
Single Isomer N-Heterocyclic Cyclodextrin Derivatives as Chiral Selectors in Capillary Electrophoresis

 , ,
, , 
Abstract
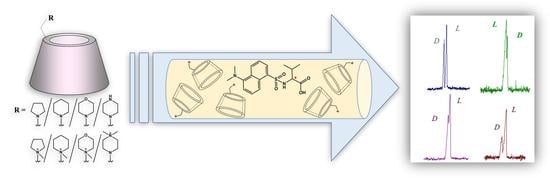
:
1. Introduction
2. Results and Discussion
2.1. Synthetic Procedures
2.2. Determination of pKa of the pH-Adjustable CD-Derivatives by 1H NMR-Titration
2.3. CE Experiments
2.3.1. Enantioseparation of the Mono-6-N-heterocycle-β-CDs (Selectors 2, 3, 4, 5)
Enantioseparation at pH 6.0
Enantioseparation at pH 4.75
Enantioseparation at pH 10.0
2.3.2. Enantioseparation of the N-Methylated Mono-6-N-heterocycle-β-CDs (Selectors 6, 7, 8, 9)
Enantioseparation at pH 6.0
Enantioseparation at pH 10.0
2.3.3. Comparison of the Enantioseparation Performance of the Native and the Acyclic Derivatives
2.3.4. Enantiomer Migration Order (EMO)
2.4. NMR Experiments
3. Materials and Methods
3.1. Materials
3.2. Synthetic Procedures
3.2.1. Synthesis of Mono-(6-N-heterocycle-6-deoxy)-β-CDs (Compounds 2, 3, 4, 5)
3.2.2. Synthesis of Mono-(6-N-(N-methylheterocycle)-6-deoxy)-β-CDs (Compounds 6, 7, 8, 9, 10)
3.3. NMR Experiments
3.4. CE Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Fanali, S.; Chankvetadze, B. Some thoughts about enantioseparations in capillary electrophoresis. Electrophoresis 2019, 40, 2420–2437. [Google Scholar] [CrossRef]
- Hancu, G.; Orlandini, S.; Papp, L.A.; Modroiu, A.; Gotti, R.; Furlanetto, S. Application of Experimental Design Methodologies in the Enantioseparation of Pharmaceuticals by Capillary Electrophoresis: A Review. Molecules 2021, 26, 4681. [Google Scholar] [CrossRef]
- Cucinotta, V.; Contino, A.; Giuffrida, A.; Maccarrone, G.; Messina, M. Application of charged single isomer derivatives of cyclodextrins in capillary electrophoresis for chiral analysis. J. Chromatogr. A 2010, 1217, 953–967. [Google Scholar] [CrossRef]
- Fejős, I.; Kalydi, E.; Malanga, M.; Benkovics, G.; Béni, S. Single isomer cyclodextrins as chiral selectors in capillary electrophoresis. J. Chromatogr. A 2020, 1627, 461375. [Google Scholar] [CrossRef]
- Chankvetadze, B. Separation of enantiomers with charged chiral selectors in CE. Electrophoresis 2009, 30, 211–221. [Google Scholar] [CrossRef]
- Nardi, A.; Eliseev, A.; Boček, P.; Fanali, S. Use of charged and neutral cyclodextrins in capillary zone electrophoresis: Enantiomeric resolution of some 2-hydroxy acids. J. Chromatogr. A 1993, 638, 247–253. [Google Scholar] [CrossRef]
- Zhou, J.; Tang, J.; Tang, W. Recent development of cationic cyclodextrins for chiral separation. TrAC-Trends Anal. Chem. 2015, 65, 22–29. [Google Scholar] [CrossRef]
- Williams, B.A.; Vigh, G. Dry look at the CHARM (charged resolving agent migration) model of enantiomer separations by capillary electrophoresis. J. Chromatogr. A 1997, 777, 295–309. [Google Scholar] [CrossRef]
