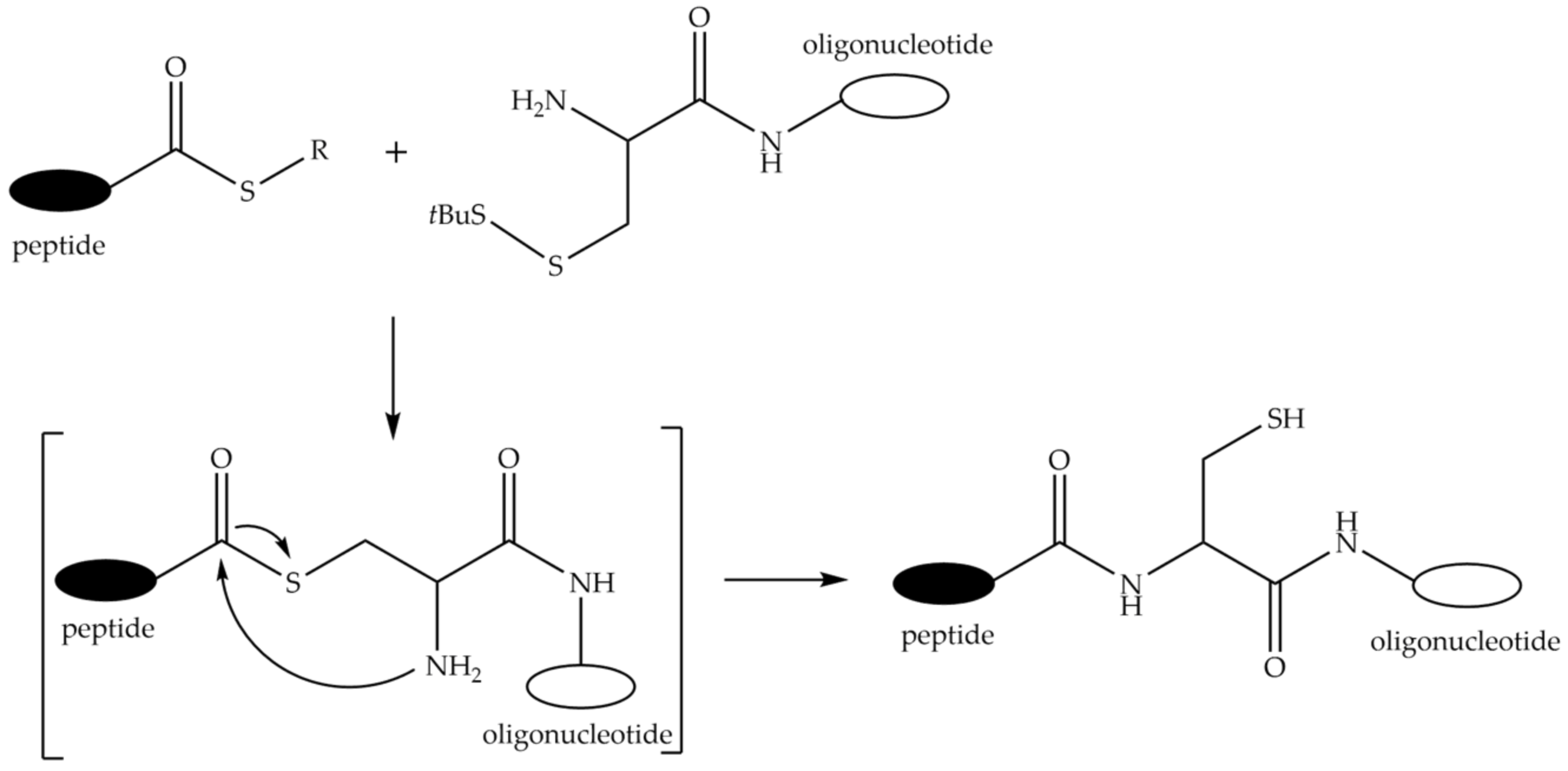
9.1. Conjugation via Thioether or Disulfide Bonds
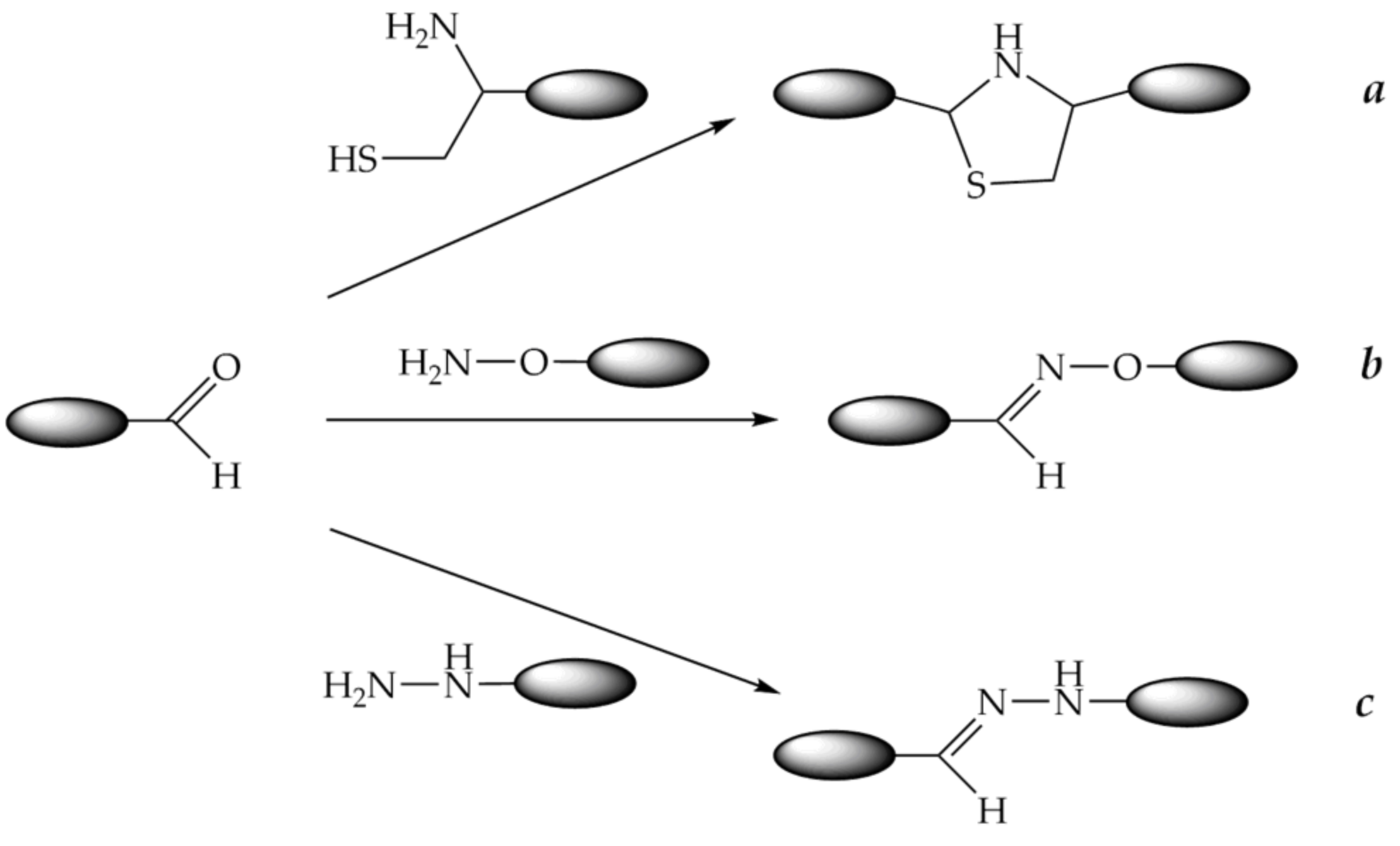
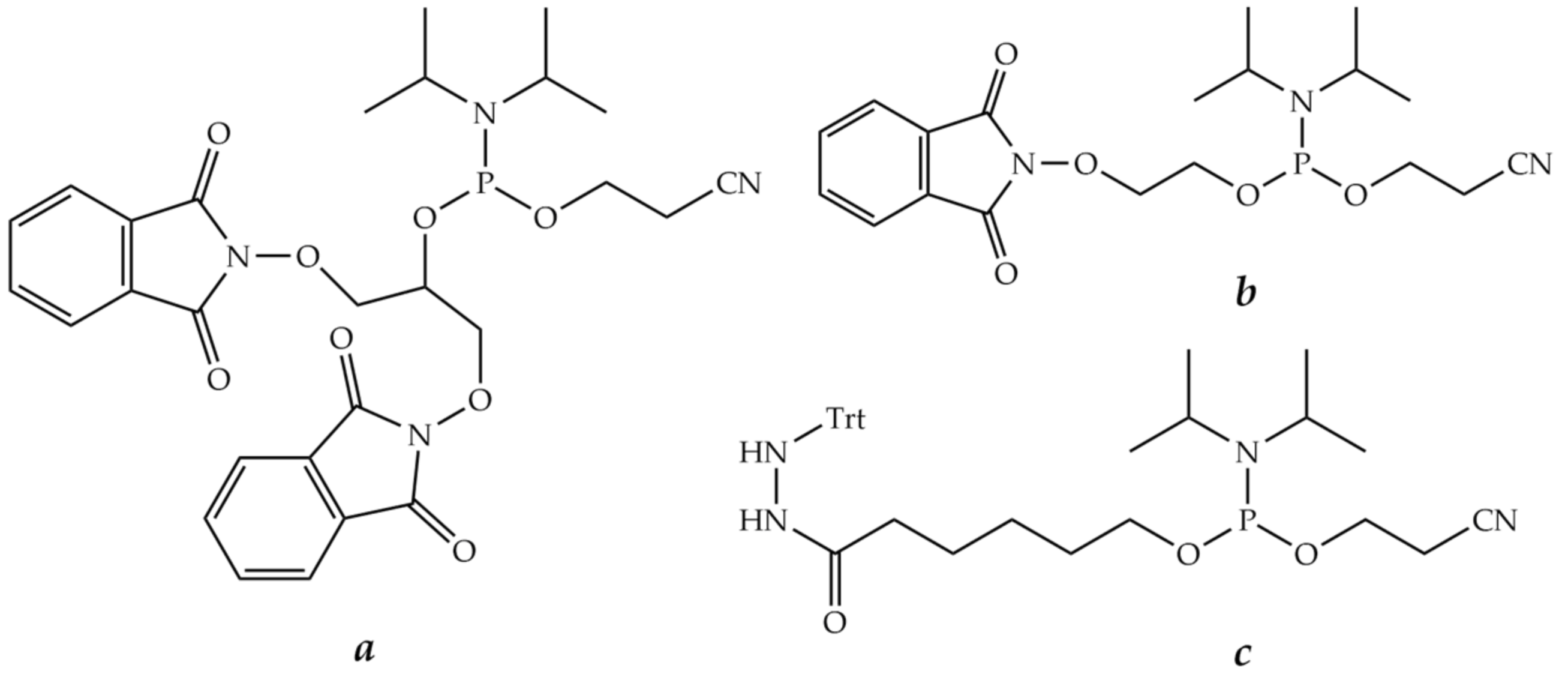
9.2. Conjugation through Oxime, Thiazolidine, or Hydrazone Bonds
9.3. Conjugation through Amide Bonds
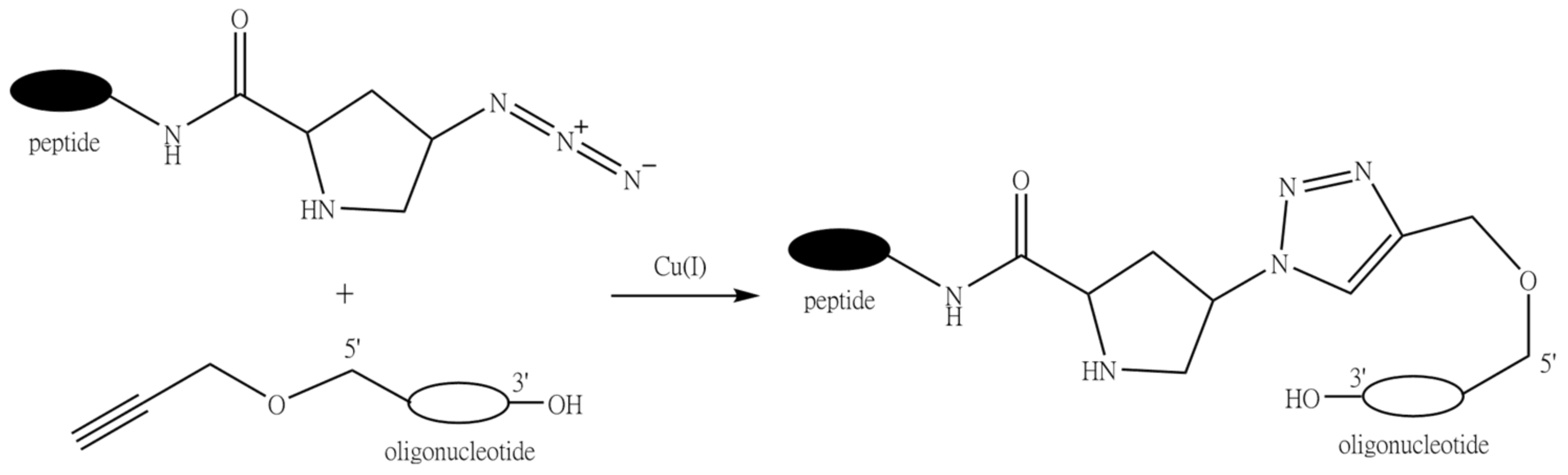
9.4. Conjugation through Click Chemistry (1,3-Dipolar Cycloaddition Reaction of Alkynes to Azides)
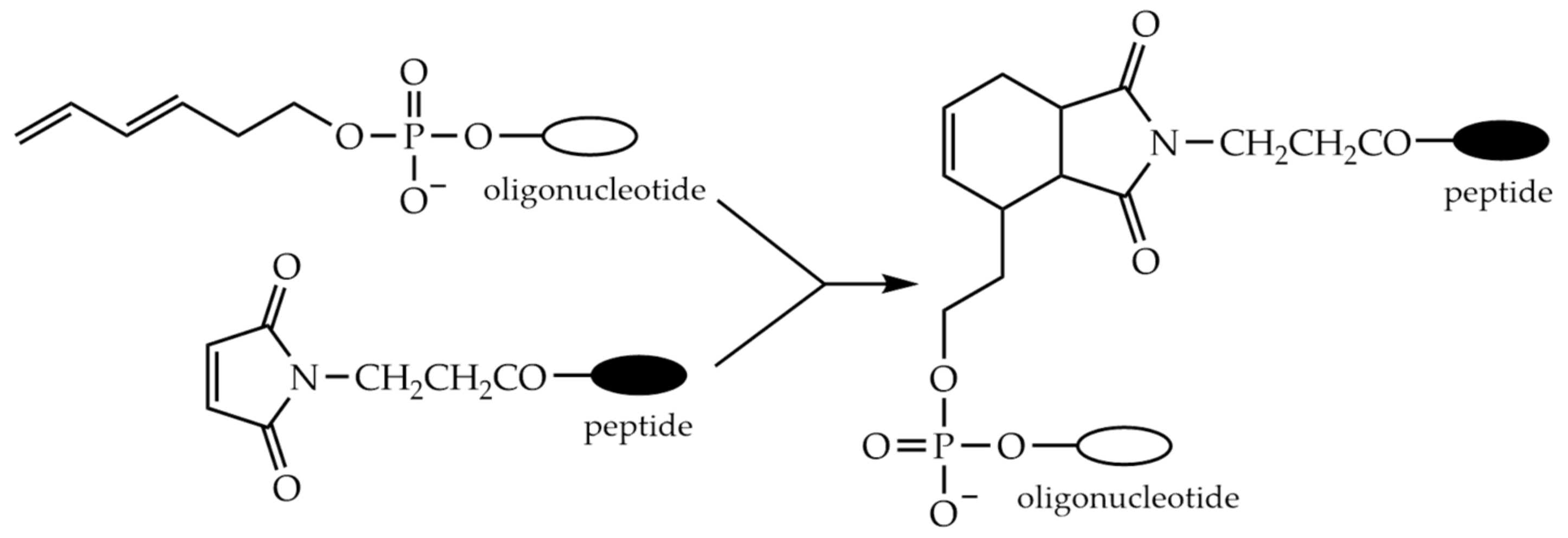
9.5. Conjugation through the Diels-Alder Reaction
10. Comparison of the Two Approaches: Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Schattenkerk, C.; Wreesmann, C.T.J.; de Graaf, M.J.; van der Marel, G.A.; van Boom, J.H. Synthesis of a naturally occurring nucleopeptide fragment via a phosphotriester approach. Tetrahedron Lett. 1984, 25, 5197–5200. [Google Scholar] [CrossRef]
- Dreef-Tromp, C.M.; van den Elst, H.; van den Boogaart, J.E.; van der Marel, G.A.; van Boom, J.H. Solid-phase synthesis of an RNA nucleopeptide fragment from the nucleoprotein of poliovirus. Nucleic Acids Res. 1992, 20, 2435–2439. [Google Scholar] [CrossRef] [Green Version]
- Robles, J.; Pedroso, E.; Grandas, A. Solid-phase synthesis of a nucleopeptide from the linking site of adenovirus-2 nucleoprotein, -Ser(p5′CATCAT)-Gly-Asp-. Convergent versus stepwise strategy. Nucleic Acids Res. 1995, 23, 4151–4161. [Google Scholar] [CrossRef] [Green Version]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlmann, E.; Peyman, A. Antisense oligonucleotides: A new therapeutic principle. Chem. Rev. 1990, 90, 543–584. [Google Scholar] [CrossRef]
- De Mesmaeker, A.; Haener, R.; Martin, P.; Moser, H.E. Antisense Oligonucleotides. Acc. Chem. Res. 1995, 28, 366–374. [Google Scholar] [CrossRef]
- Opalinska, J.B.; Gewirtz, A.M. Nucleic-acid therapeutics: Basic principles and recent applications. Nat. Rev. Drug Discov. 2002, 1, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Goodchild, J. Therapeutic oligonucleotides. Methods Mol. Biol. 2011, 764, 1–15. [Google Scholar]
- Brasseur, R.; Divita, G. Happy birthday cell penetrating peptides: Already 20 years. Biochim. Biophys. Acta 2010, 1798, 2177–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, C.M.; Balfour, J.A. Fomivirsen. Drugs 1999, 57, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Roehr, B. Fomivirsen approved for CMV retinitis. J. Int. Assoc. Physicians AIDS Care 1998, 4, 14–16. [Google Scholar]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Hoy, S.M. Patisiran: First Global Approval. Drugs 2018, 78, 1625–1631. [Google Scholar] [CrossRef] [PubMed]
- Dhuri, K.; Bechtold, C.; Quijano, E.; Pham, H.; Gupta, A.; Vikram, A.; Bahal, R. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J. Clin. Med. 2020, 9, 2004. [Google Scholar] [CrossRef]
- Xiong, H.; Veedu, R.N.; Diermeier, S.D. Recent Advances in Oligonucleotide Therapeutics in Oncology. Int. J. Mol. Sci. 2021, 22, 3295. [Google Scholar] [CrossRef]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef]
- Godfrey, C.; Desviat, L.R.; Smedsrod, B.; Pietri-Rouxel, F.; Denti, M.A.; Disterer, P.; Lorain, S.; Nogales-Gadea, G.; Sardone, V.; Anwar, R.; et al. Delivery is key: Lessons learnt from developing splice-switching antisense therapies. EMBO. Mol. Med. 2017, 9, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.M.; Toonen, L.J.; van Roon-Mom, W.M. Antisense oligonucleotides in therapy for neurodegenerative disorders. Adv. Drug Deliv. Rev. 2015, 87, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.C.; Lobo, D.D.; Martins, I.M.; Lopes, S.M.; Henriques, C.; Duarte, S.P.; Dodart, J.-C.; Nobre, R.J.; Pereira de Almeida, L. Antisense oligonucleotide therapeutics in neurodegenerative diseases: The case of polyglutamine disorders. Brain 2019, 143, 407–429. [Google Scholar] [CrossRef]
- Wan, Y.; Moyle, P.M.; Toth, I. Endosome Escape Strategies for Improving the Efficacy of Oligonucleotide Delivery Systems. Curr. Med. Chem. 2015, 22, 3326–3346. [Google Scholar] [CrossRef]
- Juliano, R.L.; Ming, X.; Nakagawa, O. Cellular uptake and intracellular trafficking of antisense and siRNA oligonucleotides. Bioconjugate Chem. 2012, 23, 147–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, T.; Weber, H.; DiMuzio, J.; Matter, A.; Dogdas, B.; Shah, T.; Thankappan, A.; Disa, J.; Jadhav, V.; Lubbers, L.; et al. Silencing Myostatin Using Cholesterol-conjugated siRNAs Induces Muscle Growth. Mol. Ther.-Nucleic Acids 2016, 5, e342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Østergaard, M.E.; Jackson, M.; Low, A.; Chappell, A.E.; Lee, R.G.; Peralta, R.Q.; Yu, J.; Kinberger, G.A.; Dan, A.; Carty, R.; et al. Conjugation of hydrophobic moieties enhances potency of antisense oligonucleotides in the muscle of rodents and non-human primates. Nucleic Acids Res. 2019, 47, 6045–6058. [Google Scholar] [CrossRef] [Green Version]
- Chernikov, I.V.; Meschaninova, M.I.; Chernolovskaya, E.L. Preparation, Determination of Activity, and Biodistribution of Cholesterol-Containing Nuclease-Resistant siRNAs In Vivo. In RNA interference and CRISPR Technologies; Humana: New York, NY, USA, 2020; pp. 57–77. [Google Scholar]
- Patwa, A.; Gissot, A.; Bestel, I.; Barthélémy, P. Hybrid lipid oligonucleotide conjugates: Synthesis, self-assemblies and biomedical applications. Chem. Soc. Rev. 2011, 40, 5844–5854. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Tian, Q.; Bagheri, Y.; You, M. Lipid-Oligonucleotide Conjugates for Simple and Efficient Cell Membrane Engineering and Bioanalysis. Curr. Opin. Biomed. Eng. 2020, 13, 76–83. [Google Scholar] [CrossRef]
- Li, X.; Feng, K.; Li, L.; Yang, L.; Pan, X.; Yazd, H.S.; Cui, C.; Li, J.; Moroz, L.; Sun, Y.; et al. Lipid–oligonucleotide conjugates for bioapplications. Natl. Sci. Rev. 2020, 7, 1933–1953. [Google Scholar] [CrossRef]
- Khan, A.; Benboubetra, M.; Sayyed, P.Z.; Ng, K.W.; Fox, S.; Beck, G.; Benter, I.F.; Akhtar, S. Sustained polymeric delivery of gene silencing antisense ODNs, siRNA, DNAzymes and ribozymes: In vitro and in vivo studies. J. Drug Target. 2004, 12, 393–404. [Google Scholar] [CrossRef]
- Reimann, E.M.; Soloff, M.S. The effect of radioactive contaminants on the estimation of binding parameters by Scatchard analysis. Biochim. Biophys. Acta 1978, 533, 130–139. [Google Scholar] [CrossRef]
- Ravina, M.; Paolicelli, P.; Seijo, B.; Sanchez, A. Knocking down gene expression with dendritic vectors. Mini-Rev. Med. Chem. 2010, 10, 73–86. [Google Scholar] [CrossRef]
- Parveen, S.; Misra, R.; Sahoo, S.K. Nanoparticles: A boon to drug delivery, therapeutics, diagnostics and imaging. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 147–166. [Google Scholar] [CrossRef]
- Hu, Q.; Li, H.; Wang, L.; Gu, H.; Fan, C. DNA Nanotechnology-Enabled Drug Delivery Systems. Chem. Rev. 2019, 119, 6459–6506. [Google Scholar] [CrossRef]
- Xu, F.; Xia, Q.; Wang, P. Rationally Designed DNA Nanostructures for Drug Delivery. Front. Chem. 2020, 8, 751. [Google Scholar] [CrossRef]
- Craig, K.; Abrams, M.; Amiji, M. Recent preclinical and clinical advances in oligonucleotide conjugates. Expert Opin. Drug Deliv. 2018, 15, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- McClorey, G.; Banerjee, S. Cell-Penetrating Peptides to Enhance Delivery of Oligonucleotide-Based Therapeutics. Biomedicines 2018, 6, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehto, T.; Kurrikoff, K.; Langel, U. Cell-penetrating peptides for the delivery of nucleic acids. Expert Opin. Drug Deliv. 2012, 9, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Boisguerin, P.; Redt-Clouet, C.; Franck-Miclo, A.; Licheheb, S.; Nargeot, J.; Barrere-Lemaire, S.; Lebleu, B. Systemic delivery of BH4 anti-apoptotic peptide using CPPs prevents cardiac ischemia-reperfusion injuries in vivo. J. Control. Release 2011, 156, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Zaro, J.L.; Fei, L.; Shen, W.C. Recombinant peptide constructs for targeted cell penetrating peptide-mediated delivery. J. Control. Release 2012, 158, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Ru, Q.; Shang, B.Y.; Miao, Q.F.; Li, L.; Wu, S.Y.; Gao, R.J.; Zhen, Y.S. A cell penetrating peptide-integrated and enediyne-energized fusion protein shows potent antitumor activity. Eur. J. Pharm. Sci. 2012, 47, 781–789. [Google Scholar] [CrossRef]
- Silva, S.; Almeida, A.J.; Vale, N. Combination of Cell-Penetrating Peptides with Nanoparticles for Therapeutic Application: A Review. Biomolecules 2019, 9, 22. [Google Scholar] [CrossRef] [Green Version]
