
Membrane Sensor Histidine Kinases: Insights from Structural, Ligand and Inhibitor Studies of Full-Length Proteins and Signalling Domains for Antibiotic Discovery


Abstract
:1. Introduction
1.1. General Scheme and Mechanism of Signal Transduction Amongst TCSs
1.2. Occurrence and Distribution of TCS Proteins amongst Prokaryotes Including Agents of Infection
2. Methodology for Producing Purified Active Forms of Full-Length Membrane SHKs
3. Identification and Characterisation of Signalling Ligands and Inhibitors Using Full-Length SHKs
3.1. Effect of Gelatinase Biosynthesis-Activating Pheromone (GBAP) on Autophosphorylation Activity of Intact FsrC
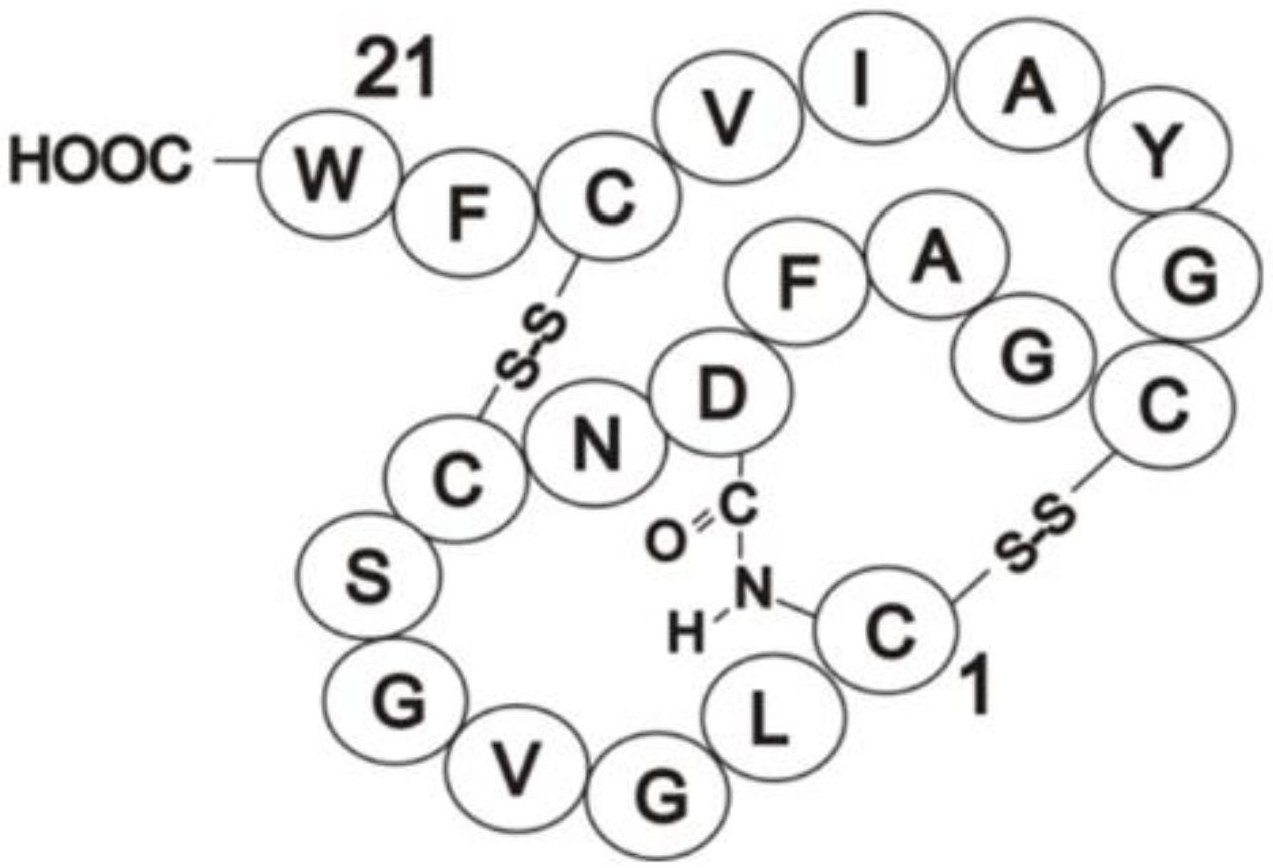
3.2. Identification of Direct Inhibitors of SHKs Using Full-Length Intact Proteins: Siamycin I Inhibition of FsrC
3.3. Binding of the GBAP Pheromone to Purified Intact FsrC
3.4. Identification of the Environmental Signalling Ligand That Activates VanS Involved in Regulating Resistance to the Antibiotic Vancomycin
3.5. Modulation of PrrB Histidine Kinase Activity by the Signalling Cbb3-Type Cytochrome C Oxidase Complex in Rhodobacter sphaeroides
3.6. Reconstitution of Full-Length SHKs into Phospholipid Liposomes to Study Ligand Interactions: Effects of YycH and YycI Signalling Membrane Proteins on WalK Activities
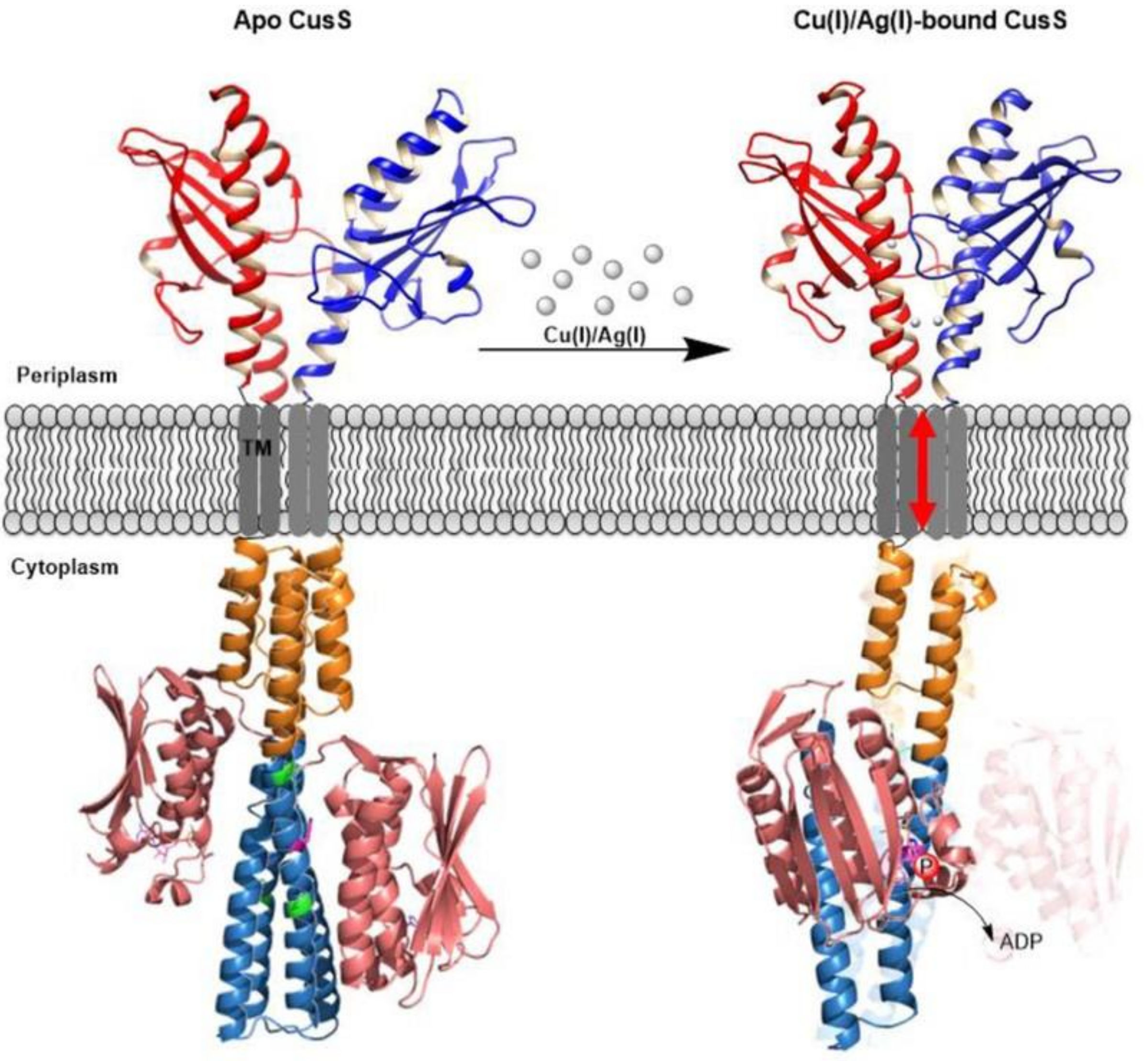
3.7. Reconstitution of Full-Length SHKs into Nanodiscs to Study Ligand Interactions and Signal Transduction: Effects of Metal Ion Ligands on CusS Kinase
4. Extracellular Sensing and Transmembrane Signalling Domains
5. Conclusions and Perspectives
- (i)
- They lend themselves particularly well to investigations aimed at identification of environmental signals together with characterisation of their downstream impacts on enzyme activities and structural conformations, especially across the membrane. In this review, we have described the advances in knowledge of ligand binding by CusS, FsrC, PrrB, VanSA, VanSSC, and WalK and their effects on activities and/or mechanisms of signal transmission. This should apply equally well to identification and characterisation of inhibitors; one study using intact FsrC successfully identified siamycin I as a direct inhibitor and therefore it should be possible to expand this approach. Quantitative data for ligand binding in the presence of the entire intact proteins is also achievable, though from the few studies to date it seems that ligand binding strengths are generally comparable to those obtained using sensing domains in isolation which are often more convenient to produce; and
- (ii)
- Intact membrane SHKs can be used to investigate ligand interactions that occur within the membrane itself. Indeed, the first description of the successful heterologous expression and purification of any intact membrane SHK came from studies of an intramembranous-sensing SHK—PrrB (RegB) from Rhodobacter sphaeroides. Intact protein studies established that the membrane-bound cbb3-type cytochrome c oxidase complex serves as the interacting regulator of PrrB kinase/phosphatase activities and that the PrrB transmembrane domain plays a crucial role in the interactions.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Nixon, B.C.; Ronson, C.W.; Ausubel, F.M. Two-component regulatory systems responsive to environmental stimuli share strongly conserved domains with the nitrogen assimilation regulatory genes ntrB and ntrC. Proc. Natl. Acad. Sci. USA 1986, 83, 7850–7854. [Google Scholar] [CrossRef] [Green Version]
- Magasanik, B. Historical Perspective. In Two-Component Signal Transduction; Hoch, J.A., Silhavy, T.J., Eds.; American Society for Microbiology: Washington, WA, USA, 1995; Chapter 1; pp. 1–5. [Google Scholar]
- Ninfa, E.G.; Atkinson, M.R.; Kamberov, E.S.; Ninfa, A.J. Mechanism of autophosphorylation of Escherichia coli nitrogen regulator II (NRII or NtrB): Transphosphorylation between subunits. J. Bacteriol. 1993, 175, 7024–7032. [Google Scholar] [CrossRef] [Green Version]
- Swanson, R.V.; Bourret, R.B.; Simon, M.I. Intermolecular complementation of the kinase activity of CheA. Mol. Microbiol. 1993, 8, 435–441. [Google Scholar] [CrossRef]
- Casino, P.; Rubio, V.; Marina, A. Structural insight into partner specificity and phosphoryl transfer in two-component signal transduction. Cell 2009, 139, 325–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pena-Sandoval, G.R.; Georgellis, D. The ArcB sensor kinase of Escherichia coli autophosphorylates by an intramolecular reaction. J. Bacteriol. 2010, 192, 1735–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casino, P.; Lopez-Redondo, M.; Marina, A. Structural basis of signal transduction and specificity in two-component systems. In Two-Component Systems in Bacteria; Gross, R., Beier, D., Eds.; Caister Academic Press: Norfolk, UK, 2012; Chapter 2; pp. 21–40. [Google Scholar]
- Groisman, E.A. Feedback control of two-component regulatory systems. Ann. Rev. Microbiol. 2016, 70, 103–124. [Google Scholar] [CrossRef] [PubMed]
- Zschiedrich, C.P.; Keidel, V.; Szurmant, H. Molecular mechanisms of two-component signal transduction. J. Mol. Biol. 2016, 428, 3752–3775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoch, J.A. A life in Bacillus subtilis signal transduction. Ann. Rev. Microbiol. 2017, 71, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padilla-Vaca, F.; Mongradon, V.; Franco, B. General aspects of two-component regulatory circuits in bacteria: Domains, signals and roles. Curr. Protein Pep. Sci. 2017, 18, 990–1004. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Stock, A.M. Quantitative analysis of intracellular response regulator phosphatase activity of histidine kinases. Meth. Enzymol. 2018, 607, 301–319. [Google Scholar]
- Gushchin, I.; Gordeliy, V. Transmembrane signal transduction in two-component systems: Piston, scissoring, or helical rotation? Bioessays 2018, 40, 1700197. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, K.; Hoch, J.A. Two-component and phosphorelay signal-transduction systems as therapeutic targets. Curr. Opin. Pharmacol. 2002, 2, 507–512. [Google Scholar] [CrossRef]