- Kanagaraj, K.; Liang, W.; Rao, M.; Yao, J.; Wu, W.; Cheng, G.; Ji, J.; Wei, X.; Peng, C.; Yang, C. pH-Controlled Chirality Inversion in Enantiodifferentiating Photocyclodimerization of 2-Antharacenecarboxylic Acid Mediated by γ-Cyclodextrin Derivatives. Org. Lett. 2020, 22, 5273–5278. [Google Scholar] [CrossRef]
- Kanagaraj, K.; Xiao, C.; Rao, M.; Fan, C.; Borovkov, V.; Cheng, G.; Zhou, D.; Zhong, Z.; Su, D.; Yu, X.; et al. A Quinoline-Appended Cyclodextrin Derivative as a Highly Selective Receptor and Colorimetric Probe for Nucleotides. iScience 2020, 23, 100927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wren, S.A.C.; Rowe, R.C. Theoretical aspects of chiral separation in capillary electrophoresis. I. Initial evaluation of a model. J. Chromatogr. A 1992, 603, 235–241. [Google Scholar] [CrossRef]
- Zhao, B.; Li, L.; Wang, Y.; Zhou, Z. Preparation of polar group derivative β-cyclodextrin bonded hydride silica chiral stationary phases and their chromatography separation performances. Chin. Chem. Lett. 2019, 30, 643–649. [Google Scholar] [CrossRef]
- Zhu, B.; Xu, S.; Guo, X.; Wei, L.; Yu, J.; Wang, T. Use of various β-cyclodextrin derivatives as chiral selectors for the enantiomeric separation of ofloxacin and its five related substances by capillary electrophoresis. J. Sep. Sci. 2017, 40, 1784–1795. [Google Scholar] [CrossRef]
- Yu, J.; Zhao, Y.; Song, J.; Guo, X. Enantioseparation of meptazinol and its three intermediate enantiomers by capillary electrophoresis using a new cationic β-cyclodextrin derivative in single and dual cyclodextrin systems. Biomed. Chromatogr. 2014, 28, 868–874. [Google Scholar] [CrossRef]
- Puglisi, A.; Spencer, J.; Clarke, J.; Milton, J. Microwave-assisted synthesis of 6-amino-β-cyclodextrins. J. Incl. Phenom. Macrocycl. Chem. 2012, 73, 475–478. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, Y.; Ong, T.T.; Ge, L.; Tan, S.N.; Young, D.J.; Tan, T.T.Y.; Ng, S.C. Chiral capillary electrophoresis with cationic pyrrolidinium-β- cyclodextrin derivatives as chiral selectors. J. Sep. Sci. 2010, 33, 1797–1805. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.M.; Yang, Z.X.; Chen, Y.; Ding, F.; Liu, Y. Molecular binding and assembly behavior of β-cyclodextrin with piperazine and 1,4-dioxane in aqueous solution and solid state. Cryst. Growth Des. 2012, 12, 1370–1377. [Google Scholar] [CrossRef]
- Zhang, M.; Xiong, Q.; Shen, W.; Zhang, Q. Facile synthesis of well-defined cyclodextrin-pendant polymer via ATRP for nanostructure fabrication. RSC Adv. 2014, 4, 30566–30572. [Google Scholar] [CrossRef]
- Budanova, N.; Shapovalova, E.; Lopatin, S.; Varlamov, V.; Shpigun, O. Heptakis(6-amino-6-deoxy)-β-cyclodextrin as a chiral selector for the separation of anionic analyte enantiomers by capillary electrophoresis. Electrophoresis 2004, 25, 2795–2800. [Google Scholar] [CrossRef]