- Falanga, A.; Vitiello, M.T.; Cantisani, M.; Tarallo, R.; Guarnieri, D.; Mignogna, E.; Netti, P.; Pedone, C.; Galdiero, M.; Galdiero, S. A peptide derived from herpes simplex virus type 1 glycoprotein H: Membrane translocation and applications to the delivery of quantum dots. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Abes, R.; Arzumanov, A.A.; Moulton, H.M.; Abes, S.; Ivanova, G.D.; Iversen, P.L.; Gait, M.J.; Lebleu, B. Cell-penetrating-peptide-based delivery of oligonucleotides: An overview. Biochem. Soc. Trans. 2007, 35, 775–779. [Google Scholar] [CrossRef] [Green Version]
- Said Hassane, F.; Saleh, A.F.; Abes, R.; Gait, M.J.; Lebleu, B. Cell penetrating peptides: Overview and applications to the delivery of oligonucleotides. Cell Mol. Life Sci. 2010, 67, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef]
- Crombez, L.; Morris, M.C.; Deshayes, S.; Heitz, F.; Divita, G. Peptide-based nanoparticle for ex vivo and in vivo drug delivery. Curr. Pharm. Des. 2008, 14, 3656–3665. [Google Scholar] [CrossRef]
- Bhardwaj, I.; Jha, D.; Admane, P.; Panda, A.K.; Haridas, V. Self-assembling tryptophan-based designer peptides as intracellular delivery vehicles. Bioorganic Med. Chem. Lett. 2016, 26, 672–676. [Google Scholar] [CrossRef]
- Lebleu, B.; Moulton, H.M.; Abes, R.; Ivanova, G.D.; Abes, S.; Stein, D.A.; Iversen, P.L.; Arzumanov, A.A.; Gait, M.J. Cell penetrating peptide conjugates of steric block oligonucleotides. Adv. Drug Deliv. Rev. 2008, 60, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Taskova, M.; Mantsiou, A.; Astakhova, K. Synthetic Nucleic Acid Analogues in Gene Therapy: An Update for Peptide-Oligonucleotide Conjugates. Chembiochem 2017, 18, 1671–1682. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, A.; Tornesello, A.L.; Tornesello, M.L.; Buonaguro, F.M. Cell Penetrating Peptides as Molecular Carriers for Anti-Cancer Agents. Molecules 2018, 23, 295. [Google Scholar] [CrossRef] [Green Version]
- Boisguerin, P.; Deshayes, S.; Gait, M.J.; O’Donovan, L.; Godfrey, C.; Betts, C.A.; Wood, M.J.; Lebleu, B. Delivery of therapeutic oligonucleotides with cell penetrating peptides. Adv. Drug Deliv. Rev. 2015, 87, 52–67. [Google Scholar] [CrossRef]
- Gait, M.J.; Arzumanov, A.A.; McClorey, G.; Godfrey, C.; Betts, C.; Hammond, S.; Wood, M.J.A. Cell-Penetrating Peptide Conjugates of Steric Blocking Oligonucleotides as Therapeutics for Neuromuscular Diseases from a Historical Perspective to Current Prospects of Treatment. Nucleic Acid Ther. 2019, 29, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Hammond, S.M.; Abendroth, F.; Gait, M.J.; Wood, M.J.A. Evaluation of Cell-Penetrating Peptide Delivery of Antisense Oligonucleotides for Therapeutic Efficacy in Spinal Muscular Atrophy. Methods Mol. Biol. 2019, 2036, 221–236. [Google Scholar]
- Levin, A.A. Treating Disease at the RNA Level with Oligonucleotides. N. Engl. J. Med. 2019, 380, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, B.C.; Ooi, J.Y.; Lin, R.C.; McMullen, J.R. miRNA therapeutics: A new class of drugs with potential therapeutic applications in the heart. Future Med. Chem. 2015, 7, 1771–1792. [Google Scholar] [CrossRef]
- Wagner, A.; Bock, C.T.; Fechner, H.; Kurreck, J. Application of modified antisense oligonucleotides and siRNAs as antiviral drugs. Future Med. Chem. 2015, 7, 1637–1642. [Google Scholar] [CrossRef]
- Hegarty, J.P.; Stewart, D.B., Sr. Advances in therapeutic bacterial antisense biotechnology. Appl. Microbiol. Biotechnol. 2018, 102, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Duca, M.; Vekhoff, P.; Oussedik, K.; Halby, L.; Arimondo, P.B. The triple helix: 50 years later, the outcome. Nucleic Acids Res. 2008, 36, 5123–5138. [Google Scholar] [CrossRef] [PubMed]
- Rad, S.M.; Langroudi, L.; Kouhkan, F.; Yazdani, L.; Koupaee, A.N.; Asgharpour, S.; Shojaei, Z.; Bamdad, T.; Arefian, E. Transcription factor decoy: A pre-transcriptional approach for gene downregulation purpose in cancer. Tumour Biol. 2015, 36, 4871–4881. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, J.R.; Roy, K.; Maremanda, N.G.; Subramanian, K.; Veedu, R.N.; Bawa, R.; Kanwar, R.K. Nucleic acid-based aptamers: Applications, development and clinical trials. Curr. Med. Chem. 2015, 22, 2539–2557. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Nakamura, Y. Aptamers: A Review of Their Chemical Properties and Modifications for Therapeutic Application. Molecules 2019, 24, 4229. [Google Scholar] [CrossRef] [Green Version]
- Alagia, A.; Eritja, R. siRNA and RNAi optimization. Wiley Interdiscip. Rev. RNA. 2016, 7, 316–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.R.; Lee, C.H.; Im, J.Y.; Kim, J.H.; Kim, J.H.; Kim, S.J.; Cho, Y.W.; Kim, E.; Kim, Y.; Ryu, J.H.; et al. Targeted suicide gene therapy for liver cancer based on ribozyme-mediated RNA replacement through post-transcriptional regulation. Mol. Ther.-Nucleic Acids 2021, 23, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Fokina, A.A.; Stetsenko, D.A.; Francois, J.C. DNA enzymes as potential therapeutics: Towards clinical application of 10–23 DNAzymes. Expert. Opin. Biol. 2015, 15, 689–711. [Google Scholar] [CrossRef]
- Fokina, A.A.; Chelobanov, B.P.; Fujii, M.; Stetsenko, D.A. Delivery of therapeutic RNA-cleaving oligodeoxyribonucleotides (deoxyribozymes): From cell culture studies to clinical trials. Expert Opin. Drug Deliv. 2017, 14, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Krutzfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef]
- Haussecker, D. Stacking up CRISPR against RNAi for therapeutic gene inhibition. FEBS. J. 2016, 283, 3249–3260. [Google Scholar] [CrossRef] [Green Version]
- Cavagnari, B.M. Gene therapy: Nucleic acids as drugs. Action mechanisms and delivery into the cell. Arch. Argent. De Pediatr. 2011, 109, 237–244. [Google Scholar]
- Yamamoto, T.; Nakatani, M.; Narukawa, K.; Obika, S. Antisense drug discovery and development. Future Med. Chem. 2011, 3, 339–365. [Google Scholar] [CrossRef]
- Smith, C.I.E.; Zain, R. Therapeutic Oligonucleotides: State of the Art. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 605–630. [Google Scholar] [CrossRef]
- Scharner, J.; Aznarez, I. Clinical Applications of Single-Stranded Oligonucleotides: Current Landscape of Approved and In-Development Therapeutics. Mol. Ther. 2021, 29, 540–554. [Google Scholar] [CrossRef]
- Kuijper, E.C.; Bergsma, A.J.; Pijnappel, W.; Aartsma-Rus, A. Opportunities and challenges for antisense oligonucleotide therapies. J. Inherit. Metab. Dis. 2021, 44, 72–87. [Google Scholar] [CrossRef] [PubMed]
- Jarver, P.; O′Donovan, L.; Gait, M.J. A chemical view of oligonucleotides for exon skipping and related drug applications. Nucleic Acid Ther. 2014, 24, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Mastaglia, F.L.; Fletcher, S.; Wilton, S.D. Precision Medicine through Antisense Oligonucleotide-Mediated Exon Skipping. Trends Pharmacol. Sci. 2018, 39, 982–994. [Google Scholar] [CrossRef] [PubMed]
- Shimo, T.; Maruyama, R.; Yokota, T. Designing Effective Antisense Oligonucleotides for Exon Skipping. Methods Mol. Biol. 2018, 1687, 143–155. [Google Scholar] [PubMed]
- Crooke, S.T. Molecular Mechanisms of Antisense Oligonucleotides. Nucleic Acid Ther. 2017, 27, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Lima, W.F.; Crooke, S.T. Binding affinity and specificity of Escherichia coli RNase H1: Impact on the kinetics of catalysis of antisense oligonucleotide-RNA hybrids. Biochemistry 1997, 36, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.P.; Kent, K.; Bird, J.; Fishback, J.; Froehler, B. Modified deoxyoligonucleotides stable to exonuclease degradation in serum. Nucleic Acids Res. 1991, 19, 747–750. [Google Scholar] [CrossRef] [Green Version]
- Ortigao, J.F.; Rosch, H.; Selter, H.; Frohlich, A.; Lorenz, A.; Montenarh, M.; Seliger, H. Antisense effect of oligodeoxynucleotides with inverted terminal internucleotidic linkages: A minimal modification protecting against nucleolytic degradation. Antisense Res. Dev. 1992, 2, 129–146. [Google Scholar] [CrossRef]
- Mansoor, M.; Melendez, A.J. Advances in antisense oligonucleotide development for target identification, validation, and as novel therapeutics. Gene Regul. Syst. Biol. 2008, 2, 275–295. [Google Scholar] [CrossRef]
- Eckstein, F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 2014, 24, 374–387. [Google Scholar] [CrossRef]
- Miller, P.S. Oligonucleoside methylphosphonates as antisense reagents. Bio/Technology 1991, 9, 358–362. [Google Scholar] [CrossRef]
- Marshall, W.S.; Caruthers, M.H. Phosphorodithioate DNA as a potential therapeutic drug. Science 1993, 259, 1564–1570. [Google Scholar] [CrossRef]
