
Seven Conformations of the Macrocycle Cyclododecanone Unveiled by Microwave Spectroscopy

Abstract
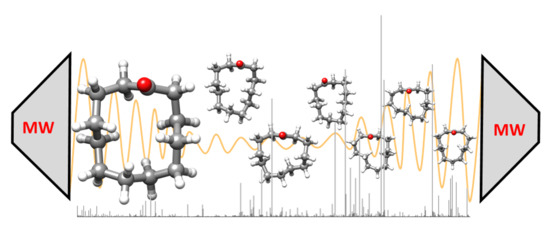
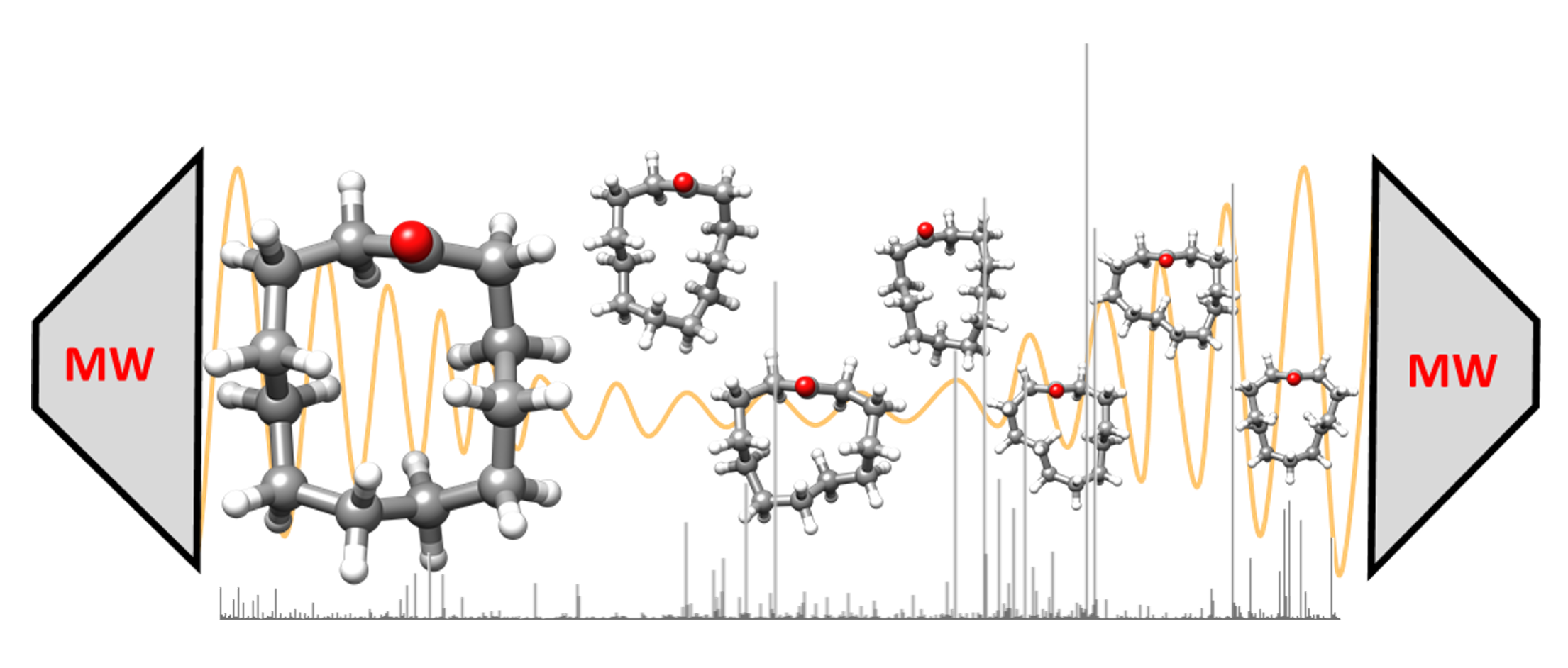
:
1. Introduction
2. Results and Discussion
2.1. Rotational Spectrum
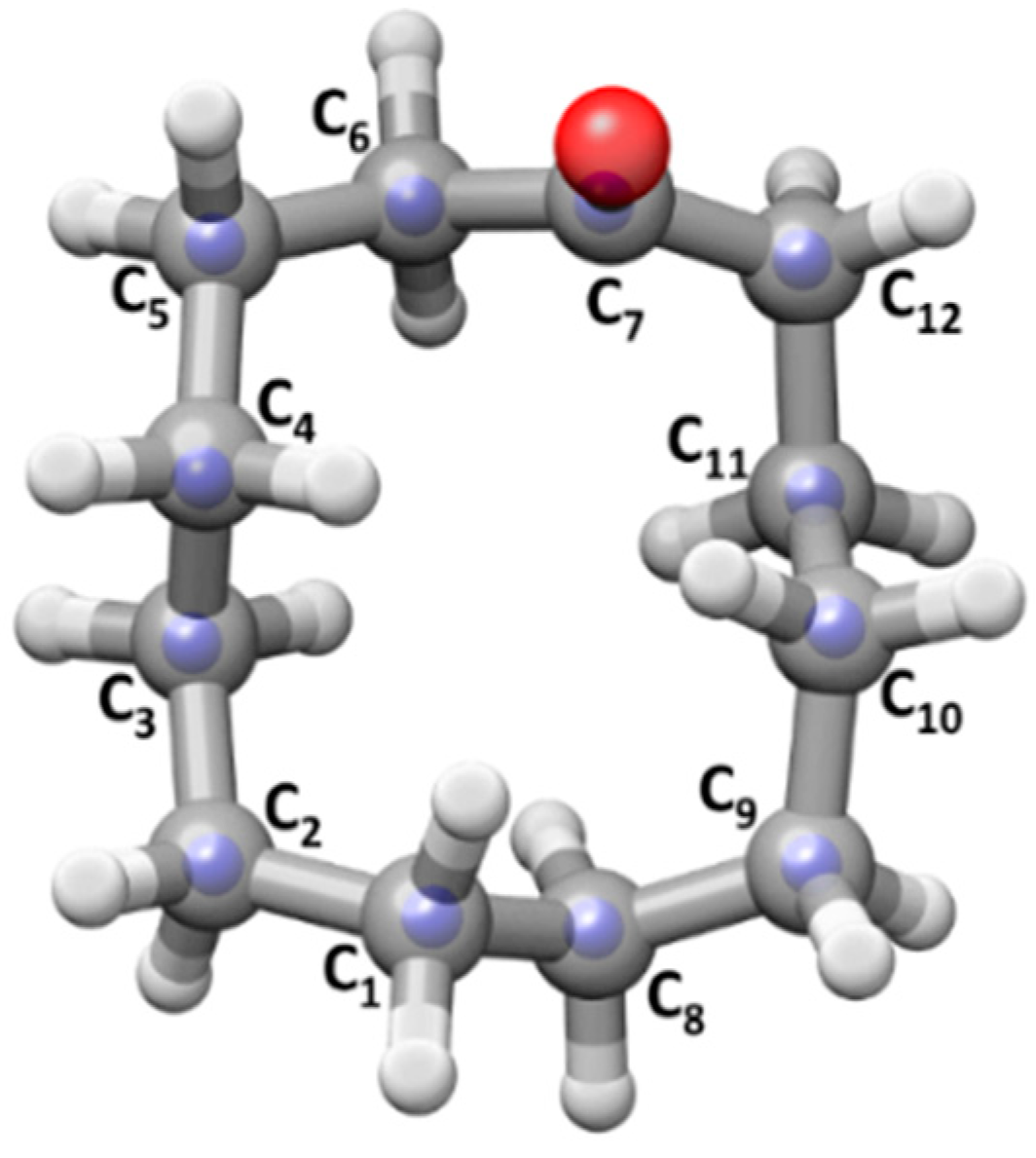
2.2. Experimental Structure of Conformer I
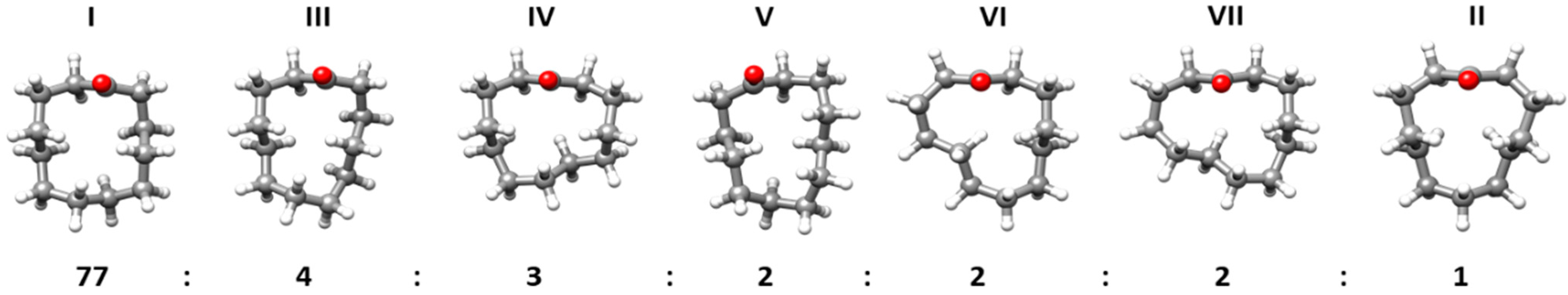
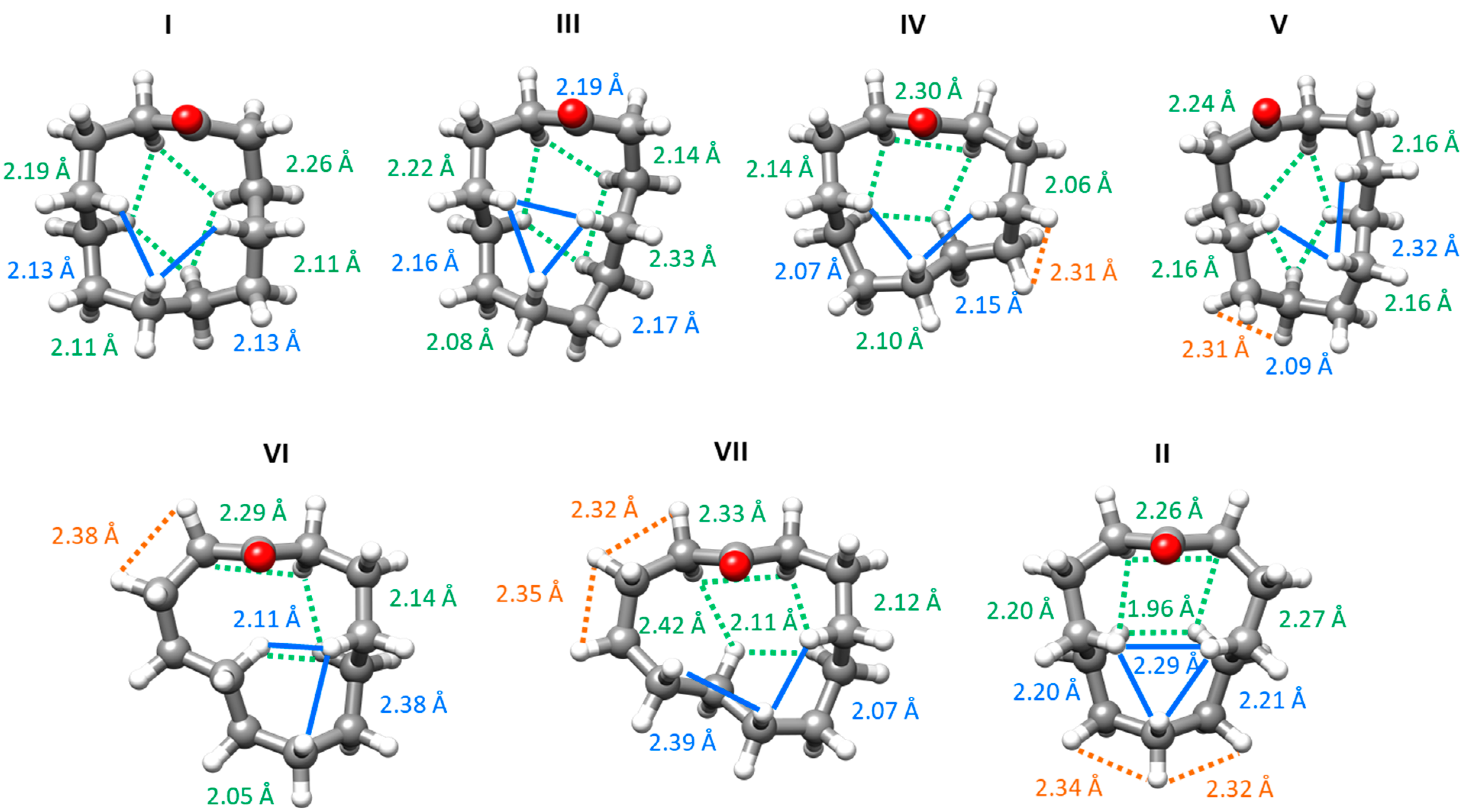
2.3. Conformations and Intramolecular Interactions
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Marsault, E.; Peterson, M.L. Macrocycles Are Great Cycles: Applications, Opportunities, and Challenges of Synthetic Macrocycles in Drug Discovery. J. Med. Chem. 2011, 54, 1961–2004. [Google Scholar] [CrossRef]
- Davis, F.; Higson, S. Macrocycles: Construction, Chemistry and Nanotechnology Applications; John Wiley & Sons: Chichester, UK, 2011; ISBN 9780470714621. [Google Scholar]
- Skinner, H.A. Structure of Cyclopropane. Nature 1947, 160, 902. [Google Scholar] [CrossRef]
- Harris, D.O.; Harrington, H.W.; Luntz, A.C.; Gwinn, W.D. Microwave Spectrum, Vibration-Rotation Interaction, and Potential Function for the Ring-Puckering Vibration of Trimethylene Sulfide. J. Chem. Phys. 1966, 44, 3467–3480. [Google Scholar] [CrossRef]
- Cotton, F.A.; Frenz, B.A. Conformations of Cyclobutane. Tetrahedron 1974, 30, 1587–1594. [Google Scholar] [CrossRef]
- Engerholm, G.G.; Luntz, A.C.; Gwinn, W.D.; Harris, D.O. Ring Puckering in Five-Membered Rings. II. The Microwave Spectrum, Dipole Moment, and Barrier to Pseudorotation in Tetrahydrofuran. J. Chem. Phys. 1969, 50, 2446–2457. [Google Scholar] [CrossRef]
- Harris, D.O.; Engerholm, G.G.; Tolman, C.A.; Luntz, A.C.; Keller, R.A.; Hyunyong, K.I.M.; Gwinn, W.D. Ring Puckering in Five-Membered Rings. I. General Theory. J. Chem. Phys. 1969, 50, 2438–2445. [Google Scholar] [CrossRef]
- Ogata, T.; Kozima, K. Microwave Spectrum of Cyclohexene. Bull. Chem. Soc. Jpn. 1969, 42, 1263–1265. [Google Scholar] [CrossRef] [Green Version]
- Meyers, R.A. (Ed.) Encyclopedia of Physical Science and Technology, 3rd ed.; Academic Press, Elsevier Science: Cambridge, MA, USA, 2001. [Google Scholar]
- Bocian, D.F.; Pickett, H.M.; Rounds, T.C.; Strauss, H.L. Conformations of Cycloheptane. J. Am. Chem. Soc. 1975, 97, 687–695. [Google Scholar] [CrossRef]