- Kiil, K.; Ferchaud, J.B.; David, C.; Binnewies, T.T.; Wu, H.; Sicheritz-Ponte, T.; Willenbrock, H.; Ussery, D.W. Genome update: Distribution of two-component transduction systems in 250 bacterial genomes. Microbiology 2005, 151, 3447–3452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, A.; Gotoh, Y.; Watanabe, T.; Furuta, E.; Yamamoto, K.; Utsumi, R. Targeting two-component signal transduction: A novel drug discovery system. Two-component signalling systems Part A. Meth. Enzymol. 2007, 422, 386–395. [Google Scholar]
- Gotoh, Y.; Eguchi, Y.; Watanabe, T.; Okamoto, S.; Doi, A.; Utsumi, R. Two-component signal transduction as potential drug targets in pathogenic bacteria. Curr. Opin. Microbiol. 2010, 13, 232–239. [Google Scholar] [CrossRef]
- Kurosu, M.; Begari, E. Bacterial protein kinase inhibitors. Drug Devel. Res. 2010, 71, 168–187. [Google Scholar] [CrossRef]
- Utsumi, R.; Igarashi, M. Two-component signal transduction as attractive drug targets in pathogenic bacteria. Yakugaku Zasshi-J. Pharmaceut. Soc. Jpn. 2012, 132, 51–58. [Google Scholar] [CrossRef]
- Wuichet, K.; Cantwell, B.J.; Zhulin, I.B. Evolution and phyletic distribution of two-component signal transduction systems. Curr. Opin. Microbiol. 2010, 13, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Worthington, R.J.; Blackledge, M.S.; Melander, C. Small-molecule inhibition of bacterial two-component systems to combat antibiotic resistance and virulence. Future Med. Chem. 2013, 5, 1265–1284. [Google Scholar] [CrossRef]
- Tiganova, I.G.; Ilyina, T.S.; Romanova, Y.M. Two-component bacterial regulation systems: Targets of a search for new antibacterial drugs. Mol. Gen. Microbiol. Virol. 2014, 29, 93–103. [Google Scholar] [CrossRef]
- Tiwari, S.; Jamal, S.B.; Hassan, S.S.; Carvalho, P.V.S.D.; Almeida, S.; Barh, D.; Ghosh, P.; Silva, A.; Castro, T.L.P.; Azevedo, V. Two-component signal transduction systems of pathogenic bacteria as targets for antimicrobial therapy: An overview. Front. Microbiol. 2017, 8, 1878. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Phyletic distribution and lineage-specific domain architectures of Archaeal two-component signal transduction systems. J. Bacteriol. 2018, 200, e00681-17. [Google Scholar] [CrossRef] [Green Version]
- De Silva, P.M.; Kumar, A. Signal transduction proteins in Acinetobacter baumannii: Role in antibiotic resistance, virulence, and potential as drug targets. Front. Microbiol. 2019, 10, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, R.; Stock, A.M. Biological insights from structures of two-component proteins. Ann. Rev. Microbiol. 2009, 63, 133–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Copley, R.R.; Schmidt, S.; Ciccarelli, F.D.; Doerks, T.; Schultz, J.; Ponting, C.P.; Bork, P. SMART 4.0: Towards genomic data integration. Nucl. Acids Res. 2004, 32, D142–D144. [Google Scholar] [CrossRef] [Green Version]
- Barakat, M.; Ortet, P.; Jourlin-Castelli, C.; Mejean, V.; Whitworth, D.E. P2CS: A two-component system resource for prokaryotic signal transduction research. BMC Genom. 2009, 10, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulrich, L.E.; Zhulin, I.B. The MiST2 database: A comprehensive genomics resource on microbial signal transduction. Nucl. Ac. Res. 2010, 38, D401–D407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galperin, M.Y. Diversity of structure and function of response regulator output domains. Curr. Opin. Microbiol. 2010, 13, 150–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitworth, D.E. Classification and organisation of two-component systems. In Two-Component Systems in Bacteria; Gross, R., Beier, D., Eds.; Caister Academic Press: Norfolk, UK, 2012; Chapter 1; pp. 1–20. [Google Scholar]
- Yamamoto, K.; Hirao, K.; Oshima, T.; Aiba, H.; Utsumi, R.; Ishihama, A. Functional characterization in vitro of all two-component signal transduction systems from Escherichia coli. J. Biol. Chem. 2005, 280, 1448–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancock, L.; Perego, M. Two-Component signal transduction in Enterococcus faecalis. J. Bacteriol. 2002, 184, 5819–5825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, P.; Yuille, H.M.; Blessie, V.; Göhring, N.; Iglói, Z.; Nishiguchi, K.; Nakayama, J.; Henderson, P.J.F.; Phillips-Jones, M.K. Expression, purification and activities of the entire family of intact membrane sensor kinases from Enterococcus faecalis. Mol. Membr. Biol. 2008, 25, 449–473. [Google Scholar] [CrossRef]
- Holden, M.; Titball, R.W.; Peacock, S.J.; Cerdeño-Tárraga, A.M.; Atkins, T.; Crossman, L.C.; Pitt, T.; Churcher, C.; Mungall, K.; Bentley, S.D.; et al. Genomic plasticity of the causative agent of melioidosis, Burkholderia pseudomallei. Proc. Natl. Acad. Sci. USA 2004, 101, 14240–14245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stover, C.K.; Pham, X.Q.; Erwin, A.L.; Mizoguchi, S.D.; Warrener, P.; Hickey, M.J.; Brinkman, F.S.L.; Hufnagle, W.O.; Kowalik, D.J.; Lagrou, M.; et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 2000, 406, 959–964. [Google Scholar] [CrossRef]
- Read, T.D.; Peterson, S.N.; Tourasse, N.; Baillie, L.W.; Paulsen, I.T.; Nelson, K.E.; Tettelin, H.; Fouts, D.E.; Eisen, J.A.; Gill, S.R.; et al. The genome sequence of Bacillus anthracis Ames and comparison to closely related bacteria. Nature 2003, 423, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Blattner, F.R.; Plunkett, G.; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The complete genome sequence of Escherichia coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef] [Green Version]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E., III; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef]