- Summers, J.S.; Shaw, B.R. Boranophosphates as mimics of natural phosphodiesters in DNA. Curr. Med. Chem. 2001, 8, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Miroshnichenko, S.K.; Patutina, O.A.; Burakova, E.A.; Chelobanov, B.P.; Fokina, A.A.; Vlassov, V.V.; Altman, S.; Zenkova, M.A.; Stetsenko, D.A. Mesyl phosphoramidate antisense oligonucleotides as an alternative to phosphorothioates with improved biochemical and biological properties. Proc. Natl. Acad. Sci. USA 2019, 116, 1229–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patutina, O.A.; Gaponova Miroshnichenko, S.K.; Sen’kova, A.V.; Savin, I.A.; Gladkikh, D.V.; Burakova, E.A.; Fokina, A.A.; Maslov, M.A.; Shmendel, E.V.; Wood, M.J.A.; et al. Mesyl phosphoramidate backbone modified antisense oligonucleotides targeting miR-21 with enhanced in vivo therapeutic potency. Proc. Natl. Acad. Sci. USA 2020, 117, 32370–32379. [Google Scholar] [CrossRef] [PubMed]
- Dellinger, D.J.; Sheehan, D.M.; Christensen, N.K.; Lindberg, J.G.; Caruthers, M.H. Solid-phase chemical synthesis of phosphonoacetate and thiophosphonoacetate oligodeoxynucleotides. J. Am. Chem. Soc. 2003, 125, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Yamada, C.M.; Dellinger, D.J.; Caruthers, M.H. Synthesis and biochemical evaluation of phosphonoformate oligodeoxyribonucleotides. J. Am. Chem. Soc. 2006, 128, 5251–5261. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, M. 2′-carbohydrate modifications in antisense oligonucleotide therapy: Importance of conformation, configuration and conjugation. Biochim. Biophys. Acta 1999, 1489, 117–130. [Google Scholar] [CrossRef]
- Prakash, T.P.; Bhat, B. 2′-Modified oligonucleotides for antisense therapeutics. Curr. Top. Med. Chem. 2007, 7, 641–649. [Google Scholar] [CrossRef]
- Prakash, T.P. An overview of sugar-modified oligonucleotides for antisense therapeutics. Chem. Biodivers. 2011, 8, 1616–1641. [Google Scholar] [CrossRef] [PubMed]
- Lamond, A.I.; Sproat, B.S. Antisense oligonucleotides made of 2′-O-alkylRNA: Their properties and applications in RNA biochemistry. FEBS. Lett. 1993, 325, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.M.; Gamper, H.B. Inhibition of tRNA aminoacylation by 2′-O-methyl oligonucleotides. Biochemistry 1996, 35, 15340–15348. [Google Scholar] [CrossRef]
- Majlessi, M.; Nelson, N.C.; Becker, M.M. Advantages of 2′-O-methyl oligoribonucleotide probes for detecting RNA targets. Nucleic Acids Res. 1998, 26, 2224–2229. [Google Scholar] [CrossRef] [PubMed]
- Altmann, K.H.; Dean, N.M.; Fabbro, D.; Freier, S.M.; Geiger, T.; Hanera, R.; Hiisken, D.A.; Martina, P.; Monia, B.P.B.; Miiller, M.; et al. Second Generation of Antisense Oligonucleotides: From Nuclease Resistance to Biological Efficacy in Animals. Chim. (Aarau) 1996, 50, 168–176. [Google Scholar]
- Chi, K.N.; Eisenhauer, E.; Fazli, L.; Jones, E.C.; Goldenberg, S.L.; Powers, J.; Tu, D.; Gleave, M.E. A phase I pharmacokinetic and pharmacodynamic study of OGX-011, a 2′-methoxyethyl antisense oligonucleotide to clusterin, in patients with localized prostate cancer. J. Natl. Cancer Inst. 2005, 97, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, A.M.; Casper, M.D.; Freier, S.M.; Lesnik, E.A.; Zounes, M.C.; Cummins, L.L.; Gonzalez, C.; Cook, P.D. Uniformly modified 2′-deoxy-2′-fluoro phosphorothioate oligonucleotides as nuclease-resistant antisense compounds with high affinity and specificity for RNA targets. J. Med. Chem. 1993, 36, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, T.; Obika, S. BNAs: Novel nucleic acid analogs with a bridged sugar moiety. Chem. Commun. 2002, 1653–1659. [Google Scholar] [CrossRef]
- Kaur, H.; Babu, B.R.; Maiti, S. Perspectives on chemistry and therapeutic applications of Locked Nucleic Acid (LNA). Chem. Rev. 2007, 107, 4672–4697. [Google Scholar] [CrossRef]
- Veedu, R.N.; Wengel, J. Locked nucleic acids: Promising nucleic acid analogs for therapeutic applications. Chem. Biodivers. 2010, 7, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Soler-Bistue, A.; Zorreguieta, A.; Tolmasky, M.E. Bridged Nucleic Acids Reloaded. Molecules 2019, 24, 2297. [Google Scholar] [CrossRef] [Green Version]
- Renneberg, D.; Bouliong, E.; Reber, U.; Schumperli, D.; Leumann, C.J. Antisense properties of tricyclo-DNA. Nucleic Acids Res. 2002, 30, 2751–2757. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, P.E. Peptide nucleic acids (PNA) in chemical biology and drug discovery. Chem. Biodivers. 2010, 7, 786–804. [Google Scholar] [CrossRef]
- Summerton, J.; Weller, D. Morpholino antisense oligomers: Design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997, 7, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Summerton, J. History and Properties of Morpholino Antisense Oligos. J. Drug Discov. Develop. Deliv. 2016, 3, 1019. [Google Scholar]
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 2017, 27, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Heo, Y.A. Golodirsen: First Approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef]
- Shirley, M. Casimersen: First Approval. Drugs 2021, 81, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.J.; Baulcombe, D.C. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science 1999, 286, 950–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Kurreck, J. RNA interference: From basic research to therapeutic applications. Angew. Chem. 2009, 48, 1378–1398. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef]
- Zhou, L.Y.; Qin, Z.; Zhu, Y.H.; He, Z.Y.; Xu, T. Current RNA-based Therapeutics in Clinical Trials. Curr. Gene Ther. 2019, 19, 172–196. [Google Scholar] [CrossRef]
- Nair, J.K.; Willoughby, J.L.; Chan, A.; Charisse, K.; Alam, M.R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’in, A.V.; Milstein, S.; et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar] [CrossRef] [Green Version]
- Scott, L.J. Givosiran: First Approval. Drugs 2020, 80, 335–339. [Google Scholar] [CrossRef]
- Scott, L.J.; Keam, S.J. Lumasiran: First Approval. Drugs 2021, 81, 277–282. [Google Scholar] [CrossRef]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.J.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. New Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mojica, F.J.; Juez, G.; Rodriguez-Valera, F. Transcription at different salinities of Haloferax mediterranei sequences adjacent to partially modified PstI sites. Mol. Microbiol. 1993, 9, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.; Embden, J.D.; Gaastra, W.; Schouls, L.M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002, 43, 1565–1575. [Google Scholar] [CrossRef]
- Mojica, F.J.; Diez-Villasenor, C.; Garcia-Martinez, J.; Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Brouns, S.J.; Jore, M.M.; Lundgren, M.; Westra, E.R.; Slijkhuis, R.J.; Snijders, A.P.; Dickman, M.J.; Makarova, K.S.; Koonin, E.V.; van der Oost, J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 2008, 321, 960–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mojica, F.J.M.; Diez-Villasenor, C.; Garcia-Martinez, J.; Almendros, C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 2009, 155, 733–740. [Google Scholar] [CrossRef] [Green Version]
- Sapranauskas, R.; Gasiunas, G.; Fremaux, C.; Barrangou, R.; Horvath, P.; Siksnys, V. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 2011, 39, 9275–9282. [Google Scholar] [CrossRef]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; East, A.; Cheng, A.; Lin, S.; Ma, E.; Doudna, J. RNA-programmed genome editing in human cells. Elife 2013, 2, e00471. [Google Scholar] [CrossRef] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Cong, L.; Zhang, F. Genome engineering using CRISPR-Cas9 system. Methods Mol. Biol. 2015, 1239, 197–217. [Google Scholar]
- Baylis, F.; McLeod, M. First-in-human Phase 1 CRISPR Gene Editing Cancer Trials: Are We Ready? Curr. Gene Ther. 2017, 17, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Stefanidakis, M.; Wilson, C.J.; Baral, R.; Barrera, L.A.; Bounoutas, G.S.; Bumcrot, D.; Chao, H.; Ciulla, D.M.; DaSilva, J.A.; et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019, 25, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.; Bauman, J.; Kang, H.; Ming, X. Biological barriers to therapy with antisense and siRNA oligonucleotides. Mol. Pharm. 2009, 6, 686–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juliano, R.L.; Ming, X.; Nakagawa, O. The chemistry and biology of oligonucleotide conjugates. Acc. Chem. Res. 2012, 45, 1067–1076. [Google Scholar] [CrossRef] [Green Version]