- Bocian, D.F.; Strauss, H.L. Conformational Structure and Energy of Cycloheptane and Some Related Oxepanes. J. Am. Chem. Soc. 1977, 99, 2876–2882. [Google Scholar] [CrossRef]
- Dragojlovic, V. Conformational Analysis of Cycloalkanes. ChemTexts 2015, 1, 14. [Google Scholar] [CrossRef]
- Dorofeeva, O.V.; Mastryukov, V.S.; Allinger, N.L.; Almenningen, A. The Molecular Structure and Conformation of Cyclooctane as Determined by Electron Diffraction and Molecular Mechanics Calculations. J. Phys. Chem. 1985, 89, 252–257. [Google Scholar] [CrossRef]
- Dorofeeva, O.V.; Mastryukov, V.S.; Allinger, N.L.; Almenningen, A. The Conformations of Cyclononane Dynamic Nuclear Magnetic Resonance and Force-field Calculations. Isr. J. Chem. 1990, 94, 8044–8048. [Google Scholar] [CrossRef]
- Pawar, D.M.; Smith, S.V.; Mark, H.L.; Odom, R.M.; Noe, E.A. Conformational Study of Cyclodecane and Substituted Cyclodecanes by Dynamic NMR Spectroscopy and Computational Methods. J. Am. Chem. Soc. 1998, 120, 10715–10720. [Google Scholar] [CrossRef]
- Pawar, D.M.; Brown, J.; Chen, K.H.; Allinger, N.L.; Noe, E.A. Conformations of Cycloundecane. J. Org. Chem. 2006, 71, 6512–6515. [Google Scholar] [CrossRef] [PubMed]
- Atavin, E.G.; Mastryukov, V.S.; Allinger, N.L.; Almenningen, A.; Seip, R. Molecular Structure of Cyclododecane, C12H24, as Determined by Electron Diffraction and Molecular Mechanics. J. Mol. Struct. 1989, 212, 87–95. [Google Scholar] [CrossRef]
- Dunitz, J.D.; Shearer, H.M.M. Die Strukturen Der Mittleren Ringverbindungen III. Die Struktur Des Cyclododecans. Helv. Chim. Acta 1960, 43, 18–35. [Google Scholar] [CrossRef]
- Anet, F.A.L.; Rawdah, T.N. Cyclododecane. Force-Field Calculations and NMR Spectra of Deuterated Isotopomers. J. Am. Chem. Soc. 1978, 100, 7166–7171. [Google Scholar] [CrossRef]
- Groth, P.; Nord, G.; Woldbye, F.; Watson, K.J.; Sandström, M. Crystal Structure of Cyclotetradecane at −157 Degrees C. Acta Chem. Scand. 1976, 30, 155–156. [Google Scholar] [CrossRef] [Green Version]
- Vajnštejn, B.K. Structure Analysis by Electron Diffraction. Il Nuovo Cim. 1956, 3, 773–797. [Google Scholar] [CrossRef]
- Thompson, A.L.; Watkin, D.J. X-Ray Crystallography and Chirality: Understanding the Limitations. Tetrahedron Asymmetry 2009, 20, 712–717. [Google Scholar] [CrossRef]