- Parkhill, J.; Sebaihia, M.; Preston, A.; Murphy, L.; Thomson, N.; E Harris, D.; Holden, M.; Churcher, C.M.; Bentley, S.D.; Mungall, K.L.; et al. Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat. Genet. 2003, 35, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.G.; Gianoulis, T.A.; Pukatzki, S.; Mekalanos, J.J.; Ornston, L.N.; Gerstein, M.; Snyder, M. New insights into Acinetobacter baumannii pathogenesis revealed by high-density pyrosequencing and transposon mutagenesis. Genes Devel. 2007, 21, 601–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, M.D.; Goglin, K.; Molyneaux, N.; Hujer, K.M.; Lavender, H.; Jamison, J.J.; MacDonald, I.J.; Martin, K.M.; Russo, T.; Campagnari, A.A.; et al. Comparative genome sequence analysis of multidrug-resistant Acinetobacter Baumannii. J. Bacteriol. 2008, 190, 8053–8064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulsen, I.T.; Banerjei, L.; Myers, G.S.A.; Nelson, K.E.; Seshadri, R.; Read, T.D.; Fouts, D.E.; Eisen, J.A.; Gill, S.R.; Heidelberg, J.F.; et al. Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science 2003, 299, 2071–2074. [Google Scholar] [CrossRef] [Green Version]
- DelVecchio, V.G.; Kapatral, V.; Redkar, R.J.; Patra, G.; Mujer, C.; Los, T.; Ivanova, N.; Anderson, I.; Bhattacharyya, A.; Lykidis, A.; et al. The genome sequence of the facultative intracellular pathogen Brucella melitensis. Proc. Natl. Acad. Sci. USA 2002, 99, 443–448. [Google Scholar] [CrossRef] [Green Version]
- Halling, S.M.; Peterson-Burch, B.D.; Bricker, B.J.; Zuerner, R.L.; Qing, Z.; Li, L.-L.; Kapur, V.; Alt, D.P.; Olsen, S.C. Completion of the genome sequence of Brucella abortus and comparison to the highly similar genomes of Brucella melitensis and Brucella suis. J. Bacteriol. 2005, 187, 2715–2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T.; Ohtani, K.; Hirakawa, H. Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater. Proc. Natl. Acad. Sci. USA 2002, 99, 996–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuroda, M.; Ohta, T.; Uchiyama, I.; Baba, T.; Yuzawa, H.; Kobayashi, I.; Cui, L.; Oguchi, A.; Aoki, K.; Nagai, Y.; et al. Whole genome sequencing of methicillin-resistant Staphylococcus aureus. Lancet 2001, 357, 1225–1240. [Google Scholar] [CrossRef]
- Glaser, P.; Rusniok, C.; Buchrieser, C. Genome sequence of Streptococcus agalactiae, a pathogen causing invasive neonatal disease. Mol. Microbiol. 2002, 45, 1499–1513. [Google Scholar] [CrossRef]
- Thomas, L.; Cook, L. Two-component signal transduction systems in the human pathogen Streptococcus agalactiae. Infect. Immun. 2020, 88, e00931-19. [Google Scholar] [CrossRef] [PubMed]
- Lange, R.; Wagner, C.; de Saizieu, A. Domain organization and molecular characterization of 13 two-component systems identified by genome sequencing of Streptococcus pneumoniae. Gene 1999, 237, 223–234. [Google Scholar] [CrossRef]
- Dopazo, J.; Mendoza, A.; Herrero, J.; Caldara, F.; Humbert, Y.; Friedli, L.; Guerrier, M.; Grand-Schenk, E.; Gandin, C.; de Francesco, M.; et al. Annotated draft genomic sequence from a Streptococcus pneumoniae type 19F clinical isolate. Microb. Drug Res. 2001, 7, 99–125. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, J.J.; McShan, W.M.; Ajdic, D.; Savic, D.J.; Savic, G.; Lyon, K.; Primeaux, C.; Sezate, S.; Suvorov, A.; Kenton, S.; et al. Complete genome sequence of an M1 strain of Streptococcus pyogenes. Proc. Natl. Acad. Sci. USA 2001, 98, 4658–4663. [Google Scholar] [CrossRef] [Green Version]
- Fleischmann, R.D.; Adams, M.D.; White, O.; Clayton, R.A.; Kirkness, E.F.; Kerlavage, A.R.; Bult, C.J.; Tomb, J.F.; Dougherty, B.A.; Merrick, J.M.; et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 1995, 269, 496–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomb, J.-F.; White, O.; Kerlavage, A.R.; Clayton, R.A.; Sutton, G.G.; Fleischmann, R.D.; Ketchum, K.A.; Klenk, H.P.; Gill, S.; Dougherty, B.A.; et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 1997, 388, 539–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkhill, J.; Wren, B.W.; Mungall, K. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 2000, 403, 665–668. [Google Scholar] [CrossRef] [Green Version]
- Fraser, C.; Gocayne, J.D.; White, O.; Adams, M.D.; Clayton, R.A.; Fleischmann, R.D.; Bult, C.J.; Kerlavage, A.R.; Sutton, G.; Kelley, J.M.; et al. The minimal gene complement of Mycoplasma genitalium. Science 1995, 270, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, Y.; Oshima, T.; Mori, H.; Aono, R.; Yamamoto, K.; Ishihama, A.; Utsumi, R. Transcriptional regulation of efflux genes by EvgAS, a two-component system in Escherichia coli. Microbiology 2003, 149, 2819–2828. [Google Scholar] [CrossRef] [PubMed]
- Beier, D.; Frank, R. Molecular characterization of two-component systems of Helicobacter pylori. J. Bacteriol. 2000, 182, 2068–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabret, C.; Hoch, J.A. A two-component signal transduction system essential for growth of Bacillus subtilis: Implications for anti-infective therapy. J. Bacteriol. 1998, 180, 6375–6383. [Google Scholar] [CrossRef]
- Takada, H.; Yoshikawa, H. Essentiality and function of WalK/WalR two-component system: The past, present, and future of research. Biosci. Biotechol. Biochem. 2018, 82, 741–751. [Google Scholar] [CrossRef]
- Rosales-Hurtado, M.; Meffre, P.; Szurmant, H.; Benfodda, Z. Synthesis of histidine kinase inhibitors and their biological properties. Med. Res. Rev. 2020, 40, 1440–1495. [Google Scholar] [CrossRef]
- Lakshmi, S.A.; Bhaskar, J.P.; Krishnan, V.; Sethupathy, S.; Pandipriya, S.; Aruni, W.; Pandian, S.K. Inhibition of biofilm and biofilm-associated virulence factor production in methicillin-resistant Staphylococcus aureus by docosanol. J. Biotechol. 2020, 317, 59–69. [Google Scholar] [CrossRef]
- Tsai, C.N.; MacNair, C.R.; Cao, M.P.T.; Perry, J.N.; Magolan, J.; Brown, E.D.; Coombes, B.K. Targeting two-component systems uncovers a small-molecule inhibitor of Salmonella virulence. Cell Chem. Biol. 2020, 27, 793–805. [Google Scholar] [CrossRef]
- Zheng, H.; Abramovitch, R.B. Inhibiting DosRST as a new approach to tuberculosis therapy. Future Med. Chem. 2020, 12, 457–467. [Google Scholar] [CrossRef]
- Ayelén Carabajal, M.; Asquith, C.R.M.; Laitinen, T.; Tizzard, G.J.; Yim, L.; Rial, A.; Chabalgoity, J.A.; Zuercher, W.J.; Garcia Véscovi, E. Quinazoline-based antivirulence compounds selectively target Salmonella PhoP/PhoQ signal transduction system. Antimicrob. Agents Chemother. 2020, 64, e01744-19. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, H.; Kurushima, J.; Hashimoto, Y.; Tomita, H. Progress overview of bacterial two-component regulatory systems as potential targets for antimicrobial chemotherapy. Antibiotics 2020, 9, 635. [Google Scholar] [CrossRef]