- Juliano, R.L.; Carver, K.; Cao, C.; Ming, X. Receptors, endocytosis, and trafficking: The biological basis of targeted delivery of antisense and siRNA oligonucleotides. J. Drug Target. 2013, 21, 27–43. [Google Scholar] [CrossRef] [Green Version]
- Juliano, R.L.; Ming, X.; Carver, K.; Laing, B. Cellular uptake and intracellular trafficking of oligonucleotides: Implications for oligonucleotide pharmacology. Nucleic Acid Ther. 2014, 24, 101–113. [Google Scholar] [CrossRef]
- Crooke, S.T.; Wang, S.; Vickers, T.A.; Shen, W.; Liang, X. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017, 35, 230–237. [Google Scholar] [CrossRef]
- Ming, X. Cellular delivery of siRNA and antisense oligonucleotides via receptor-mediated endocytosis. Expert. Opin. Drug Deliv. 2011, 8, 435–449. [Google Scholar] [CrossRef]
- Juliano, R.; Alam, M.R.; Dixit, V.; Kang, H. Mechanisms and strategies for effective delivery of antisense and siRNA oligonucleotides. Nucleic Acids Res. 2008, 36, 4158–4171. [Google Scholar] [CrossRef] [Green Version]
- Deprey, K.; Batistatou, N.; Kritzer, J.A. A critical analysis of methods used to investigate the cellular uptake and subcellular localization of RNA therapeutics. Nucleic Acids Res. 2020, 48, 7623–7639. [Google Scholar] [CrossRef] [PubMed]
- Maier, M.; Bleicher, K.; Kalthoff, H.; Bayer, E. Enzymatic degradation of various antisense oligonucleotides: Monitoring and fragment identification by MECC and ES-MS. Biomed. Pept. Proteins Nucleic Acids. 1995, 1, 235–242. [Google Scholar]
- Monia, B.P.; Johnston, J.F.; Sasmor, H.; Cummins, L.L. Nuclease resistance and antisense activity of modified oligonucleotides targeted to Ha-ras. J. Biol. Chem. 1996, 271, 14533–14540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilanowska, A.; Studzińska, S. In vivo and in vitro studies of antisense oligonucleotides—A review. RSC Adv. 2020, 10, 34501–34516. [Google Scholar] [CrossRef]
- He, X.; Urip, B.A.; Zhang, Z.; Ngan, C.C.; Feng, B. Evolving AAV-delivered therapeutics towards ultimate cures. J. Mol. Med. 2021, 99, 593–617. [Google Scholar] [CrossRef]
- Tremblay, J.P.; Annoni, A.; Suzuki, M. Three Decades of Clinical Gene Therapy: From Experimental Technologies to Viable Treatments. Mol. Ther. 2021, 29, 411–412. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.C.; Wang, Z.L.; Xu, T.; He, Z.Y.; Wei, Y.Q. The approved gene therapy drugs worldwide: From 1998 to 2019. Biotechnol. Adv. 2020, 40, 107502. [Google Scholar] [CrossRef]
- Loring, H.S.; ElMallah, M.K.; Flotte, T.R. Development of rAAV2-CFTR: History of the First rAAV Vector Product to be Used in Humans. Hum. Gene Ther. Methods 2016, 27, 49–58. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, J.A.; Witzigmann, D.; Chen, S.; Cullis, P.R.; van der Meel, R. Lipid Nanoparticle Technology for Clinical Translation of siRNA Therapeutics. Acc. Chem. Res. 2019, 52, 2435–2444. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L. Intracellular delivery of oligonucleotide conjugates and dendrimer complexes. Ann. N. Y. Acad. Sci. 2006, 1082, 18–26. [Google Scholar] [CrossRef]
- Hawner, M.; Ducho, C. Cellular Targeting of Oligonucleotides by Conjugation with Small Molecules. Molecules 2020, 25, 5963. [Google Scholar] [CrossRef] [PubMed]
- Langel, Ü. (Ed.) Cell-Penetrating Peptides. In Methods and Protocols, 2nd ed.; Humana Press: Totowa, NJ, USA, 2015; Volume 1324. [Google Scholar]
- Derakhshankhah, H.; Jafari, S. Cell penetrating peptides: A concise review with emphasis on biomedical applications. Biomed. Pharmacother. 2018, 108, 1090–1096. [Google Scholar] [CrossRef]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar] [CrossRef]
- Vives, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef] [Green Version]
- Pooga, M.; Soomets, U.; Hallbrink, M.; Valkna, A.; Saar, K.; Rezaei, K.; Kahl, U.; Hao, J.X.; Xu, X.J.; Wiesenfeld-Hallin, Z.; et al. Cell penetrating PNA constructs regulate galanin receptor levels and modify pain transmission in vivo. Nat. Biotechnol. 1998, 16, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In vivo protein transduction: Delivery of a biologically active protein into the mouse. Science 1999, 285, 1569–1572. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Li, F.; Huang, Y. Smart Cell-Penetrating Peptide-Based Techniques for Intracellular Delivery of Therapeutic Macromolecules. Adv. Protein Chem. Struct. Biol. 2018, 112, 183–220. [Google Scholar]
- Kardani, K.; Milani, A.; Shabani, S.H.; Bolhassani, A. Cell penetrating peptides: The potent multi-cargo intracellular carriers. Expert Opin. Drug Deliv. 2019, 16, 1227–1258. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.J.; Kim, D.T.; Steinman, L.; Fathman, C.G.; Rothbard, J.B. Polyarginine enters cells more efficiently than other polycationic homopolymers. J. Pept. Res. 2000, 56, 318–325. [Google Scholar] [CrossRef]
- Rothbard, J.B.; Jessop, T.C.; Lewis, R.S.; Murray, B.A.; Wender, P.A. Role of membrane potential and hydrogen bonding in the mechanism of translocation of guanidinium-rich peptides into cells. J. Am. Chem. Soc. 2004, 126, 9506–9507. [Google Scholar] [CrossRef] [PubMed]
- Futaki, S.; Suzuki, T.; Ohashi, W.; Yagami, T.; Tanaka, S.; Ueda, K.; Sugiura, Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 2001, 276, 5836–5840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milletti, F. Cell-penetrating peptides: Classes, origin, and current landscape. Drug Discov. Today 2012, 17, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Zaro, J.L.; Shen, W.-C. Cationic and amphipathic cell-penetrating peptides (CPPs): Their structures and in vivo studies in drug delivery. Front. Chem. Sci. Eng. 2015, 9, 407–427. [Google Scholar] [CrossRef]
- Morris, M.C.; Vidal, P.; Chaloin, L.; Heitz, F.; Divita, G. A new peptide vector for efficient delivery of oligonucleotides into mammalian cells. Nucleic Acids Res. 1997, 25, 2730–2736. [Google Scholar] [CrossRef]
- Morris, M.C.; Depollier, J.; Mery, J.; Heitz, F.; Divita, G. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat. Biotechnol. 2001, 19, 1173–1176. [Google Scholar] [CrossRef]
- Johansson, H.J.; El-Andaloussi, S.; Holm, T.; Mae, M.; Janes, J.; Maimets, T.; Langel, U. Characterization of a novel cytotoxic cell-penetrating peptide derived from p14ARF protein. Mol. Ther. 2008, 16, 115–123. [Google Scholar] [CrossRef]
- Lundberg, P.; Magzoub, M.; Lindberg, M.; Hallbrink, M.; Jarvet, J.; Eriksson, L.E.; Langel, U.; Graslund, A. Cell membrane translocation of the N-terminal (1–28) part of the prion protein. Biochem. Biophys. Res. Commun. 2002, 299, 85–90. [Google Scholar] [CrossRef]
- Magzoub, M.; Sandgren, S.; Lundberg, P.; Oglęcka, K.; Lilja, J.; Wittrup, A.; Göran Eriksson, L.E.; Langel, Ü.; Belting, M.; Gräslund, A. N-terminal peptides from unprocessed prion proteins enter cells by macropinocytosis. Biochem. Biophys. Res. Commun. 2006, 348, 379–385. [Google Scholar] [CrossRef]
- Oehlke, J.; Scheller, A.; Wiesner, B.; Krause, E.; Beyermann, M.; Klauschenz, E.; Melzig, M.; Bienert, M. Cellular uptake of an alpha-helical amphipathic model peptide with the potential to deliver polar compounds into the cell interior non-endocytically. Biochim. Biophys. Acta 1998, 1414, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Soomets, U.; Lindgren, M.; Gallet, X.; Hallbrink, M.; Elmquist, A.; Balaspiri, L.; Zorko, M.; Pooga, M.; Brasseur, R.; Langel, U. Deletion analogues of transportan. Biochim. Biophys. Acta 2000, 1467, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Crombez, L.; Aldrian-Herrada, G.; Konate, K.; Nguyen, Q.N.; McMaster, G.K.; Brasseur, R.; Heitz, F.; Divita, G. A new potent secondary amphipathic cell-penetrating peptide for siRNA delivery into mammalian cells. Mol. Ther. 2009, 17, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Christov, K.; Shilkaitis, A.; Bratescu, L.; Green, A.; Santini, S.; Bizzarri, A.R.; Cannistraro, S.; Gupta, T.K.; Beattie, C.W. p28, a first in class peptide inhibitor of cop1 binding to p53. Br. J. Cancer 2013, 108, 2495–2504. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.N.; Mehta, R.R.; Yamada, T.; Lekmine, F.; Christov, K.; Chakrabarty, A.M.; Green, A.; Bratescu, L.; Shilkaitis, A.; Beattie, C.W.; et al. Noncationic peptides obtained from azurin preferentially enter cancer cells. Cancer Res. 2009, 69, 537–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujals, S.; Giralt, E. Proline-rich, amphipathic cell-penetrating peptides. Adv. Drug Deliv. Rev. 2008, 60, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Sadler, K.; Eom, K.D.; Yang, J.L.; Dimitrova, Y.; Tam, J.P. Translocating proline-rich peptides from the antimicrobial peptide bactenecin 7. Biochemistry 2002, 41, 14150–14157. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.S.; Schepartz, A. Intrinsically cell-permeable miniature proteins based on a minimal cationic PPII motif. J. Am. Chem. Soc. 2007, 129, 14578–14579. [Google Scholar] [CrossRef] [PubMed]