- Tormena, C.F. Conformational Analysis of Small Molecules: NMR and Quantum Mechanics Calculations. Prog. Nucl. Magn. Reson. Spectrosc. 2016, 96, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Skibinski, M.; Wang, Y.; Slawin, A.M.Z.; Lebl, T.; Kirsch, P.; O’Hagan, D. Alicyclic Ring Structure: Conformational Influence of the CF 2 Group in Cyclododecanes. Angew. Chem.-Int. Ed. 2011, 50, 10581–10584. [Google Scholar] [CrossRef] [PubMed]
- Groth, P. On the Crystal Conformations of Cyclic Oximes, (CH2)(n-1) CNOH with n = 10, …, 14. Acta Chem. Scand. 1979, 33, 503–513. [Google Scholar] [CrossRef] [Green Version]
- Dehli, J.; Groth, P.; Bruun, H.H.; Smidsrød, O.; Lindberg, A.A.; Jansen, G.; Lamm, B.; Samuelsson, B. Crystal Structure of 2,12-Dibromo-Cyclododecanone. Acta Chem. Scand. 1969, 23, 587–596. [Google Scholar] [CrossRef] [Green Version]
- Anet, F.A.L.; Cheng, A.K.; Wagner, J.J. Determination of Conformational Energy Barriers in Medium- and Large-Ring Cycloalkanes by 1H and 13C Nuclear Magnetic Resonance. J. Am. Chem. Soc. 1972, 94, 9250–9252. [Google Scholar] [CrossRef]
- Rubin, B.H.; Williamson, M.; Takeshita, M.; Menger, F.M.; Anet, F.A.L.; Bacon, B.; Allinger, N.L. Conformation of a Saturated 13-Membered Ring. J. Am. Chem. Soc. 1984, 106, 2088–2092. [Google Scholar] [CrossRef]
- Wang, M.A.; Zhang, N.; Lu, H.Z.; Wang, D.Q. Conformation of α,α,A′-Trisubstituted Cyclododecanone. Chin. J. Chem. 2007, 25, 1196–1201. [Google Scholar] [CrossRef]
- Groth, P.; Haaland, A.; Novak, D.P.; Andresen, A.F.; Southern, J.T.; Edlund, K.; Eliasen, M.; Herskind, C.; Laursen, T.; Pedersen, P.M. On the Crystal Structure of Cyclotetradecanone at −157 Degrees C. Acta Chem. Scand. 1975, 29, 374–375. [Google Scholar] [CrossRef]
- Peña, I.; Sanz, M.E.; Alonso, E.R.; Alonso, J.L. The Multiple Hydrogen-Bonding Networks of Polyol Ribitol. Chem. Eur. J. 2018, 24, 13408–13412. [Google Scholar] [CrossRef] [Green Version]
- Loru, D.; Quesada-Moreno, M.M.; Avilés-Moreno, J.R.; Jarman, N.; Huet, T.R.; López-González, J.J.; Sanz, M.E. Conformational Flexibility of Limonene Oxide Studied by Microwave Spectroscopy. ChemPhysChem 2017, 18, 274–280. [Google Scholar] [CrossRef] [Green Version]