- Diensthuber, R.P.; Bommer, M.; Gleichmann, T.; Möglich, A. Full-length structure of a sensor histidine kinase pinpoints coaxial coiled coils as signal transducers and modulators. Structure 2013, 21, 1127–1136. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Cancel, G.; Ko, W.-h.; Tomchick, D.R.; Correa, F.; Gardner, K.H. Full-length structure of a monomeric histidine kinase reveals basis for sensory regulation. Proc. Natl. Acad. Sci. USA 2014, 111, 17839–17844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinaldi, J.; Fernandez, I.; Poth, L.M.; Shepard, W.E.; Savko, M.; Goldbaum, F.A.; Klinke, S. Crystallization and initial X-ray diffraction analysis of the multidomain Brucella blue light-activated histidine kinase LOV-HK in its illuminated state. Biochem. Biophys. Rep. 2018, 16, 39–43. [Google Scholar] [PubMed]
- Wright, G.S.A.; Saeki, A.; Hikima, T.; Nishizono, Y.; Hisano, T.; Kamaya, M.; Nukina, K.; Nishitani, H.; Nakamura, H.; Yamamoto, M.; et al. Architecture of the complete oxygen-sensing FixL-FixJ two-component signal transduction system. Sci. Signal. 2018, 11, eaaq0825. [Google Scholar] [CrossRef] [Green Version]
- Moeglich, A. Signal transduction in photoreceptor histidine kinases. Protein Sci. 2019, 28, 1923–1946. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, J.; Fernández, I.; Shin, H.; Sycz, G.; Gunawardana, S.; Kumarapperuma, I.; Paz, J.M.; Otero, L.H.; Cerutti, M.L.; Zorreguieta, A.; et al. Dimer asymmetry and light activation mechanism in Brucella blue-light sensor histidine kinase. MBio 2021, 12, e00264-21. [Google Scholar] [CrossRef] [PubMed]
- Moraes, I.; Evans, G.; Sanchez-Weatherby, J.; Newstead, S.; Shaw Stewart, P.D. Membrane protein structure determination-the next generation. Biochim. Biophys. Acta Biomembr. 2014, 1838, 78–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patching, S.G.; Edara, S.; Ma, P.; Nakayama, J.; Hussain, R.; Siligardi, G.; Phillips-Jones, M.K. Interactions of the intact FsrC membrane histidine kinase with its pheromone ligand GBAP revealed through synchrotron radiation circular dichroism. Biochim. Biophys. Acta Biomembr. 2012, 1818, 1595–1602. [Google Scholar] [CrossRef] [Green Version]
- Phillips-Jones, M.K.; Channell, G.; Kelsall, C.J.; Hughes, C.S.; Ashcroft, A.E.; Patching, S.G.; Dinu, V.; Gillis, R.B.; Adams, G.A.; Harding, S.E. Hydrodynamics of the VanA-type VanS histidine kinase: An extended solution conformation and first evidence for interactions with vancomycin. Sci. Rep. 2017, 7, 46180. [Google Scholar] [CrossRef]
- Ma, P.; Nishiguchi, K.; Yuille, H.M.; Davis, L.M.; Nakayama, J.; Phillips-Jones, M.K. Anti-HIV siamycin I directly inhibits autophosphorylation activity of the bacterial FsrC quorum sensor and other ATP-dependent enzyme activities. FEBS Lett. 2011, 585, 2660–2664. [Google Scholar] [CrossRef] [Green Version]
- Chase, O.M.; Espinasse, A.; Wilke, K.E.; Carlson, E.E. Exploration of the effects of γ-phosphate-modified ATP analogues on histidine kinase autophosphorylation. Biochemistry 2018, 57, 4368–4373. [Google Scholar] [CrossRef]
- Kinoshita-Kikuta, E.; Maruta, S.; Eguchi, Y.; Igarashi, M.; Okajima, T.; Utsumi, R.; Kinoshita, E.; Koike, T. An immuno-dot blot assay for screening histidine kinase inhibitors. Anal. Biochem. 2020, 600, 113765. [Google Scholar] [CrossRef]
- Rasko, D.A.; Moreira, C.G.; Li, D.R.; Reading, N.C.; Ritchie, J.M.; Waldor, M.K.; Williams, N.; Taussig, R.; Wei, S.; Roth, M.; et al. Targeting QseC signaling and virulence for antibiotic development. Science 2008, 321, 1078–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, B.K.; Colvin, C.J.; Needle, D.B.; Mba Medie, F.; DiGiuseppe Champion, P.A.; Abramovitch, R.B. The carbonic anhydrase inhibitor ethoxzolamide inhibits the Mycobacterium tuberculosis PhoPR regulon and Esx-1 secretion and attenuates virulence. Antimicrob. Agents Chemother. 2015, 59, 4436–4445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.; Colvin, C.H.; Johnson, B.K.; Kirchhoff, P.D.; Wilson, M.; Jorgensen-Muga, K.; Larsen, S.D.; Abramovitch, R.B. Inhibitors of Mycobacterium tuberculosis DosRST signaling and persistence. Nat. Chem. Biol. 2017, 13, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Wilke, K.E.; Fihn, C.A.; Carlson, E.E. Screening serine/threonine and tyrosine kinase inhibitors for histidine kinase inhibition. Bioorg. Med. Chem. 2018, 26, 5322–5326. [Google Scholar] [CrossRef]
- Zheng, H.; Williams, J.T.; Aleiwi, B.; Ellsworth, E.; Abramovitch, R.B. Inhibiting Mycobacterium tuberculosis DosRST signaling by targeting response regulator DNA binding and sensor kinase heme. ACS Chem. Biol. 2019, 15, 52–62. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, J.; Tanaka, E.; Kariyama, R.; Nagata, K.; Nishiguchi, K.; Mitsuhata, R.; Uemura, Y.; Tanokura, M.; Kumon, H.; Sonomoto, K. Siamycin attenuates fsr quorum sensing mediated by a gelatinase biosynthesis-activating pheromone in Enterococcus faecalis. J. Bacteriol. 2007, 189, 1358–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goswami, M.; Espinasse, A.; Carlson, E.E. Disarming the virulence arsenal of Pseudomonas aeruginosa by blocking two-component system signalling. Chem. Sci. 2018, 9, 7332–7337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Z.; Zhang, J.; Xu, B.; Chen, L.; Wu, Y.; Yang, X.; Shen, X.; Molin, S.; Danchin, A.; Jiang, H.; et al. Structure-based discovery of inhibitors of the YycG histidine kinase: New chemical leads to combat Staphylococcus epidermidis infections. BMC Microbiol. 2006, 6, 96. [Google Scholar]
- Potter, C.A.; Ward, A.; Laguri, C.; Williamson, M.P.; Henderson, P.J.F.; Phillips-Jones, M.K. Expression, purification and characterisation of full-length heterologously expressed histidine protein kinase RegB from Rhodobacter sphaeroides. J. Mol. Biol. 2002, 320, 201–213. [Google Scholar] [CrossRef]