- Béni, S.; Sohajda, T.; Neumajer, G.; Iványi, R.; Szente, L.; Noszál, B. Separation and characterization of modified pregabalins in terms of cyclodextrin complexation, using capillary electrophoresis and nuclear magnetic resonance. J. Pharm. Biomed. Anal. 2010, 51, 842–852. [Google Scholar] [CrossRef]
- Chankvetadze, B. Enantiomer migration order in chiral capillary electrophoresis. Electrophoresis 2002, 23, 4022–4035. [Google Scholar] [CrossRef] [PubMed]
- Salgado, A.; Chankvetadze, B. Applications of nuclear magnetic resonance spectroscopy for the understanding of enantiomer separation mechanisms in capillary electrophoresis. J. Chromatogr. A 2016, 1467, 95–144. [Google Scholar] [CrossRef] [PubMed]
- Lomsadze, K.; Martínez-Girón, A.B.; Castro-Puyana, M.; Chankvetadze, L.; Crego, A.L.; Salgado, A.; Marina, M.L.; Chankvetadze, B. About the role of enantioselective selector-selectand interactions and the mobilities of diastereomeric associates in enantiomer separations using CE. Electrophoresis 2009, 30, 2803–2811. [Google Scholar] [CrossRef] [PubMed]
- Gogolashvili, A.; Tatunashvili, E.; Chankvetadze, L.; Sohajda, T.; Szeman, J.; Salgado, A.; Chankvetadze, B. Separation of enilconazole enantiomers in capillary electrophoresis with cyclodextrin-type chiral selectors and investigation of structure of selector-selectand complexes by using nuclear magnetic resonance spectroscopy. Electrophoresis 2017, 38, 1851–1859. [Google Scholar] [CrossRef]
- Hwang, T.L.; Shaka, A.J. Water suppression that works. J. Magn. Reson. Ser. A 1995, 112, 275–279. [Google Scholar] [CrossRef]
- Szakács, Z.; Kraszni, M.; Noszál, B. Determination of microscopic acid-base parameters from NMR-pH titrations. Anal. Bioanal. Chem. 2004, 378, 1428–1448. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Predicted pKa | Measured pKa |
|---|---|---|
| PYR-β-CD | 9.50 | 8.78 ± 0.01 |
| PIP-β-CD | 9.37 | 8.43 ± 0.01 |
| MO-β-CD | 7.15 | 5.86 ± 0.01 |
| PIPA-β-CD | 4.50 9.26 | 3.05 ± 0.02 9.08 ± 0.01 |
| PYR-β-CD | PIP-β-CD | MO-β-CD | PIPA-β-CD | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.5 mM | 1 mM | 2.5 mM | 5 mM | 0.5 mM | 1 mM | 2.5 mM | 0.5 mM | 1 mM | 2.5 mM | 0.5 mM | 1 mM | 2.5 mM | 5 mM | |
| MA | - | 0.41 | 1.00 | 1.93 | - | - | 0.54 | - | - | - | - | - | - | 0.36 |
| 2-ClMA | - | 0.55 | 1.22 | 2.36 | - | - | 0.60 | - | - | - | - | - | 0.36 | 0.50 |
| TA | - | - | 0.49 | 0.85 | - | 0.25 | 0.64 | - | - | - | - | - | - | - |
| cCA | 1.80 | 8.57 | 6.26 | 5.37 | 0.91 | 3.50 | 4.77 | 1.40 | 2.56 | 3.14 | 0.42 | 1.46 | 1.51 | 0.98 |
| tPA | 1.09 | 4.47 | 2.63 | 2.45 | 0.54 | 1.50 | 1.65 | 0.64 | 1.50 | 1.69 | 0.53 | 0.53 | 0.67 | 0.45 |
| 2-PPA | 0.78 | 1.96 | 1.89 | - | 0.51 | 0.99 | - | 0.97 | 1.28 | - | 0.43 | 0.82 | plateau | |