- Shipman, S.T.; Neill, J.L.; Suenram, R.D.; Muckle, M.T.; Pate, B.H. Structure Determination of Strawberry Aldehyde by Broadband Microwave Spectroscopy: Conformational Stabilization by Dispersive Interactions. J. Phys. Chem. Lett. 2011, 2, 443–448. [Google Scholar] [CrossRef]
- Domingos, S.R.; Pérez, C.; Medcraft, C.; Pinacho, P.; Schnell, M. Flexibility Unleashed in Acyclic Monoterpenes: Conformational Space of Citronellal Revealed by Broadband Rotational Spectroscopy. Phys. Chem. Chem. Phys. 2016, 18, 16682–16689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gámez, F.; Martínez-Haya, B.; Blanco, S.; López, J.C.; Alonso, J.L. Microwave Spectroscopy and Quantum Chemical Investigation of Nine Low Energy Conformers of the 15-Crown-5 Ether. Phys. Chem. Chem. Phys. 2012, 14, 12912–12918. [Google Scholar] [CrossRef] [PubMed]
- Burevschi, E.; Peña, I.; Sanz, M.E. Medium-Sized Rings: Conformational Preferences in Cyclooctanone Driven by Transannular Repulsive Interactions. Phys. Chem. Chem. Phys. 2019, 21, 4331–4338. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Kawakami, K.; Seto, H.; Furihata, K. Structure of a New Antibiotic, Roseophilin. Tetrahedron Lett. 1992, 33, 2701–2704. [Google Scholar] [CrossRef]
- Boger, D.L.; Hong, J. Asymmetric Total Synthesis of Ent-(−)-Roseophilin: Assignment of Absolute Configuration. J. Am. Chem. Soc. 2001, 123, 8515–8519. [Google Scholar] [CrossRef]
- Zoorob, H.H.; Elsherbini, M.S.; Hamama, W.S. Reactivity Features of Cyclododecanone. Arkivoc 2011, 2011, 429–495. [Google Scholar] [CrossRef]
- Bollbuck, B.; Kraft, P.; Tochtermann, W. Nature-like Odorants by Stereoselective Ring Enlargement of Cyclohexanone and Cyclododecanone. Tetrahedron 1996, 52, 4581–4592. [Google Scholar] [CrossRef]
- Scafato, P.; Larocca, A.; Rosini, C. An Alternative Stereoselective Synthesis of the Macrocyclic Fragrances (R)-12-Methyltridecanolide and (S)-Muscolide by Means of an Asymmetric Catalytic Conjugate Addition/Baeyer-Villiger Oxidation. Tetrahedron Asymmetry 2006, 17, 2511–2515. [Google Scholar] [CrossRef]
- Nowicki, J. Claisen, Cope and Related Rearrangements in the Synthesis of Flavour and Fragrance Compounds. Molecules 2000, 5, 1033–1050. [Google Scholar] [CrossRef] [Green Version]
- Dowd, P.; Choi, S.C. Homologation of Large Rings. Tetrahedron 1992, 48, 4773–4792. [Google Scholar] [CrossRef]