- Ward, A.; Sanderson, N.M.; O’Reilly, J.; Rutherford, N.G.; Poolman, B.; Henderson, P.J.F. The amplified expression, identification, purification, assay and properties of hexahistidine-tagged bacterial membrane transport proteins. In Membrane Transport—A Practical Approach; Baldwin, S.A., Ed.; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Phillips-Jones, M.K.; Harding, S.E. Antimicrobial resistance (AMR) nanomachines-mechanisms for fluoroquinolone and glycopeptide recognition, efflux and deactivation. Biophys. Rev. 2018, 10, 347–362. [Google Scholar] [CrossRef] [Green Version]
- Azam, A.A.; Kinder, J.M.; Khan, G.N.; Alase, A.; Ma, P.; Liu, Y.; Ault, J.R.; Henderson, P.J.F.; Chowdhry, B.; Alexander, B.D.; et al. Production of membrane proteins for characterisation of their pheromone-sensing and antimicrobial resistance functions. Eur. Biophys. J. 2018, 47, 723–737. [Google Scholar] [CrossRef] [Green Version]
- Potter, C.A.; Jeong, E.-L.; Williamson, M.P.; Henderson, P.J.F.; Phillips-Jones, M.K. Redox-responsive in vitro modulation of the signalling state of the isolated PrrB sensor kinase of Rhodobacter sphaeroides NCIB 8253. FEBS Lett. 2006, 580, 3206–3210. [Google Scholar] [CrossRef]
- Oh, J.-I.; Ko, I.-J.; Kaplan, S. Reconstitution of the Rhodobacter sphaeroides cbb3-PrrBA signal transduction pathway in vitro. Biochemistry 2004, 43, 7915–7923. [Google Scholar] [CrossRef]
- Tabor, S.; Richardson, C.C. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc. Natl. Acad. Sci. USA 1985, 82, 1074–1078. [Google Scholar] [CrossRef] [Green Version]
- Lockey, C.; Edwards, R.J.; Roper, D.I.; Dixon, A.M. The extracellular domain of two-component system sensor kinase VanS from Streptomyces coelicolor binds vancomycin at a newly identified binding site. Sci. Rep. 2020, 10, 5727. [Google Scholar] [CrossRef]
- Upton, E.C.; Maciunas, L.J.; Loll, P.J. Vancomycin does not affect the enzymatic activities of purified VanSA. PLoS ONE 2019, 14, e0210627. [Google Scholar] [CrossRef] [PubMed]
- Weeks, S.D.; Drinker, M.; Loll, P.J. Ligation independent cloning vectors for expression of SUMO fusions. Protein Expr. Purif. 2007, 53, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Saidijam, M.; Bettaney, K.E.; Szakonyi, G.; Psakis, G.; Shibayama, K.; Suzuki, S.; Clough, J.L.; Blessie, V.; Abu-bakr, A.; Baumberg, S.; et al. Active membrane transport and receptor proteins from bacteria. Biochem. Soc. Trans. 2005, 33, 867–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, S.; Henderson, P.J.F. The hydantoin transport protein from Microbacterium liquefaciens. J. Bacteriol. 2006, 188, 3329–3336. [Google Scholar] [CrossRef] [Green Version]
- Surade, S.; Klein, M.; Stolt-Bergner, P.C.; Muenke, C.; Roy, A.; Michel, H. Comparative analysis and ‘‘expression space’’ coverage of the production of prokaryotic membrane proteins for structural genomics. Protein Sci. 2006, 15, 2178–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, P. Structure-Activity Relationships of Membrane Proteins: The NCS1 Family of Transporters and Sensor Kinases of Two-Component Systems. Ph.D. Thesis, University of Leeds, Leeds, UK, 2010. [Google Scholar]
- Rigaud, J.-L.; Lévy, D. Reconstitution of membrane proteins into liposomes. Meth. Enzymol. 2003, 372, 65–86. [Google Scholar]
- Türck, M.; Bierbaum, G. Purification and activity testing of the full-length YycFGHI proteins of Staphylococcus aureus. PLoS ONE 2012, 7, e30403. [Google Scholar] [CrossRef] [PubMed]
- Pocanschi, C.L.; Dahmane, T.; Gohon, Y.; Rappaport, F.; Apell, H.-J.; Kleinschmidt, J.H.; Popot, J.-L. Amphipathic polymers: Tools to fold integral membrane proteins to their active form. Biochemistry 2006, 45, 13954–13961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boldog, T.; Li, M.; Hazelbauer, G.L. Using nanodiscs to create water-soluble transmembrane chemoreceptors inserted in lipid bilayers. Meth. Enzymol. 2007, 423, 317–335. [Google Scholar]
- Pollock, N.L.; Lee, S.C.; Patel, J.H.; Gulamhussein, A.A.; Rothnie, A.J. Structure and function of membrane proteins encapsulated in a polymer-bound lipid bilayer. Biochim. Biophys. Acta Biomembr. 2018, 1860, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Trieber, C.; Overduin, M. Structural biology of endogenous membrane protein assemblies in native nanodiscs. Curr. Opin. Struct. Biol. 2021, 69, 70–77. [Google Scholar] [CrossRef]
- Wang, B.; Zhao, A.; Novick, R.P.; Muir, T.W. Activation and inhibition of the receptor histidine kinase AgrC occurs through opposite helical transduction motions. Mol. Cell. 2014, 53, 929–940. [Google Scholar] [CrossRef] [Green Version]
- Hoernschemeyer, P.; Liss, V.; Heermann, R.; Jung, K.; Hunke, S. Interaction analysis of a two-component system using nanodiscs. PLoS ONE 2016, 11, e0149187. [Google Scholar] [CrossRef] [Green Version]
- Affandi, T.; McEvoy, M.M. Mechanism of metal ion-induced activation of a two-component sensor kinase. Biochem. J. 2019, 476, 115–135. [Google Scholar] [CrossRef]
- Su, C.C.; Morgan, C.E.; Kambakam, S.; Rajavel, M.; Scott, H.; Huang, W.; Emerson, C.C.; Taylor, D.J.; Stewart, P.L.; Bonomo, R.A.; et al. Cryo-electron microscopy structure of an Acinetobacter baumannii multidrug efflux pump. MBio 2019, 10, e01295-19. [Google Scholar] [CrossRef] [Green Version]
- Yardeni, E.H.; Mishra, S.; Stein, R.A.; Bibi, E.; Mchaourab, H.S. The multidrug transporter MdfA deviates from the canonical model of alternating access of MFS transporters. J. Mol. Biol. 2020, 432, 5665–5680. [Google Scholar] [CrossRef] [PubMed]
- Hernando, M.; Orriss, G.; Perodeau, J.; Lei, S.; Ferens, F.G.; Patel, T.R.; Stetefeld, J.; Nieuwkoop, A.J.; O’Neil, J.D. Solution structure and oligomeric state of the E. coli glycerol facilitator. Biochim. Biophys. Acta. Biomembr. 2020, 1862, 183191. [Google Scholar] [CrossRef]
- Asamoto, D.K.; Kang, G.; Kim, J.E. Folding of the β-Barrel membrane protein OmpA into nanodiscs. Biophys. J. 2020, 118, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Bibow, S.; Böhm, R.; Modaresi, S.M.; Hiller, S. Detergent titration as an efficient method for NMR resonance assignments of membrane proteins in lipid-bilayer nanodiscs. Anal. Chem. 2020, 92, 7786–7793. [Google Scholar] [CrossRef] [PubMed]