| Chlorprop | 0.49 | 1.66 | 3.31 | 0.52 | 0.35 | 1.33 | 2.67 | - | 1.06 | 1.70 | 0.49 | 0.70 | 1.50 | plateau |
| Mecoprop | 0.35 | 0.21 | 1.34 | 0.67 | 0.17 | 0.62 | 0.90 | 1.15 | 1.53 | - | 0.28 | 0.47 | plateau | |
| Dichlorprop | - | 0.41 | 0.67 | 0.94 | - | - | - | - | - | 1.01 | - | - | - | plateau |
| Trichlorprop | 0.63 | n.a. | 1.05 | 0.53 | - | 1.52 | 1.65 | - | 1.09 | 0.94 | 1.04 | 0.64 | 1.42 | plateau |
| Dns-Ser | 0.72 | 1.68 | 2.57 | 3.14 | 0.81 | 1.77 | 2.49 | 0.87 | 1.08 | 2.30 | 0.56 | 0.82 | 0.96 | 1.11 |
| Dns-Thr | 0.37 | 1.21 | 1.87 | 1.16 | - | 1.12 | 1.51 | - | 1.19 | 0.56 | 0.28 | 1.05 | 1.56 | 1.01 |
| Dns-Trp | - | - | 1.09 | 1.13 | - | - | - | - | - | 0.99 | - | - | - | - |
| Dns-Val | - | 0.67 | 2.07 | 2.08 | - | - | 1.15 | - | 0.15 | 0.65 | 0.08 | 0.63 | 0.79 | 1.08 |
| Ibuprofen | - | - | - | - | 0.17 | 0.20 | 0.28 | - | - | 1.02 | - | - | - | - |
| Fexofenadine | - | - | - | - | - | - | - | - | - | - | 0.77 | n.a. | - | - |
| Gatifloxacin | - | - | - | - | 0.39 | - | - | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | - |
| MePYR-β-CD | MePIP-β-CD | MeMO-β-CD | diMePIPA-β-CD | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.5 mM | 1 mM | 2.5 mM | 5 mM | 0.5 mM | 1 mM | 2.5 mM | 5 mM | 0.5 mM | 1 mM | 2.5 mM | 5 mM | 0.5 mM | 1 mM | 2.5 mM | 5 mM | |
| MA | - | - | - | - | - | - | 0.35 | 0.57 | - | 0.16 | 0.17 | n.a. | - | - | - | - |
| TA | - | - | - | - | - | - | - | 0.62 | - | - | - | n.a. | - | - | - | - |
| cCA | - | 0.42 | 0.95 | 1.32 | - | 0.56 | 0.63 | 0.70 | 0.32 | 0.42 | n.a. | n.a. | - | n.a. | n.a. | n.a. |
| tPA | 0.41 | 0.50 | 0.44 | 0.51 | 0.32 | 0.46 | 0.51 | 0.48 | 0.62 | n.a. | n.a. | n.a. | 0.29 | n.a. | - | - |
| 2-PPA | - | - | 0.49 | 0.67 | - | - | 0.41 | 0.60 | - | - | 0.34 | 0.35 | - | n.a. | n.a. | n.a. |
| Chlorprop | - | - | 0.67 | 0.54 | - | - | - | 0.37 | - | 0.15 | 0.42 | 0.56 | 0.19 | n.a. | 0.46 | - |
| Mecoprop | - | 0.38 | 0.82 | 1.17 | 0.36 | 0.52 | 0.81 | 1.10 | 0.31 | 0.46 | 0.62 | 0.97 | 0.09 | n.a. | 0.47 | 1.57 |
| Dichlorprop | - | - | - | - | - | - | 0.24 | 0.14. | - | - | - | - | - | - | - | - |
| Trichlorprop | - | - | 0.82 | 1.11 | 0.42 | 0.58 | 0.78 | 0.89 | 0.56 | 0.68 | 1.07 | 0.84 | 0.37 | - | - | - |
| Dns-Ser | 0.67 | 1.22 | 1.56 | 1.66 | 0.74 | 0.94 | 1.65 | 1.77 | 0.73 | 0.86 | 1.13 | n.a. | 0.41 | n.a. | n.a. | n.a. |
| Dns-Thr | - | 0.45 | 0.93 | 1.27 | - | 0.46 | 0.97 | 1.36 | 0.22 | 0.36 | 0.60 | 0.83 | 0.21 | n.a. | 0.52 | 1.75 |
| Dns-Trp | - | - | - | 0.51 | - | - | - | - | - | - | - | - | - | - | - | - |
| Dns-Val | 0.39 | 0.55 | 0.62 | 0.93 | 0.32 | 0.81 | 0.77 | 0.92 | 0.22 | 0.25 | 0.56 | 0.57 | 0.25 | 0.41 | 0.50 | 1.27 |
| Flurbiprofen | - | - | - | - | - | - | - | - | 0.29 | 0.82 | n.a. | n.a. | - | - | - | - |