- Groth, P.; Larsen, E.; Bjerrum, J.; Wahlbeck, P.G.; Hoyer, E.; Spiridonov, V.P.; Strand, T.G. On the Crystal Structure of Cyclododecanone. Acta Chem. Scand. 1979, 33, 203–205. [Google Scholar] [CrossRef]
- Froimowitz, M. HyperChem(TM): A Software Package for Computational Chemistry and Molecular Modeling. BioTechniques 1993, 14, 1010–1013. [Google Scholar]
- Pracht, P.; Bohle, F.; Grimme, S. Automated Exploration of the Low-Energy Chemical Space with Fast Quantum Chemical Methods. Phys. Chem. Chem. Phys. 2020, 22, 7169–7192. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.K.G. Vibrational Spectra and Structure; Elsevier: Amsterdam, The Netherlands, 1977; Volume 6, pp. 1–89. [Google Scholar]
- Pickett, H.M. The Fitting and Prediction of Vibration-Rotation Spectra with Spin Interactions. J. Mol. Spectrosc. 1991, 148, 371–377. [Google Scholar] [CrossRef]
- Western, C.M. PGOPHER: A Program for Simulating Rotational, Vibrational and Electronic Spectra. J. Quant. Spectrosc. Radiat. Transf. 2017, 186, 221–242. [Google Scholar] [CrossRef] [Green Version]
- Western, C.M.; Billinghurst, B.E. Automatic and Semi-Automatic Assignment and Fitting of Spectra with PGOPHER. Phys. Chem. Chem. Phys. 2019, 21, 13986–13999. [Google Scholar] [CrossRef] [Green Version]
- Kraitchman, J. Determination of Molecular Structure from Microwave Spectroscopic Data. Am. J. Phys. 1953, 21, 17–24. [Google Scholar] [CrossRef]
- Pauling, L. The Nature of the Chemical Bond and the Structure of Molecules and Crystals: An Introduction to Modern Structural Chemistry, 3rd ed.; Cornell University Press: New York, NY, USA, 1960. [Google Scholar]
- Bondi, A. Van Der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Pérez, C.; López, J.C.; Blanco, S.; Schnell, M. Water-Induced Structural Changes in Crown Ethers from Broadband Rotational Spectroscopy. J. Phys. Chem. Lett. 2016, 7, 4053–4058. [Google Scholar] [CrossRef] [Green Version]
- Gámez, F.; Martínez-Haya, B.; Blanco, S.; López, J.C.; Alonso, J.L. High-Resolution Rotational Spectroscopy of a Cyclic Ether. J. Phys. Chem. Lett. 2012, 3, 482–485. [Google Scholar] [CrossRef] [PubMed]
- López, J.C.; Pérez, C.; Blanco, S.; Shubert, V.A.; Temelso, B.; Shields, G.C.; Schnell, M. Water Induces the Same Crown Shapes as Li+ or Na+ in 15-Crown-5 Ether: A Broadband Rotational Study. Phys. Chem. Chem. Phys. 2019, 21, 2875–2881. [Google Scholar] [CrossRef] [PubMed]