- Fiori, M.C.; Jiang, Y.; Zheng, W.; Anzaldua, M.; Borgnia, M.J.; Altenberg, G.A.; Liang, H. Polymer nanodiscs: Discoidal amphiphilic block copolymer membranes as a new platform for membrane proteins. Sci. Rep. 2017, 7, 15227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ter Beek, J.; Kahle, M.; Ädelroth, P. Modulation of protein function in membrane mimetics: Characterization of P. denitrificans cNOR in nanodiscs or liposomes. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1951–1961. [Google Scholar] [CrossRef]
- Liu, Y.; Moura, E.C.C.M.; Dörr, J.M.; Scheidelaar, S.; Heger, M.; Egmond, M.R.; Killian, J.A.; Mohammadi, T.; Breukink, E. Bacillus subtilis MraY in detergent-free system of nanodiscs wrapped by styrene-maleic acid copolymers. PLoS ONE 2018, 13, e0206692. [Google Scholar] [CrossRef]
- Cuozzo, J.W.; Soutter, H.H. Overview of recent progress in protein-expression technologies for small-molecule screening. J. Biomol. Screen. 2014, 19, 1000–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, J.; Cao, Y.; Horii, T.; Sakuda, S.; Akkermans, A.D.L.; De Vos, W.M.; Nagasawa, H. Gelatinase biosynthesis-activating pheromone: A peptide lactone that mediates a quorum sensing in Enterococcus faecalis. Mol. Microbiol. 2001, 41, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Singh, K.V.; Weinstock, G.M.; Murray, B.E. Effects of Enterococcus faecalis fsr genes on production of gelatinase and a serine protease and virulence. Infect. Immun. 2000, 68, 2579–2586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, X.; Singh, K.V.; Weinstock, G.M.; Murray, B.E. Characterization of fsr, a regulator controlling expression of gelatinase and serine protease in Enterococcus faecalis OG1RF. J. Bacteriol. 2001, 183, 3372–3382. [Google Scholar] [CrossRef] [Green Version]
- Hancock, L.E.; Perego, M. The Enterococcus faecalis fsr two-component system controls biofilm development through production of gelatinase. J. Bacteriol. 2004, 186, 5629–5639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillai, S.K.; Sakoulas, G.M.; Eliopoulos, G.M.; Moellering, R.C.; Murray, B.E.; Inouye, R.T. Effects of glucose on fsr-mediated biofilm formation in Enterococcus faecalis. J. Infect. Dis. 2004, 190, 967–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips-Jones, M.K.; Patching, S.G.; Edara, S.; Nakayama, J.; Hussain, R.; Siligardi, G. Interactions of the intact FsrC membrane histidine kinase with the tricyclic peptide siamycin I revealed through synchrotron radiation circular dichroism. Phys. Chem. Chem. Phys. 2013, 15, 444–447. [Google Scholar] [CrossRef]
- Lin, P.F.; Samanta, H.; Bechtold, C.M.; Deminie, C.A.; Patick, A.K.; Alam, M.; Riccardi, K.; Rose, R.E.; White, R.J.; Colonno, R.J. Characterization of siamycin I, a human immunodeficiency virus fusion inhibitor. Antimicr. Agents Chemother. 1996, 40, 133–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsunakawa, M.; Hu, S.-L.; Hoshino, Y.; Detlefson, D.J.; Hill, S.E.; Furumai, T.; White, R.J.; Nishio, M.; Kawano, K.; Yamamoto, S.; et al. Siamycins I and II, new anti-HIV peptides: I. Fermentation, isolation, biological activity and initial characterization. J. Antibiot. 1995, 48, 433–434. [Google Scholar] [CrossRef] [Green Version]
- Gerharz, T.; Reinelt, S.; Kaspar, S.; Scapozza, L.; Bott, M. Identification of basic amino acids important for citrate binding by the periplasmic receptor domain of the sensor kinase CitA. Biochem. 2003, 42, 5917–5924. [Google Scholar] [CrossRef] [Green Version]
- Arthur, M.; Molinas, C.; Courvalin, P. The VanS-VanR two-component regulatory system controls synthesis of depsipeptide peptidoglycan precursors in Enterococcus faecium BM4147. J. Bacteriol. 1992, 174, 2582–2591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, H.-J.; Hutchings, M.I.; Buttner, M.J. Vancomycin resistance VanS/VanR two-component systems. In Bacterial Signal Transduction: Networks and Drug Targets; Utsumi, R., Ed.; Landes Bioscience & Springer Science & Business Media: New York, NY, USA, 2008. [Google Scholar]
- Cetinkaya, Y.; Falk, P.; Mayhall, G.C. Vancomycin-resistant enterococci. Clin. Microbiol. Rev. 2000, 13, 686–707. [Google Scholar] [CrossRef] [PubMed]
- Courvalin, P. Genetics of glycopeptide resistance in Gram-positive pathogens. Int. J. Med. Microbiol. 2005, 294, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.H.; Kirsch, D.R. Induction signals for vancomycin resistance encoded by the vanA gene cluster in Enterococcus faecium. Antimicrob. Agents Chemother. 1996, 40, 1645–1648. [Google Scholar] [CrossRef] [Green Version]
- Baptista, M.; Depardieu, F.; Courvalin, P.; Arthur, M. Specificity of induction of glycopeptide resistance genes in Enterococcus faecalis. Antimicrob. Agents Chemother. 1996, 40, 2291–2295. [Google Scholar] [CrossRef] [Green Version]
- Hughes, C.S.; Longo, E.; Phillips-Jones, M.K.; Hussain, R. Characterisation of the selective binding of antibiotics vancomycin and teicoplanin by the VanS receptor regulating type A vancomycin resistance in the enterococci. Biochim. Biophys. Acta 2017, 1861, 1951–1959. [Google Scholar] [CrossRef]
- Arthur, M.; Depardieu, F.; Gerbaud, G.; Galimand, M.; Leclercq, R.; Courvalin, P. The VanS sensor negatively controls VanR-mediated transcriptional activation of glycopeptide resistance genes of Tn1546 and related elements in the absence of induction. J. Bacteriol. 1997, 179, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Baptista, M.; Rodrigues, P.; Depardieu, F.; Courvalin, P.; Arthur, M. Single-cell analysis of glycopeptide resistance gene expression in teicoplanin-resistant mutants of a VanB-type Enterococcus faecalis. Mol. Microbiol. 1999, 32, 17–28. [Google Scholar] [CrossRef]
- Arthur, M.; Quintiliani (Jnr), R. Regulation of VanA- and VanB-type glycopeptide resistance in enterococci. Antimicrob. Agents Chemother. 2001, 45, 375–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koteva, K.; Hong, H.-J.; Wang, X.D.; Nazi, I.; Hughes, D.; Naldrett, M.J.; Buttner, M.J.; Wright, G.D. A vancomycin photoprobe identifies the histidine kinase VanSsc as a vancomycin receptor. Nat. Chem. Biol. 2010, 6, 327–329. [Google Scholar] [CrossRef] [PubMed]