- Rhee, M.; Davis, P. Mechanism of uptake of C105Y, a novel cell-penetrating peptide. J. Biol. Chem. 2006, 281, 1233–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C.; Mao, S.; Ditzel, H.J.; Farnaes, L.; Wirsching, P.; Lerner, R.A.; Janda, K.D. A cell-penetrating peptide from a novel pVII-pIX phage-displayed random peptide library. Bioorg. Med. Chem. 2002, 10, 4057–4065. [Google Scholar] [CrossRef]
- Abushahba, M.F.; Mohammad, H.; Thangamani, S.; Hussein, A.A.; Seleem, M.N. Impact of different cell penetrating peptides on the efficacy of antisense therapeutics for targeting intracellular pathogens. Sci. Rep. 2016, 6, 20832. [Google Scholar] [CrossRef]
- Moulton, H.M.; Hase, M.C.; Smith, K.M.; Iversen, P.L. HIV Tat peptide enhances cellular delivery of antisense morpholino oligomers. Antisense Nucleic Acid Drug Dev. 2003, 13, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Astriab-Fisher, A.; Sergueev, D.; Fisher, M.; Shaw, B.R.; Juliano, R.L. Conjugates of antisense oligonucleotides with the Tat and antennapedia cell-penetrating peptides: Effects on cellular uptake, binding to target sequences, and biologic actions. Pharm. Res. 2002, 19, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; He, Y.; Xia, Y.; Wang, H.; Wang, L.; Gao, R.; Zhang, M. Inhibiting the growth of methicillin-resistant Staphylococcus aureus in vitro with antisense peptide nucleic acid conjugates targeting the ftsZ gene. Int. J. Infect. Dis. 2015, 30, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Lin, Y.; Xuan, W.; Iversen, P.L.; Smith, L.J.; Benchimol, S. Inhibition of p53 expression by peptide-conjugated phosphorodiamidate morpholino oligomers sensitizes human cancer cells to chemotherapeutic drugs. Oncogene 2012, 31, 1024–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, J.; Da, F.; Ma, X.; Wang, N.; Wang, Y.; Zhang, H.; Li, M.; Zhou, Y.; Xue, X.; Hou, Z.; et al. Antisense growth inhibition of methicillin-resistant Staphylococcus aureus by locked nucleic acid conjugated with cell-penetrating peptide as a novel FtsZ inhibitor. Antimicrob. Agents Chemother. 2015, 59, 914–922. [Google Scholar] [CrossRef] [Green Version]
- Betts, C.; Saleh, A.F.; Arzumanov, A.A.; Hammond, S.M.; Godfrey, C.; Coursindel, T.; Gait, M.J.; Wood, M.J. Pip6-PMO, A New Generation of Peptide-oligonucleotide Conjugates With Improved Cardiac Exon Skipping Activity for DMD Treatment. Mol. Ther.-Nucleic Acids 2012, 1, e38. [Google Scholar] [CrossRef]
- El-Andaloussi, S.; Johansson, H.J.; Lundberg, P.; Langel, U. Induction of splice correction by cell-penetrating peptide nucleic acids. J. Gene Med. 2006, 8, 1262–1273. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Nie, D.; Hu, Y.; Li, M.; Hou, Z.; Mao, X.; Luo, X.; Xue, X. Efficient Delivery of Antisense Oligonucleotides by an Amphipathic Cell-Penetrating Peptide in Acinetobacter baumannii. Curr. Drug Deliv. 2019, 16, 728–736. [Google Scholar] [CrossRef]
- Vaissiere, A.; Aldrian, G.; Konate, K.; Lindberg, M.F.; Jourdan, C.; Telmar, A.; Seisel, Q.; Fernandez, F.; Viguier, V.; Genevois, C.; et al. A retro-inverso cell-penetrating peptide for siRNA delivery. J. Nanobiotechnol. 2017, 15, 34. [Google Scholar] [CrossRef]
- Alexander-Bryant, A.A.; Zhang, H.; Attaway, C.C.; Pugh, W.; Eggart, L.; Sansevere, R.M.; Andino, L.M.; Dinh, L.; Cantini, L.P.; Jakymiw, A. Dual peptide-mediated targeted delivery of bioactive siRNAs to oral cancer cells in vivo. Oral. Oncol. 2017, 72, 123–131. [Google Scholar] [CrossRef]
- Cantini, L.; Attaway, C.C.; Butler, B.; Andino, L.M.; Sokolosky, M.L.; Jakymiw, A. Fusogenic-oligoarginine peptide-mediated delivery of siRNAs targeting the CIP2A oncogene into oral cancer cells. PLoS ONE 2013, 8, e73348. [Google Scholar] [CrossRef] [Green Version]
- Jirka, S.M.; Heemskerk, H.; Tanganyika-de Winter, C.L.; Muilwijk, D.; Pang, K.H.; de Visser, P.C.; Janson, A.; Karnaoukh, T.G.; Vermue, R.; ‘t Hoen, P.A.; et al. Peptide conjugation of 2′-O-methyl phosphorothioate antisense oligonucleotides enhances cardiac uptake and exon skipping in mdx mice. Nucleic Acid Ther. 2014, 24, 25–36. [Google Scholar] [CrossRef]
- Marks, J.R.; Placone, J.; Hristova, K.; Wimley, W.C. Spontaneous Membrane-Translocating Peptides by Orthogonal High-Throughput Screening. J. Am. Chem. Soc. 2011, 133, 8995–9004. [Google Scholar] [CrossRef] [Green Version]
- Mueller, J.; Kretzschmar, I.; Volkmer, R.; Boisguerin, P. Comparison of cellular uptake using 22 CPPs in 4 different cell lines. Bioconjugate Chem. 2008, 19, 2363–2374. [Google Scholar] [CrossRef]
- Kosuge, M.; Takeuchi, T.; Nakase, I.; Jones, A.T.; Futaki, S. Cellular internalization and distribution of arginine-rich peptides as a function of extracellular peptide concentration, serum, and plasma membrane associated proteoglycans. Bioconjugate Chem. 2008, 19, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Fretz, M.M.; Penning, N.A.; Al-Taei, S.; Futaki, S.; Takeuchi, T.; Nakase, I.; Storm, G.; Jones, A.T. Temperature-, concentration- and cholesterol-dependent translocation of L- and D-octa-arginine across the plasma and nuclear membrane of CD34+ leukaemia cells. Biochem. J. 2007, 403, 335–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palm-Apergi, C.; Lonn, P.; Dowdy, S.F. Do cell-penetrating peptides actually “penetrate” cellular membranes? Mol. Ther. 2012, 20, 695–697. [Google Scholar] [CrossRef] [Green Version]
- Hirose, H.; Takeuchi, T.; Osakada, H.; Pujals, S.; Katayama, S.; Nakase, I.; Kobayashi, S.; Haraguchi, T.; Futaki, S. Transient focal membrane deformation induced by arginine-rich peptides leads to their direct penetration into cells. Mol. Ther. 2012, 20, 984–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundberg, M.; Wikstrom, S.; Johansson, M. Cell surface adherence and endocytosis of protein transduction domains. Mol. Ther. 2003, 8, 143–150. [Google Scholar] [CrossRef]
- Herce, H.D.; Garcia, A.E. Molecular dynamics simulations suggest a mechanism for translocation of the HIV-1 TAT peptide across lipid membranes. Proc. Natl. Acad. Sci. USA 2007, 104, 20805–20810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herce, H.D.; Garcia, A.E.; Cardoso, M.C. Fundamental molecular mechanism for the cellular uptake of guanidinium-rich molecules. J. Am. Chem. Soc. 2014, 136, 17459–17467. [Google Scholar] [CrossRef] [PubMed]
- Thennarasu, S.; Tan, A.; Penumatchu, R.; Shelburne, C.E.; Heyl, D.L.; Ramamoorthy, A. Antimicrobial and membrane disrupting activities of a peptide derived from the human cathelicidin antimicrobial peptide LL37. Biophys. J. 2010, 98, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Pouny, Y.; Rapaport, D.; Mor, A.; Nicolas, P.; Shai, Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogues with phospholipid membranes. Biochemistry 1992, 31, 12416–12423. [Google Scholar] [CrossRef] [PubMed]
- Alves, I.D.; Goasdoue, N.; Correia, I.; Aubry, S.; Galanth, C.; Sagan, S.; Lavielle, S.; Chassaing, G. Membrane interaction and perturbation mechanisms induced by two cationic cell penetrating peptides with distinct charge distribution. Biochim. Biophys. Acta 2008, 1780, 948–959. [Google Scholar] [CrossRef]
- Koren, E.; Torchilin, V.P. Cell-penetrating peptides: Breaking through to the other side. Trends Mol. Med. 2012, 18, 385–393. [Google Scholar] [CrossRef]
- Maiolo, J.R.; Ferrer, M.; Ottinger, E.A. Effects of cargo molecules on the cellular uptake of arginine-rich cell-penetrating peptides. Biochim. Biophys. Acta 2005, 1712, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Berg, A.; Dowdy, S.F. Protein transduction domain delivery of therapeutic macromolecules. Curr. Opin. Biotechnol. 2011, 22, 888–893. [Google Scholar] [CrossRef]
- Hitz, T.; Iten, R.; Gardiner, J.; Namoto, K.; Walde, P.; Seebach, D. Interaction of alpha-and beta-oligoarginine-acids and amides with anionic lipid vesicles: A mechanistic and thermodynamic study. Biochemistry 2006, 45, 5817–5829. [Google Scholar] [CrossRef]