- Loru, D.; Bermúdez, M.A.; Sanz, M.E. Structure of Fenchone by Broadband Rotational Spectroscopy. J. Chem. Phys. 2016, 145, 074311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loru, D.; Peña, I.; Sanz, M.E. Ethanol Dimer: Observation of Three New Conformers by Broadband Rotational Spectroscopy. J. Mol. Spectrosc. 2017, 335, 93–101. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| I | II | III | IV | V | VI | VII | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| B3LYP | MP2 | B3LYP | MP2 | B3LYP | MP2 | B3LYP | MP2 | B3LYP | MP2 | B3LYP | MP2 | B3LYP | MP2 | |
| A[b] (MHz) | 872.3 | 888.5 | 896.1 | 918.6 | 967.3 | 979.6 | 858.0 | 880.7 | 988.5 | 1002.0 | 914.3 | 925.0 | 943.7 | 951.8 |
| B (MHz) | 691.1 | 693.2 | 693.2 | 697.0 | 632.2 | 641.2 | 740.9 | 741.3 | 630.1 | 635.2 | 673.7 | 680.3 | 682.2 | 687.6 |
| C (MHz) | 447.0 | 453.0 | 450.7 | 459.1 | 450.4 | 459.4 | 461.2 | 468.1 | 445.6 | 451.7 | 442.3 | 447.9 | 453.3 | 458.3 |
| μa (D) | 0.9 | 0.8 | 0.8 | 0.6 | 0.9 | 0.7 | −0.2 | −0.2 | 1.3 | 1.1 | 0.3 | 0.2 | −0.1 | −0.1 |
| μb (D) | 0.2 | 0.2 | 0.2 | 0.4 | −0.6 | −0.5 | 0.5 | 0.5 | 0.4 | 0.4 | −0.3 | −0.3 | −0.3 | −0.2 |
| μc (D) | −2.6 | −2.4 | −2.3 | −2.2 | 2.6 | 2.4 | 2.4 | 2.2 | −2.5 | −2.3 | −2.4 | −2.2 | 2.4 | 2.2 |
| ΔE [c] (cm−1) | 0.0 | 0.0 | 469.1 | 516.4 | 461.6 | 401.9 | 583.8 | 574.0 | 495.3 | 452.6 | 645.6 | 604.9 | 698.0 | 726.3 |
| ΔE0 [d] (cm−1) | 0.0 | 0.0 | 446.4 | 527.8 | 455.8 | 443.3 | 575.0 | 585.1 | 590.6 | 508.3 | 592.1 | 572.0 | 611.2 | 673.8 |
| ΔG [e] (cm−1) | 0.0 | 0.0 | 269.7 | 566.5 | 537.7 | 579.2 | 521.7 | 649.4 | 780.9 | 663.3 | 583.4 | 538.2 | 594.6 | 696.4 |
| Parameter | I | II | III | IV | V | VI | VII |
|---|---|---|---|---|---|---|---|
| A[a] (MHz) | 865.65313(23) [f] | 894.30343(70) | 966.10552(37) | 868.61009(42) | 987.56269(41) | 915.21738(48) | 942.67936(52) |
| B (MHz) | 700.47778(27) | 677.77615(98) | 638.54650(36) | 739.83405(53) | 634.23052(50) | 677.88681(41) | 684.66240(45) |
| C (MHz) | 449.17010(22) | 451.3106(34) | 453.92123(43) | 463.7153(46) | 447.67053(47) | 444.2827(29) | 454.18205(29) |
| ∆J (kHz) | 0.1378(71) | 0.080(17) | 0.0295(52) | 0.144(10) | 0.0305(58) | - | - |
| ∆JK (kHz) | −0.336(25) | - | - | −0.275(30) | - | - | - |
| ∆K (kHz) | 0.205(19) | - | - | - | - | - | - |