- Jung Kwun, M.; Novotna, G.; Hesketh, A.R.; Hill, L.; Hong, H.-J. In vivo studies suggest that induction of VanS-dependent vancomycin resistance requires binding of the drug to D-Ala-D-Ala termini in the peptidoglycan cell wall. Antimicrob. Agents Chemother. 2013, 57, 4470–4480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evers, S.; Courvalin, P. Regulation of VanB-type vancomycin resistance gene expression by the VanSB-VanRB two-component regulatory system in Enterococcus faecalis V583. J. Bacteriol. 1996, 178, 1302–1309. [Google Scholar] [CrossRef] [Green Version]
- Eraso, J.M.; Roh, J.H.; Zeng, X.; Callister, S.J.; Lipton, M.S.; Kaplan, S. Role of the global transcriptional regulator PrrA in Rhodobacter sphaeroides 2.4.1: Combined transcriptome and proteome analysis. J. Bacteriol. 2008, 190, 4831–4848. [Google Scholar] [CrossRef] [Green Version]
- Gibson, J.L.; Dubbs, J.M.; Tabita, F.R. Differential expression of the CO2 fixation operons of Rhodobacter sphaeroides by the Prr/Reg two-component system during chemoautotrophic growth. J. Bacteriol. 2002, 184, 6654–6664. [Google Scholar] [CrossRef] [Green Version]
- Laguri, C.; Phillips-Jones, M.K.; Williamson, M.P. Solution structure and DNA binding of the effector domain from the global regulator PrrA (RegA) from Rhodobacter sphaeroides: Insights into DNA binding specificity. Nucl. Acids Res. 2003, 31, 6778–6787. [Google Scholar] [CrossRef] [Green Version]
- Jeong, E.-L.; Broad, S.J.; Moody, R.G.; Phillips-Jones, M.K. The adherence-associated Fdp fasciclin I domain protein of the biohydrogen producer Rhodobacter sphaeroides is regulated by the global Prr pathway. Int. J. Hydr. Energ. 2020, 45, 26840–26854. [Google Scholar] [CrossRef] [PubMed]
- O’Gara, J.P.; Eraso, J.M.; Kaplan, S. A redox-responsive pathway for aerobic regulation of photosynthesis gene expression in Rhodobacter sphaeroides 2.4.1. J. Bacteriol. 1998, 180, 4044–4050. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.I.; Kaplan, S. The cbb(3) terminal oxidase of Rhodobacter sphaeroides 2.4.1: Structural and functional implications for the regulation of spectral complex formation. Biochem. 1999, 38, 2688–2696. [Google Scholar] [CrossRef]
- Oh, J.I.; Kaplan, S. Redox signalling: Globalization of gene expression. EMBO J. 2000, 19, 4237–4247. [Google Scholar] [CrossRef] [Green Version]
- Gajdiss, M.; Monk, I.R.; Bertsche, U.; Kienemund, J.; Funk, T.; Dietrich, A.; Hort, M.; Sib, E.; Stinear, T.P.; Bierbaum, G. YycH and YycI regulate expression of Staphylococcus aureus autolysins by activation of WalRK phosphorylation. Microorganisms 2020, 8, 870. [Google Scholar] [CrossRef] [PubMed]
- Fukuchi, K.; Kasahara, Y.; Asai, K.; Kobayashi, K.; Moriya, S.; Ogasawara, N. The essential two-component regulatory system encoded by yycF and yycG modulates expression of the ftsAZ operon in Bacillus subtilis. Microbiology 2000, 146, 1573–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisicchia, P.; Noone, D.; Lioliou, E.; Howell, A.; Quigley, S.; Jensen, T.; Jarmer, H.; Devine, K.M. The essential YycFG two-component system controls cell wall metabolism in Bacillus subtilis. Mol. Microbiol. 2007, 65, 180–200. [Google Scholar] [CrossRef] [PubMed]
- Howell, A.; Dubrac, S.; Andersen, K.K.; Noone, D.; Fert, J.; Msadek, T.; Devine, K. Genes controlled by the essential YycG/YycF two-component system of Bacillus subtilis revealed through a novel hybrid regulator approach. Mol. Microbiol. 2003, 49, 1639–1655. [Google Scholar] [CrossRef] [Green Version]
- Martin, P.K.; Li, T.; Sun, D.; Biek, D.P.; Schmid, M.B. Role in cell permeability of an essential two-component system in Staphylococcus aureus. J. Bacteriol. 1999, 181, 3666–3673. [Google Scholar] [CrossRef] [Green Version]
- Dubrac, S.; Boneca, I.G.; Poupel, O.; Msadek, T. New insights into the WalK/WalR (YycG/YycF) essential signal transduction pathway reveal a major role in controlling cell wall metabolism and biofilm formation in Staphylococcus aureus. J. Bacteriol. 2007, 189, 8257–8269. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, T.; Szurmant, H.; Kim, E.-J.; Perego, M.; Hoch, J.A. A sensor histidine kinase coordinates cell wall architecture with cell division in Bacillus subtilis. Mol. Microbiol. 2008, 69, 621–632. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, T.; Furihata, I.; Emmins, R.; Daniel, R.A.; Hoch, J.A.; Szurmant, H. A role for the essential YycG sensor histidine kinase in sensing cell division. Mol. Microbiol. 2011, 79, 503–522. [Google Scholar] [CrossRef] [Green Version]
- Szurmant, H.; Nelson, K.; Kim, E.-J.; Perego, M.; Hoch, J.A. YycH regulates the activity of the essential YycFG two-component system in Bacillus subtilis. J. Bacteriol. 2005, 187, 5419–5426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szurmant, H.; Mohan, M.A.; Imus, P.M.; Hoch, J.A. YycH and YycI interact to regulate the essential YycFG two-component system in Bacillus subtilis. J. Bacteriol. 2007, 189, 3280–3289. [Google Scholar] [CrossRef] [Green Version]
- Cameron, D.R.; Jiang, J.-H.; Kostoulias, X.; Foxwell, D.J.; Peleg, A.Y. Vancomycin susceptibility in methicillin-resistant Staphylococcus aureus is mediated by YycHI activation of the WalRK essential two-component regulatory system. Sci. Rep. 2016, 6, 30823. [Google Scholar] [CrossRef]
- Poupel, O.; Moyat, M.; Groizeleau, J.; Antunes, L.C.S.; Gribaldo, S.; Msadek, T.; Dubrac, S. Transcriptional analysis and subcellular protein localization reveal specific features of the essential WalKR system in Staphylococcus aureus. PLoS ONE 2016, 11, e0151449. [Google Scholar] [CrossRef] [PubMed]
- Denisov, I.G.; Grinkova, Y.V.; Lazarides, A.A.; Sligar, S.G. Directed self-assembly of monodisperse phospholipid bilayer Nanodiscs with controlled size. J. Amer. Chem. Soc. 2004, 126, 3477–3487. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Gaupp, R.; Moriyama, H.; Eskridge, K.M.; Moriyama, E.N.; Somerville, G.A. Reductive evolution and the loss of PDC/PAS domains from the genus Staphylococcus. BMC Genom. 2013, 14, 524–540. [Google Scholar] [CrossRef] [Green Version]