- El-Sayed, A.; Futaki, S.; Harashima, H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: Ways to overcome endosomal entrapment. AAPS. J. 2009, 11, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Sirsi, S.R.; Schray, R.C.; Guan, X.; Lykens, N.M.; Williams, J.H.; Erney, M.L.; Lutz, G.J. Functionalized PEG-PEI copolymers complexed to exon-skipping oligonucleotides improve dystrophin expression in mdx mice. Hum. Gene Ther. 2008, 19, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Sugita, T.; Yoshikawa, T.; Mukai, Y.; Yamanada, N.; Imai, S.; Nagano, K.; Yoshida, Y.; Shibata, H.; Yoshioka, Y.; Nakagawa, S.; et al. Improved cytosolic translocation and tumor-killing activity of Tat-shepherdin conjugates mediated by co-treatment with Tat-fused endosome-disruptive HA2 peptide. Biochem. Biophys. Res. Commun. 2007, 363, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, P.; El-Andaloussi, S.; Sutlu, T.; Johansson, H.; Langel, U. Delivery of short interfering RNA using endosomolytic cell-penetrating peptides. FASEB. J. 2007, 21, 2664–2671. [Google Scholar] [CrossRef] [Green Version]
- Wadia, J.S.; Stan, R.V.; Dowdy, S.F. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat. Med. 2004, 10, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.L.; Kay, L.E. Measurement of histidine pKa values and tautomer populations in invisible protein states. Proc. Natl. Acad. Sci. USA 2014, 111, E1705–E1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, S.L.; Wang, S. An endosomolytic Tat peptide produced by incorporation of histidine and cysteine residues as a nonviral vector for DNA transfection. Biomaterials 2008, 29, 2408–2414. [Google Scholar] [CrossRef]
- Midoux, P.; Pichon, C.; Yaouanc, J.J.; Jaffres, P.A. Chemical vectors for gene delivery: A current review on polymers, peptides and lipids containing histidine or imidazole as nucleic acids carriers. Br. J. Pharm. 2009, 157, 166–178. [Google Scholar] [CrossRef] [Green Version]
- Leng, Q.; Chou, S.-T.; Scaria, P.V.; Woodle, M.C.; Mixson, A.J. Increased tumor distribution and expression of histidine-rich plasmid polyplexes. J. Gene Med. 2014, 16, 317–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, R.; Hufnagel, H.; Brock, R. A doubly labeled penetratin analogue as a ratiometric sensor for intracellular proteolytic stability. Bioconjugate Chem. 2010, 21, 64–73. [Google Scholar] [CrossRef]
- Cordier, C.; Boutimah, F.; Bourdeloux, M.; Dupuy, F.; Met, E.; Alberti, P.; Loll, F.; Chassaing, G.; Burlina, F.; Saison-Behmoaras, T.E. Delivery of antisense peptide nucleic acids to cells by conjugation with small arginine-rich cell-penetrating peptide (R/W)9. PLoS ONE 2014, 9, e104999. [Google Scholar] [CrossRef] [PubMed]
- Bawa, R.; Fung, S.Y.; Shiozaki, A.; Yang, H.; Zheng, G.; Keshavjee, S.; Liu, M. Self-assembling peptide-based nanoparticles enhance cellular delivery of the hydrophobic anticancer drug ellipticine through caveolae-dependent endocytosis. Nanomedicine 2012, 8, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Wickstrom, E. Solid phase synthesis of a d-peptide-phosphorothioate oligodeoxynucleotide conjugate from two arms of a polyethylene glycol-polystyrene support. Tetrahedron Lett. 1995, 36, 4943–4946. [Google Scholar]
- Avitabile, C.; Saviano, M.; D’Andrea, L.; Bianchi, N.; Fabbri, E.; Brognara, E.; Gambari, R.; Romanelli, A. Targeting pre-miRNA by peptide nucleic acids: A new strategy to interfere in the miRNA maturation. Artif. Dna Pna Xna 2012, 3, 88–96. [Google Scholar] [CrossRef] [Green Version]
- Piras, L.; Avitabile, C.; D’Andrea, L.D.; Saviano, M.; Romanelli, A. Detection of oligonucleotides by PNA-peptide conjugates recognizing the biarsenical fluorescein complex FlAsH-EDT2. Biochem. Biophys. Res. Commun. 2017, 493, 126–131. [Google Scholar] [CrossRef]
- Soudah, T.; Mogilevsky, M.; Karni, R.; Yavin, E. CLIP6-PNA-Peptide Conjugates: Non-Endosomal Delivery of Splice Switching Oligonucleotides. Bioconjugate Chem. 2017, 28, 3036–3042. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.K.; Kim, C.W.; Kawanami, H.; Kishimura, A.; Niidome, T.; Mori, T.; Katayama, Y. Utilization of a PNA-peptide conjugate to induce a cancer protease-responsive RNAi effect. RSC Adv. 2015, 5, 85816–85821. [Google Scholar] [CrossRef]
- Tetzlaff, C.N.; Schwope, I.; Bleczinski, C.F.; Steinberg, J.A.; Richert, C. A convenient synthesis of 5′-amino-5′-deoxythymidine and preparation of peptide-DNA hybrids. Tetrahedron Lett. 1998, 39, 4215–4218. [Google Scholar] [CrossRef]
- Guzzo-Pernell, N.; Tregear, G.W. Triple helical DNA formation by a hydrophobic oligonucleotide-peptide hybrid molecule. Aust. J. Chem. 2000, 53, 699–705. [Google Scholar] [CrossRef]
- Frieden, M.; Avino, A.; Tarrason, G.; Escorihuela, M.; Piulats, J.; Eritja, R. Synthesis of oligonucleotide-peptide conjugates carrying the c-myc peptide epitope as recognition system. Chem. Biodivers. 2004, 1, 930–938. [Google Scholar] [CrossRef] [Green Version]
- Aviñó, A.; Grijalvo, S.; Pérez-Rentero, S.; Garibotti, A.; Terrazas, M.; Eritja, R. Synthesis of oligonucleotide-peptide conjugates for biomedical and technological applications. In Bioconjugation Protocols; Mark, S., Ed.; Springer Protocols; Humana Press: New York, NY, USA, 2011. [Google Scholar] [CrossRef] [Green Version]
- Marchán, V.; Debéthune, L.; Beltrán, M.; Robles, J.; Travesset, I.; Fábregas, G.; Pedroso, E.; Grandas, A. The Stepwise SolidPhase Synthesis Methodology is Suitable for the Preparation of a Great Variety of Nucleopeptides. Nucleosides Nucleotides 1999, 18, 1493–1494. [Google Scholar] [CrossRef]
- Robles, J.; Beltrán, M.; Marchán, V.; Pérez, Y.; Travesset, I.; Pedroso, E.; Grandas, A. Towards nucleopeptides containing any trifunctional amino acid. Tetrahedron 1999, 55, 13251–13264. [Google Scholar] [CrossRef]
- Stetsenko, D.A.; Malakhov, A.D.; Gait, M.J. Total stepwise solid-phase peptide-oligonucleotide conjugate synthesis on macroporous polystyrene. Nucleosides Nucleotides Nucleic Acids 2003, 22, 1379–1382. [Google Scholar] [CrossRef] [PubMed]
- Grijalvo, S.; Terrazas, M.; Avino, A.; Eritja, R. Stepwise synthesis of oligonucleotide-peptide conjugates containing guanidinium and lipophilic groups in their 3′-termini. Bioorg. Med. Chem. Lett. 2010, 20, 2144–2147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Torre, B.G.; Aviñó, A.; Tarrason, G.; Piulats, J.; Albericio, F.; Eritja, R. Stepwise solid-phase synthesis of oligonucleotide-peptide hybrids. Tetrahedron Lett. 1994, 35, 2733–2736. [Google Scholar] [CrossRef]
- Truffert, J.; Lorthioir, O.; Asseline, U.; Thuong, N.T.; Brack, A. On-line solid phase synthesis of oligonucleotide-peptide hybrids using silica supports. Tetrahedron Lett. 1994, 35, 2353–2356. [Google Scholar] [CrossRef]
- Truffert, J.-C.; Asseline, U.; Brack, A.; Thuong, N.T. Synthesis, purification and characterization of two peptide-oligonucleotide conjugates as potential artificial nucleases. Tetrahedron 1996, 52, 3005–3016. [Google Scholar] [CrossRef]
- Robles, J.; Pedroso, E.; Grandas, A. Stepwise solid-phase synthesis of the nucleopeptide Phac-Phe-Val-Ser(p3′ACT)-Gly-OH. J. Org. Chem. 1994, 59, 2482–2486. [Google Scholar] [CrossRef]
- Haralambidis, J.; Duncan, L.; Angus, K.; Tregear, G.W. The synthesis of polyamide-oligonucleotide conjugate molecules. Nucleic Acids Res. 1990, 18, 493. [Google Scholar] [CrossRef] [Green Version]
- Beltrán, M.; Pedroso, E.; Grandas, A. A comparison of histidine protecting groups in the synthesis of peptide-oligonucleotide conjugates. Tetrahedron Lett. 1998, 39, 4115–4118. [Google Scholar] [CrossRef]
- De Champdoré, M.; De Napoli, L.; Di Fabio, G.; Messere, A.; Montesarchio, D.; Piccialli, G. New nucleoside based solid supports. Synthesis of 5′,3′-derivatized thymidine analogues. Chem. Commun. 2001, 2598–2599. [Google Scholar] [CrossRef]
- Robles, J.; Pedroso, E.; Grandas, A. Solid phase synthesis of a model nucleopeptide with a phosphodiester bond between the 5′ end of a trinucleotide and a serine residue. Tetrahedron Lett. 1991, 32, 4389–4392. [Google Scholar] [CrossRef]
- Bergmann, F.; Bannwarth, W. Solid phase synthesis of directly linked peptide-oligodeoxynucleotide hybrids using standard synthesis protocols. Tetrahedron Lett. 1995, 36, 1839–1842. [Google Scholar] [CrossRef]
- Haralambidis, J.; Duncan, L.; Tregear, G.W. The solid phase synthesis of oligonucleotides containing a 3′-peptide moiety. Tetrahedron Lett. 1987, 28, 5199–5202. [Google Scholar] [CrossRef]