| δJ (kHz) | - | - | - | - | 0.0148(37) | - | - |
| δK (kHz) | 0.176(22) | - | - | - | - | - | - |
| κ [b] | 0.21 | 0.02 | −0.28 | 0.36 | −0.31 | −0.01 | −0.06 |
| a/b/c[c] (D) | y/y/y | n/n/y | y/y/y | n/n/y | y/n/y | n/n/y | n/n/y |
| σ[d] (kHz) | 4.4 | 5.4 | 6.7 | 3.5 | 6.2 | 4.9 | 5.4 |
| N [e] | 85 | 15 | 49 | 21 | 43 | 19 | 19 |
| Conformer | I | I | I | III | IV | V | VI | VII | II |
|---|---|---|---|---|---|---|---|---|---|
| Parameter | rs[a] | r0[b] | re | re | re | re | re | re | re |
| r(C2-C1) | 1.577(20) | 1.528(15) | 1.537 | 1.541 | 1.538 | 1.541 | 1.542 | 1.532 | 1.540 |
| r(C3-C2) | 1.486(14) | 1.524(9) | 1.537 | 1.540 | 1.538 | 1.542 | 1.530 | 1.532 | 1.537 |
| r(C4-C3) | 1.479(19) | 1.527(16) | 1.533 | 1.533 | 1.537 | 1.539 | 1.527 | 1.539 | 1.532 |
| r(C5-C4) | 1.579(28) | 1.541(14) | 1.535 | 1.536 | 1.536 | 1.532 | 1.527 | 1.539 | 1.536 |
| r(C6-C5) | 1.554(22) | 1.523(12) | 1.533 | 1.535 | 1.535 | 1.543 | 1.540 | 1.534 | 1.534 |
| r(C7-C6) | 1.437(24) | 1.526(10) [c] | 1.525 | 1.525 | 1.525 | 1.520 | 1.529 | 1.526 | 1.524 |
| r(C12-C7) | 1.468(15) | 1.526(10) | 1.519 | 1.522 | 1.523 | 1.524 | 1.524 | 1.524 | 1.524 |
| r(C12-C11) | 1.541(9) | 1.537(13) | 1.544 | 1.541 | 1.532 | 1.536 | 1.534 | 1.534 | 1.538 |
| r(C11-C10) | 1.526(11) | 1.533(14) | 1.533 | 1.532 | 1.539 | 1.536 | 1.538 | 1.536 | 1.536 |
| r(C10-C9) | 1.533(10) | 1.534(14) | 1.537 | 1.531 | 1.541 | 1.532 | 1.534 | 1.538 | 1.531 |
| r(C9-C8) | 1.537(10) | 1.527(10) | 1.537 | 1.536 | 1.538 | 1.533 | 1.537 | 1.539 | 1.538 |
| r(C8-C1) | 1.522(10) | 1.542(13) | 1.541 | 1.540 | 1.533 | 1.535 | 1.542 | 1.540 | 1.540 |
| ∠(C3-C2-C1) | 113.8(4) | 114.3(2) | 114.1 | 117.0 | 114.6 | 116.4 | 112.7 | 113.2 | 117.4 |
| ∠(C4-C3-C2) | 116.8(17) | 114.4(7) | 113.9 | 114.3 | 113.9 | 117.1 | 116.3 | 114.7 | 114.0 |
| ∠(C5-C4-C3) | 115.4(17) | 114.5(8) [c] | 114.5 | 114.6 | 114.8 | 114.1 | 112.7 | 116.5 | 114.6 |
| ∠(C6-C5-C4) | 113.0(4) | 113.5(2) | 113.9 | 114.6 | 113.5 | 113.4 | 114.0 | 116.4 | 113.6 |
| ∠(C7-C6-C5) | 116.7(20) | 114.2(8) [c] | 114.1 | 114.3 | 113.9 | 113.1 | 114.4 | 115.6 | 113.3 |
| ∠(C12-C7-C6) | 125.2(27) | 116.5(6) [c] | 116.8 | 117.6 | 117.2 | 117.2 | 117.0 | 116.1 | 118.2 |
| ∠(C11-C12-C7) | 111.1(2) | 111.5(2) | 111.6 | 112.7 | 114.3 | 114.5 | 115.4 | 115.4 | 112.7 |