| Ibuprofen | - | - | - | - | 0.64 | 0.64 | n.a. | n.a. | - | - | 0.85 | n.a. | - | - | - | - |
| Ketoprofen | - | - | - | 0.24 | 0.09 | 0.23 | 0.30 | 0.36 | 0.18 | 0.22 | 0.27 | n.a. | - | - | - | - |
| MePYR-β-CD | MePIP-β-CD | MeMO-β-CD | diMePIPA-β-CD | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.5 mM | 1 mM | 2.5 mM | 5 mM | 0.5 mM | 1 mM | 2.5 mM | 5 mM | 0.5 mM | 1 mM | 2.5 mM | 5 mM | 0.5 mM | 1 mM | 2.5 mM | 5 mM | |
| MA | - | - | - | - | - | - | - | - | - | - | - | 0.66 | - | - | - | n.a. |
| 2-ClMA | - | - | - | - | - | - | - | - | - | - | - | - | 0.25 | - | - | - |
| TA | - | - | - | - | - | - | - | - | - | - | - | - | - | 0.56 | n.a. | 0.76 |
| cCA | - | 0.31 | 0.32 | 1.41 | - | - | 0.26 | 0.30 | 0.10 | 0.35 | 0.50 | 0.85 | - | - | - | n.a. |
| tPA | 0.56 | 0.47 | 0.52 | 0.79 | n.a. | n.a. | n.a. | 0.72 | 0.43 | 0.51 | 0.76 | 1.34 | - | 0.44 | 0.53 | 0.50 |
| 2-PPA | - | - | 0.58 | 1.80 | - | - | - | - | - | - | 0.13 | 0.54 | - | 0.45 | n.a. | n.a. |
| Chlorprop | - | - | 0.07 | n.a. | - | - | - | - | - | n.a. | 0.47 | 0.63 | - | 0.31 | 0.76 | n.a. |
| Mecoprop | - | 0.32 | 0.59 | 1.11 | - | - | 0.35 | 0.44 | 0.10 | 0.41 | 0.54 | 0.84 | - | 0.41 | 0.42 | n.a. |
| Trichlorprop | 0.31 | 0.53 | 0.72 | 1.06 | - | 0.43 | 0.50 | 0.55 | 0.26 | 0.43 | 0.66 | 0.70 | - | 0.21 | 0.57 | n.a. |
| Dns-Ser | 0.33 | 0.60 | 0.57 | n.a. | - | - | 0.85 | 1.50 | 0.39 | 0.56 | 1.16 | 2.64 | - | 0.55 | n.a. | n.a. |
| Dns-Thr | - | 0.36 | 0.57 | 1.51 | - | - | 0.45 | 0.54 | - | 0.17 | 0.34 | 1.75 | - | 0.44 | 2.07 | 1.69 |
| Dns-Trp | - | - | 0.34 | 0.59 | - | - | - | - | - | - | - | 0.24 | - | - | - | n.a. |
| Dns-Val | - | 0.28 | 0.57 | 1.63 | - | - | 0.19 | 0.32 | - | - | 0.48 | 1.22 | - | 0.17 | 0.70 | n.a. |
| Flurbiprofen | 0.11 | 0.19 | 0.08 | 0.17 | n.a. | n.a. | n.a. | n.a. | 0.17 | 0.2 | 0.68 | n.a. | - | - | n.a. | n.a. |
| Ketoprofen | - | - | 0.28 | 0.30 | - | - | 0.17 | 0.20 | - | 0.15 | 0.13 | 0.35 | - | - | 0.19 | n.a. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fejős, I.; Kalydi, E.; Kukk, E.L.; Seggio, M.; Malanga, M.; Béni, S. Single Isomer N-Heterocyclic Cyclodextrin Derivatives as Chiral Selectors in Capillary Electrophoresis. Molecules 2021, 26, 5271. https://doi.org/10.3390/molecules26175271
Fejős I, Kalydi E, Kukk EL, Seggio M, Malanga M, Béni S. Single Isomer N-Heterocyclic Cyclodextrin Derivatives as Chiral Selectors in Capillary Electrophoresis. Molecules. 2021; 26(17):5271. https://doi.org/10.3390/molecules26175271
Chicago/Turabian StyleFejős, Ida, Eszter Kalydi, Edit Luca Kukk, Mimimorena Seggio, Milo Malanga, and Szabolcs Béni. 2021. "Single Isomer N-Heterocyclic Cyclodextrin Derivatives as Chiral Selectors in Capillary Electrophoresis" Molecules 26, no. 17: 5271. https://doi.org/10.3390/molecules26175271