- Avino, A.; Gomara, M.J.; Malakoutikhah, M.; Haro, I.; Eritja, R. Oligonucleotide-peptide conjugates: Solid-phase synthesis under acidic conditions and use in ELISA assays. Molecules 2012, 17, 13825–13843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antopolsky, M.; Azhayeva, E.; Tengvall, U.; Azhayev, A. Towards a general method for the stepwise solid-phase synthesis of peptide–oligonucleotide conjugates. Tetrahedron Lett. 2002, 43, 527–530. [Google Scholar] [CrossRef]
- Juby, C.D.; Richardson, C.D.; Brousseau, R. Facile preparation of 3′oligonucleotide-peptide conjugates. Tetrahedron Lett. 1991, 32, 879–882. [Google Scholar] [CrossRef]
- Terenzi, S.; Biala, E.; Nguyen-Trung, N.Q.; Strazewski, P. Amphiphilic 3′-peptidyl-RNA conjugates. Angew. Chem. Int. Ed. 2003, 42, 2909–2912. [Google Scholar] [CrossRef]
- Nielsen, J.; Brenner, S.; Janda, K.D. Synthetic methods for the implementation of encoded combinatorial chemistry. J. Am. Chem. Soc. 1993, 115, 9812–9813. [Google Scholar] [CrossRef]
- Antopolsky, M.; Azhayev, A. Stepwise Solid-Phase Synthesis of Peptide-Oligonucleotide Conjugates on New Solid Supports. Helv. Chim. Acta 1999, 82, 2130–2140. [Google Scholar] [CrossRef]
- Aubert, Y.; Bourgerie, S.; Meunier, L.; Mayer, R.; Roche, A.C.; Monsigny, M.; Thuong, N.T.; Asseline, U. Optimized synthesis of phosphorothioate oligodeoxyribonucleotides substituted with a 5′-protected thiol function and a 3′-amino group. Nucleic Acids Res. 2000, 28, 818–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Tung, C.-H.; Zhu, T.; Stein, S. Synthesis of Oligoarginine-Oligonucleotide Conjugates and Oligoarginine-Bridged Oligonucleotide Pairs. Bioconjugate Chem. 1994, 5, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Arar, K.; Aubertin, A.M.; Roche, A.C.; Monsigny, M.; Mayer, R. Synthesis and antiviral activity of peptide-oligonucleotide conjugates prepared by using N alpha—(bromoacetyl)peptides. Bioconjugate Chem. 1995, 6, 573–577. [Google Scholar] [CrossRef]
- Ede, N.J.; Tregear, G.W.; Haralambidis, J. Routine Preparation of Thiol Oligonucleotides: Application to the Synthesis of Oligonucleotide-Peptide Hybrids. Bioconjugate Chem. 1994, 5, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Soukchareun, S.; Haralambidis, J.; Tregear, G. Use of Nα-Fmoc-cysteine(S-thiobutyl) Derivatized Oligodeoxynucleotides for the Preparation of Oligodeoxynucleotide−Peptide Hybrid Molecules. Bioconjugate Chem. 1998, 9, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Zanta, M.A.; Belguise-Valladier, P.; Behr, J.P. Gene delivery: A single nuclear localization signal peptide is sufficient to carry DNA to the cell nucleus. Proc. Natl. Acad. Sci. USA 1999, 96, 91–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hangeland, J.J.; Levis, J.T.; Lee, Y.C.; Tso, P.O.P. Cell-Type Specific and Ligand Specific Enhancement of Cellular Uptake of Oligodeoxynucleoside Methylphosphonates Covalently Linked with a Neoglycopeptide, YEE(ah-GalNAc)3. Bioconjugate Chem. 1995, 6, 695–701. [Google Scholar] [CrossRef]
- Mier, W.; Eritja, R.; Mohammed, A.; Haberkorn, U.; Eisenhut, M. Preparation and Evaluation of Tumor-Targeting Peptide−Oligonucleotide Conjugates. Bioconjugate Chem. 2000, 11, 855–860. [Google Scholar] [CrossRef] [Green Version]
- Arar, K.; Monsigny, M.; Mayer, R. Synthesis of oligonucleotide-peptide conjugates containing a KDEL signal sequence. Tetrahedron Lett. 1993, 34, 8087–8090. [Google Scholar] [CrossRef]
- de la Torre, B.G.; Albericio, F.; Saison-Behmoaras, E.; Bachi, A.; Eritja, R. Synthesis and Binding Properties of Oligonucleotides Carrying Nuclear Localization Sequences. Bioconjugate Chem. 1999, 10, 1005–1012. [Google Scholar] [CrossRef]
- Harrison, J.G.; Balasubramanian, S. Synthesis and hybridization analysis of a small library of peptide-oligonucleotide conjugates. Nucleic Acids Res. 1998, 26, 3136–3145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eritja, R.; Pons, A.; Escarceller, M.; Giralt, E.; Albericio, F. Synthesis of Defined Peptide-Oligonucleotide Hybrids Containing a Nuclear Transport Signal Sequence. Tetrahedron 1991, 47, 4113–4120. [Google Scholar] [CrossRef]
- Chaloin, L.; Vidal, P.; Lory, P.; Méry, J.; Lautredou, N.; Divita, G.; Heitz, F. Design of Carrier Peptide-Oligonucleotide Conjugates with Rapid Membrane Translocation and Nuclear Localization Properties. Biochem. Biophys. Res. Commun. 1998, 243, 601–608. [Google Scholar] [CrossRef]
- Corey, D.R. Synthesis of oligonucleotide-peptide and oligonucleotide-protein conjugates. Methods Mol. Biol. 2004, 283, 197–206. [Google Scholar] [PubMed]
- Turner, J.J.; Williams, D.; Owen, D.; Gait, M.J. Disulfide conjugation of peptides to oligonucleotides and their analogs. Curr. Protoc. Nucleic Acid Chem. 2006, 24, 4–28. [Google Scholar] [CrossRef] [PubMed]
- Tennila, T.; Antopolsky, M.; Azhayev, A.; Azhayeva, E. Peptide-oligonucleotide conjugates form stable and selective complexes with antibody and DNA. Bioconjugate Chem. 2008, 19, 1361–1367. [Google Scholar] [CrossRef]
- Vivès, E.; Lebleu, B. Selective coupling of a highly basic peptide to an oligonucleotide. Tetrahedron Lett. 1997, 38, 1183–1186. [Google Scholar] [CrossRef]
- Antopolsky, M.; Azhayeva, E.; Tengvall, U.; Auriola, S.; Jaaskelainen, I.; Ronkko, S.; Honkakoski, P.; Urtti, A.; Lonnberg, H.; Azhayev, A. Peptide-oligonucleotide phosphorothioate conjugates with membrane translocation and nuclear localization properties. Bioconjugate Chem. 1999, 10, 598–606. [Google Scholar] [CrossRef]
- Dirin, M.; Urban, E.; Lachmann, B.; Noe, C.R.; Winkler, J. Concise postsynthetic preparation of oligonucleotide-oligopeptide conjugates through facile disulfide bond formation. Future Med. Chem. 2015, 7, 1657–1673. [Google Scholar] [CrossRef] [PubMed]
- Dirin, M.; Urban, E.; Noe, C.R.; Winkler, J. Fragment-based solid-phase assembly of oligonucleotide conjugates with peptide and polyethylene glycol ligands. Eur. J. Med. Chem. 2016, 121, 132–142. [Google Scholar] [CrossRef]
- Podyminogin, M.A.; Lukhtanov, E.A.; Reed, M.W. Attachment of benzaldehyde-modified oligodeoxynucleotide probes to semicarbazide-coated glass. Nucleic Acids Res. 2001, 29, 5090–5098. [Google Scholar] [CrossRef]
- Singh, Y.; Defrancq, E.; Dumy, P. New method to prepare peptide-oligonucleotide conjugates through glyoxylic oxime formation. J. Org. Chem. 2004, 69, 8544–8546. [Google Scholar] [CrossRef]
- Forget, D.; Boturyn, D.; Defrancq, E.; Lhomme, J.; Dumy, P. Highly efficient synthesis of peptide-oligonucleotide conjugates: Chemoselective oxime and thiazolidine formation. Chemistry 2001, 7, 3976–3984. [Google Scholar] [CrossRef]
- Forget, D.; Renaudet, O.; Boturyn, D.; Defrancq, E.; Dumy, P. 3′-Oligonucleotides Conjugation via Chemoselective Oxime Bond Formation. Tetrahedron Lett. 2001, 42, 9171–9174. [Google Scholar] [CrossRef]
- Villien, M.; Defrancq, E.; Dumy, P. Chemoselective oxime and thiazolidine bond formation: A versatile and efficient route to the preparation of 3′-peptide-oligonucleotide conjugates. Nucleosides Nucleotides Nucleic Acids 2004, 23, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, N.; Edupuganti, O.P.; Defrancq, E.; Dumy, P. New solid support for the synthesis of 3′-oligonucleotide conjugates through glyoxylic oxime bond formation. Org. Lett. 2007, 9, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Defrancq, E.; Lhomme, J.H.; Dumy, P.; Bhattacharya, S. Efficient conjugation and characterization of distamycin-based peptides with selected oligonucleotide stretches. Bioconjugate Chem. 2004, 15, 520–529. [Google Scholar] [CrossRef]
- Singh, Y.; Edupuganti, O.P.; Villien, M.; Defrancq, É.; Dumy, P. The oxime bond formation as a useful tool for the preparation of oligonucleotide conjugates. Comptes Rendus Chim. 2005, 8, 789–796. [Google Scholar] [CrossRef]
- Spinelli, N.; Singh, Y.; Defrancq, E.; Dumy, P. Aldehydic oligonucleotide: A key intermediate for the preparation of oligonucleotide conjugates through oxime bond formation. Nucleosides Nucleotides Nucleic Acids 2007, 26, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Ollivier, N.; Olivier, C.; Gouyette, C.; Huynh-Dinh, T.; Gras-Masse, H.; Melnyk, O. Synthesis of oligonucleotide–peptide conjugates using hydrazone chemical ligation. Tetrahedron Lett. 2002, 43, 997–999. [Google Scholar] [CrossRef]
- Edupuganti, O.P.; Renaudet, O.; Defrancq, E.; Dumy, P. The oxime bond formation as an efficient chemical tool for the preparation of 3′,5′-bifunctionalised oligodeoxyribonucleotides. Bioorg. Med. Chem. Lett. 2004, 14, 2839–2842. [Google Scholar] [CrossRef]
- Edupuganti, O.P.; Singh, Y.; Defrancq, E.; Dumy, P. New Strategy for the Synthesis of 3′,5′-Bifunctionalized Oligonucleotide Conjugates through Sequential Formation of Chemoselective Oxime Bonds. Chem. Eur. J. 2004, 10, 5988–5995. [Google Scholar] [CrossRef]