| ∠(C12-C11-C10) | 112.7(5) | 113.2(7) | 113.5 | 113.0 | 116.2 | 114.1 | 114.7 | 114.2 | 114.1 |
| ∠(C11-C10-C9) | 114.5(11) | 113.8(10) | 113.7 | 113.4 | 116.2 | 114.7 | 114.3 | 115.0 | 114.6 |
| ∠(C10-C9-C8) | 113.8(3) | 114.1(2) | 114.1 | 114.0 | 115.9 | 113.1 | 115.1 | 114.1 | 114.2 |
| ∠(C9-C8-C1) | 113.2(9) | 114.1(8) | 114.1 | 116.7 | 114.1 | 114.3 | 117.4 | 114.4 | 117.5 |
| ∠(C8-C1-C2) | 111.2(14) | 114.8(7) | 113.9 | 116.5 | 114.2 | 116.6 | 116.1 | 114.6 | 116.7 |
| τ(C4-C3-C2-C1) | −63.0(21) | −68.1(7) [c] | −68.1 | −64.3 | −61.7 | 84.0 | 177.1 | 172.8 | −62.2 |
| τ(C5-C4-C3-C2) | 170.8(2) | 171.4(3) | 172.8 | 178.3 | 156.9 | 64.4 | −176.4 | −84.6 | 175.9 |
| τ(C6-C5-C4-C3) | −66.0(23) | −68.6(10) | −66.1 | −61.5 | −64.4 | −178.3 | 64.1 | 68.0 | −64.4 |
| τ(C7-C6-C5-C4) | −66.0(24) | −63.8(10) [c] | −65.1 | −63.5 | −59.3 | 60.4 | −75.2 | −87.2 | −62.7 |
| τ(C12-C7-C6-C5) | 152.0(9) | 153.8(3) [c] | 151.8 | 150.6 | 152.5 | 70.2 | 134.3 | 162.4 | 146.2 |
| τ(C11-C12-C7-C6) | −71.1(13) | −75.0(5) [c] | −75.2 | −62.7 | −159.8 | −149.5 | −159.6 | −164.9 | −138.1 |
| τ(C10-C11-C12-C7) | −70.6(16) | −65.1(6) [c] | −65.1 | −55.1 | 62.3 | 56.3 | 67.1 | 59.2 | 57.3 |
| τ(C12-C11-C10-C9) | 170.1(2) | 170.1(2) | 171.5 | 164.0 | 63.8 | 56.5 | 60.6 | 61.3 | 58.2 |
| τ(C11-C10-C9-C8) | −68.1(13) | −69.1(4) | −68.3 | −166.6 | −86.5 | −165.5 | −173.8 | −155.3 | 179.3 |
| τ(C10-C9-C8-C1) | −70.3(14) | −68.8(8) [c] | −68.8 | 56.2 | −68.9 | 164.6 | 62.3 | 55.1 | 62.9 |
| τ(C9-C8-C1-C2) | 150.2(4) | 149.5(4) | 148.0 | 59.5 | 173.2 | −56.7 | 54.4 | 51.5 | 78.4 |
| τ(C8-C1-C2-C3) | −74.0(21) | −68.2(3) | −69.0 | −83.6 | −59.6 | −58.3 | −85.9 | −168.8 | −72.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burevschi, E.; Sanz, M.E. Seven Conformations of the Macrocycle Cyclododecanone Unveiled by Microwave Spectroscopy. Molecules 2021, 26, 5162. https://doi.org/10.3390/molecules26175162
Burevschi E, Sanz ME. Seven Conformations of the Macrocycle Cyclododecanone Unveiled by Microwave Spectroscopy. Molecules. 2021; 26(17):5162. https://doi.org/10.3390/molecules26175162
Chicago/Turabian StyleBurevschi, Ecaterina, and M. Eugenia Sanz. 2021. "Seven Conformations of the Macrocycle Cyclododecanone Unveiled by Microwave Spectroscopy" Molecules 26, no. 17: 5162. https://doi.org/10.3390/molecules26175162