- Sevvana, M.; Vijayan, V.; Zweckstetter, M.; Reinelt, S.; Madden, D.R.; Herbst-Irmer, R.; Sheldrick, G.M.; Bott, M.; Griesinger, C.; Becker, S. A ligand-induced switch in the periplasmic domain of sensor histidine kinase CitA. J. Mol. Biol. 2008, 377, 512–523. [Google Scholar] [CrossRef] [Green Version]
- Salvi, M.; Schomburg, B.; Giller, K.; Graf, S.; Unden, G.; Becker, S.; Lange, A.; Griesinger, C. Sensory domain contraction in histidine kinase CitA triggers transmembrane signalling in the membrane-bound sensor. Proc. Natl. Acad. Sci. USA 2017, 114, 3115–3120. [Google Scholar] [CrossRef] [Green Version]
- Cheung, J.; Hendrickson, W.A. Structural analysis of ligand stimulation of the histidine kinase NarX. Structure 2009, 17, 190–201. [Google Scholar] [CrossRef] [Green Version]
- Gushchin, I.; Melnikov, I.; Polovinkin, V.; Ishchenko, A.; Yuzhakova, A.; Buslaev, P.; Bourenkov, G.; Grudinin, S.; Round, E.; Balandin, T.; et al. Mechanism of transmembrane signaling by sensor histidine kinases. Science 2017, 356, eaah6345. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Guo, Q.; Zhu, K.; Fang, B.; Yang, Y.; Teng, M.; Li, X.; Tao, Y. Interface switch mediates signal transmission in a two-component system. Proc. Natl. Acad. Sci. USA 2020, 117, 30433–30440. [Google Scholar] [CrossRef] [PubMed]
- Bhate, M.P.; Molnar, K.S.; Goulian, M.; DeGrado, W.F. Signal transduction in histidine kinases: Insights from new structures. Structure 2015, 23, 981–994. [Google Scholar] [CrossRef] [Green Version]
- Gushchin, I.; Orekhov, P.; Melnikov, I.; Polovinkin, V.; Yuzhakova, A.; Gordeliy, V. Sensor histidine kinase NarQ activates via helical rotation, diagonal scissoring, and eventually piston-like shifts. Int. J. Mol. Sci. 2020, 21, 3110. [Google Scholar] [CrossRef] [PubMed]
- Cybulski, L.E.; Martίn, M.; Mansilla, M.C.; Fernández, A.; de Mendoza, D. Membrane thickness cue for cold sensing in a bacterium. Curr. Biol. 2010, 20, 1539–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, P.; Porrini, L.; Albanesi, D.; Abriata, L.A.; Dal Peraro, M.; de Mendoza, D.; Mansilla, M.C. Transmembrane prolines mediate signal sensing and decoding in Bacillus subtilis DesK histidine kinase. MBio 2019, 10, e02564-19. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, S.D.; Clinthorne, G.D.; Goulian, M.; DeGrado, W.F. Transmembrane polar interactions are required for signaling in the Escherichia coli sensor kinase PhoQ. Proc. Natl. Acad. Sci. USA 2010, 107, 8141–8146. [Google Scholar] [CrossRef] [Green Version]
- Lemmin, T.; Soto, C.S.; Clinthorne, G.; DeGrado, W.F.; Dal Peraro, M. Assembly of the transmembrane domain of E. coli PhoQ histidine kinase: Implications for signal transduction from molecular simulations. PLoS Comput. Biol. 2013, 9, e1002878. [Google Scholar] [CrossRef]
- Szurmant, H.; Bu, L.; Brooks, C.L., III; Hoch, J.A. An essential sensor histidine kinase controlled by transmembrane helix interactions with its auxiliary proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 5891–5896. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Genome Size (Mb) | SHKs * | RRs * | Reference |
|---|---|---|---|---|
| Burkholderia pseudomallei NCTC13179 | 7.24 | 42 + 7 ** | 62 | [35]; http://www.p2cs.org (accessed on 1 August 2021) |
| Pseudomonas aeruginosa PAO1 | 6.26 | 53 | 83 | [15,36] |
| Bacillus anthracis Ames Ancestor | 5.50 | 47 | 47 | [37]; http://www.p2cs.org (accessed on 1 August 2021) |
| Escherichia coli K12 W3110 | 4.64 | 30 | 34 | [32,38] |
| Mycobacterium tuberculosis H37Rv | 4.41 | 16 | 13 | [39]; http://www.p2cs.org (accessed on 1 August 2021) |
| Bordetella pertussis Tohama I | 4.09 | 19 | 22 | [40]; http://www.p2cs.org (accessed on 1 August 2021) |
| Acinetobacter baumanii ATCC17978 | 3.98 | 19 | 19 | [41,42] |
| Enterococcus faecalis V583 | 3.36 | 17 | 18 | [33,34,43] |
| Brucella melitensis 16M | 3.29 | 22 | 24 | [44] |
| Brucella abortus 9-941 | 3.28 | 22 | 24 | [45] |
| Clostridium perfringens 13 | 3.03 | 28 | 20 | [46] |
| Staphylococcus aureus Mu50 (methicillin- and vancomycin-resistant) | 2.90 | 17 | 17 | [47] |
| Staphylococcus aureus N315 (methicillin-resistant) | 2.84 | 17 | 17 | [47] |
| Streptococcus agalactiae serogroup III strain NEM316 | 2.21 | 21 | 22 | [48,49] |
| Streptococcus pneumoniae 19F | 2.10 | 13 | 13 | [50,51] |
| Streptococcus pyogenes M1 | 1.85 | 13 | 13 | [52] |
| Haemophilus influenzae Rd KW20 | 1.83 | 3 | 6 | [53]; http://www.p2cs.org (accessed on 1 August 2021) |
| Helicobacter pylori 26695 | 1.67 | 4 | 7 | [54] |
| Campylobacter jejuni subsp. jejuni NCTC 11168 | 1.64 | 7 | 12 | [55]; http://www.p2cs.org (accessed on 1 August 2021) |
| Mycoplasma genitalium ATCC 33530 | 0.58 | 0 | 0 | [56] |
| Success Rate * (Number) | Success Rate * (%) | |
|---|---|---|
| Expression in E. coli | 15 | 94 |
| Active when assayed in E. coli membranes † | 11 | 73 |
| Detergent solubilisation | 14 | 93 |
| Purification (using hexa-His tag) | 13 | 87 |
| Purification and confirmation of intact protein | 12 | 80 |
| Purified as active protein † | 13 | 87 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, P.; Phillips-Jones, M.K. Membrane Sensor Histidine Kinases: Insights from Structural, Ligand and Inhibitor Studies of Full-Length Proteins and Signalling Domains for Antibiotic Discovery. Molecules 2021, 26, 5110. https://doi.org/10.3390/molecules26165110
Ma P, Phillips-Jones MK. Membrane Sensor Histidine Kinases: Insights from Structural, Ligand and Inhibitor Studies of Full-Length Proteins and Signalling Domains for Antibiotic Discovery. Molecules. 2021; 26(16):5110. https://doi.org/10.3390/molecules26165110
Chicago/Turabian StyleMa, Pikyee, and Mary K. Phillips-Jones. 2021. "Membrane Sensor Histidine Kinases: Insights from Structural, Ligand and Inhibitor Studies of Full-Length Proteins and Signalling Domains for Antibiotic Discovery" Molecules 26, no. 16: 5110. https://doi.org/10.3390/molecules26165110