- Pujari, S.S.; Zhang, Y.; Ji, S.; Distefano, M.D.; Tretyakova, N.Y. Site-specific cross-linking of proteins to DNA via a new bioorthogonal approach employing oxime ligation. Chem. Commun 2018, 54, 6296–6299. [Google Scholar] [CrossRef] [PubMed]
- Crisalli, P.; Hernandez, A.R.; Kool, E.T. Fluorescence quenchers for hydrazone and oxime orthogonal bioconjugation. Bioconjugate Chem. 2012, 23, 1969–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stetsenko, D.A.; Gait, M.J. Chemical methods for peptide-oligonucleotide conjugate synthesis. Methods Mol. Biol. 2005, 288, 205–224. [Google Scholar] [CrossRef]
- Khomyakova, E.A.; Zubin, E.M.; Smirnov, I.P.; Pozmogova, G.E.; Stetsenko, D.A.; Oretskaya, T.S. DNA or RNA oligonucleotide 2′-hydrazides for chemoselective click-type ligation with carbonyl compounds. Nucleosides Nucleotides Nucleic Acids 2011, 30, 577–584. [Google Scholar] [CrossRef]
- Zatsepin, T.S.; Stetsenko, D.A.; Gait, M.J.; Oretskaya, T.S. 2′-Hydrazine oligonucleotides: Synthesis and efficient conjugation with aldehydes. Nucleic Acids Symp. Ser. 2005, 49, 133–134. [Google Scholar] [CrossRef]
- Zatsepin, T.S.; Stetsenko, D.A.; Arzumanov, A.A.; Romanova, E.A.; Gait, M.J.; Oretskaya, T.S. Synthesis of peptide-oligonucleotide conjugates with single and multiple peptides attached to 2′-aldehydes through thiazolidine, oxime, and hydrazine linkages. Bioconjugate Chem. 2002, 13, 822–830. [Google Scholar] [CrossRef]
- Aho, A.; Sulkanen, M.; Korhonen, H.; Virta, P. Conjugation of Oligonucleotides to Peptide Aldehydes via a pH-Responsive N-Methoxyoxazolidine Linker. Org. Lett. 2020, 22, 6714–6718. [Google Scholar] [CrossRef]
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B. Synthesis of proteins by native chemical ligation. Science 1994, 266, 776–779. [Google Scholar] [CrossRef]
- Stetsenko, D.A.; Gait, M.J. A new “native ligation” procedure for peptide-oligonucleotide conjugation. Nucleosides Nucleotides Nucleic Acids 2001, 20, 801–804. [Google Scholar] [CrossRef]
- Stetsenko, D.A.; Gait, M.J. Efficient conjugation of peptides to oligonucleotides by “native ligation”. J. Org. Chem. 2000, 65, 4900–4908. [Google Scholar] [CrossRef] [PubMed]
- Diezmann, F.; Eberhard, H.; Seitz, O. Native chemical ligation in the synthesis of internally modified oligonucleotide-peptide conjugates. Biopolymers 2010, 94, 397–404. [Google Scholar] [CrossRef]
- Jang, E.K.; Koike, Y.; Ide, Y.; Tajima, K.; Kanaori, K.; Pack, S.P. Nucleobase-involved native chemical ligation: A novel reaction between an oxanine nucleobase and N-terminal cysteine for oligonucleotide-peptide conjugation. Chem. Commun. 2020, 56, 5508–5511. [Google Scholar] [CrossRef]
- Kachalova, A.V.; Stetsenko, D.A.; Romanova, E.A.; Tashlitsky, V.N.; Gait, M.J.; Oretskaya, T.S. A New and Efficient Method for Synthesis of 5′-Conjugates of Oligonucleotides through Amide-Bond Formation on Solid Phase. Helv. Chim. Acta. 2002, 85, 2409–2416. [Google Scholar] [CrossRef]
- Kachalova, A.; Zubin, E.; Stetsenko, D.; Gait, M.; Oretskaya, T. Oligonucleotides with 2′-O-carboxymethyl group: Synthesis and 2′-conjugation via amide bond formation on solid phase. Org. Biomol. Chem. 2004, 2, 2793–2797. [Google Scholar] [CrossRef] [Green Version]
- Kachalova, A.V.; Stetsenko, D.A.; Gait, M.J.; Oretskaya, T.S. Synthesis of oligonucleotide 2′-conjugates via amide bond formation in solution. Bioorganic Med. Chem. Lett. 2004, 14, 801–804. [Google Scholar] [CrossRef]
- Viladkar, S. Guanine rich oligonucleotide-amino acid/peptide conjugates: Preparation and characterization. Tetrahedron 2002, 58, 495–502. [Google Scholar] [CrossRef]
- Abes, S.; Moulton, H.M.; Clair, P.; Prevot, P.; Youngblood, D.S.; Wu, R.P.; Iversen, P.L.; Lebleu, B. Vectorization of morpholino oligomers by the (R-Ahx-R)4 peptide allows efficient splicing correction in the absence of endosomolytic agents. J. Control. Release 2006, 116, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Tilley, L.D.; Hine, O.S.; Kellogg, J.A.; Hassinger, J.N.; Weller, D.D.; Iversen, P.L.; Geller, B.L. Gene-specific effects of antisense phosphorodiamidate morpholino oligomer-peptide conjugates on Escherichia coli and Salmonella enterica serovar typhimurium in pure culture and in tissue culture. Antimicrob. Agents Chemother. 2006, 50, 2789–2796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michael, A. Ueber die Einwirkung von Diazobenzolimid auf Acetylendicarbonsäuremethylester. J. Prakt. Chem. 1893, 48, 94–95. [Google Scholar] [CrossRef] [Green Version]
- Huisgen, R. 1,3-Dipolar Cycloadditions. Past and Future. Angew. Chem. Int. Ed. Engl. 1963, 2, 565–598. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Tornoe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1–3]-triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Gogoi, K.; Mane, M.V.; Kunte, S.S.; Kumar, V.A. A versatile method for the preparation of conjugates of peptides with DNA/PNA/analog by employing chemo-selective click reaction in water. Nucleic Acids Res. 2007, 35, e139. [Google Scholar] [CrossRef] [Green Version]
- Shabanpoor, F.; McClorey, G.; Saleh, A.F.; Jarver, P.; Wood, M.J.; Gait, M.J. Bi-specific splice-switching PMO oligonucleotides conjugated via a single peptide active in a mouse model of Duchenne muscular dystrophy. Nucleic Acids Res. 2015, 43, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Astakhova, I.K.; Hansen, L.H.; Vester, B.; Wengel, J. Peptide-LNA oligonucleotide conjugates. Org. Biomol. Chem. 2013, 11, 4240–4249. [Google Scholar] [CrossRef] [Green Version]
- Honcharenko, M.; Honcharenko, D.; Stromberg, R. Efficient Conjugation to Phosphorothioate Oligonucleotides by Cu-Catalyzed Huisgen 1,3-Dipolar Cycloaddition. Bioconjugate Chem. 2019, 30, 1622–1628. [Google Scholar] [CrossRef]
- Kye, M.; Lim, Y.B. Synthesis and purification of self-assembling peptide-oligonucleotide conjugates by solid-phase peptide fragment condensation. J. Pept. Sci. 2018, 24, e3092. [Google Scholar] [CrossRef] [PubMed]
- Marchan, V.; Ortega, S.; Pulido, D.; Pedroso, E.; Grandas, A. Diels-Alder cycloadditions in water for the straightforward preparation of peptide-oligonucleotide conjugates. Nucleic Acids Res. 2006, 34, e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paris, C.; Brun, O.; Pedroso, E.; Grandas, A. Exploiting protected maleimides to modify oligonucleotides, peptides and peptide nucleic acids. Molecules 2015, 20, 6389–6408. [Google Scholar] [CrossRef] [Green Version]
- Agramunt, J.; Ginesi, R.; Pedroso, E.; Grandas, A. Inverse Electron-Demand Diels-Alder Bioconjugation Reactions Using 7-Oxanorbornenes as Dienophiles. J. Org. Chem. 2020, 85, 6593–6604. [Google Scholar] [CrossRef] [PubMed]
- Anno, Y.; Kubo, T.; Ueki, R.; Yano, M.; Sasaki, K.; Ohba, H.; Fujii, M. Synthesis of DNA conjugates by solid phase fragment condensation. Nucleosides Nucleotides Nucleic Acids 2003, 22, 1451–1453. [Google Scholar] [CrossRef] [PubMed]
- Grandas, A.; Robles, J.; Pedroso, E. Phosphitylation of Primary Carboxamides. Synthesis of Peptide-Oligonucleotide Conjugates with Acylphosphoramidate Linkages. Nucleosides Nucleotides 1995, 14, 825–828. [Google Scholar] [CrossRef]
- Kubo, T.; Zhelev, Z.; Rumiana, B.; Ohba, H.; Doi, K.; Fujii, M. Controlled intracellular localization and enhanced antisense effect of oligonucleotides by chemical conjugation. Org. Biomol. Chem. 2005, 3, 3257–3259. [Google Scholar] [CrossRef]



















| Stepwise Solid-Phase Synthesis | ||
|---|---|---|
| Conjugation via | Advantages | Disadvantages/Limitations |
| Bifunctional or trifunctional linker |
|
|
| Post-Synthetic Conjugation | ||
| Conjugation via | Advantages | Disadvantages/Limitations |
| Thioether or disulfide bond |
|
|
| Native ligation | ||
| Oxime, thiazolidine, or hydrazone linkage | ||
| Amide bond formation | ||
| Click chemistry | ||
| Diels-Alder reaction | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klabenkova, K.; Fokina, A.; Stetsenko, D. Chemistry of Peptide-Oligonucleotide Conjugates: A Review. Molecules 2021, 26, 5420. https://doi.org/10.3390/molecules26175420
Klabenkova K, Fokina A, Stetsenko D. Chemistry of Peptide-Oligonucleotide Conjugates: A Review. Molecules. 2021; 26(17):5420. https://doi.org/10.3390/molecules26175420
Chicago/Turabian StyleKlabenkova, Kristina, Alesya Fokina, and Dmitry Stetsenko. 2021. "Chemistry of Peptide-Oligonucleotide Conjugates: A Review" Molecules 26, no. 17: 5420. https://doi.org/10.3390/molecules26175420