
The Uncommon Active Site of D-Amino Acid Transaminase from Haliscomenobacter hydrossis: Biochemical and Structural Insights into the New Enzyme

 , , , ,
, , , ,
Abstract
:
1. Introduction
2. Results
2.1. Enzyme Identification, Expression, and Purification
2.2. Enzyme Activity, Substrate Specificity, and Enantioselectivity
2.3. Effect of Temperature and pH on Enzyme Activity and Stability
2.4. Steady-State Kinetic Parameters of the Overall Transamination Reactions
2.5. The Overall Structure of Halhy
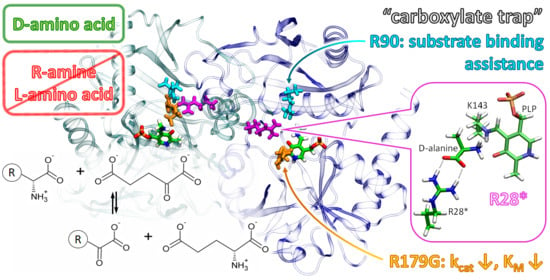
2.6. The Active Site of Halhy
2.7. Characterization of the R179G Variant
2.8. D-Alanine Binding in the Active Site of the WT Halhy and Its R179G Variant
3. Discussion
4. Materials and Methods
4.1. Cloning, Expression, and Purification of the Recombinant Halhy and Its R179G Variant
4.2. Enzyme Activity Assay
4.3. Effect of pH and Temperature on the Transamination Reaction
4.4. Analysis of Halhy Thermal Stability
4.5. Analysis of the Product Yield and Enantiomeric Excess in the Transamination Reaction
4.6. Crystallization and Data Collection
4.7. Structure Solution and Refinement
4.8. Molecular Modeling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Appendix A

References
- Eliot, A.C.; Kirsch, J.F. Pyridoxal phosphate enzymes: Mechanistic, structural, and evolutionary considerations. Annu. Rev. Biochem. 2004, 73, 383–415. [Google Scholar] [CrossRef] [Green Version]
- Toney, M.D. Controlling reaction specificity in pyridoxal phosphate enzymes. Biochim. Biophys. Acta 2011, 1814, 1407–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peisach, D.; Chipman, D.M.; Van Ophem, P.W.; Manning, J.M.; Ringe, D. Crystallographic study of steps along the reaction pathway of D-amino acid aminotransferase. Biochemistry 1998, 37, 4958–4967. [Google Scholar] [CrossRef] [PubMed]
- Bezsudnova, E.Y.; Popov, V.O.; Boyko, K.M. Structural insight into the substrate specificity of PLP fold type IV transaminases. Appl. Microbiol. Biotechnol. 2020, 104, 2343–2357. [Google Scholar] [CrossRef] [PubMed]
- Steffen-Munsberg, F.; Vickers, C.; Kohls, H.; Land, H.; Mallin, H.; Nobili, A.; Skalden, L.; van den Bergh, T.; Joosten, H.-J.; Berglund, P.; et al. Bioinformatic analysis of a PLP-dependent enzyme superfamily suitable for biocatalytic applications. Biotechnol. Adv. 2015, 33, 566–604. [Google Scholar] [CrossRef]
- Höhne, M.; Schätzle, S.; Jochens, H.; Robins, K.; Bornscheuer, U.T. Rational assignment of key motifs for function guides in silico enzyme identification. Nat. Chem. Biol. 2010, 6, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Pavkov-Keller, T.; Strohmeier, G.A.; Diepold, M.; Peeters, W.; Smeets, N.; Schürmann, M.; Gruber, K.; Schwab, H.; Steiner, K. Discovery and structural characterisation of new fold type IV-transaminases exemplify the diversity of this enzyme fold. Sci. Rep. 2016, 6, 38183. [Google Scholar] [CrossRef] [Green Version]
- Bezsudnova, E.Y.; Boyko, K.M.; Nikolaeva, A.Y.; Zeifman, Y.S.; Rakitina, T.V.; Suplatov, D.A.; Popov, V.O. Biochemical and structural insights into PLP fold type IV transaminase from Thermobaculum terrenum. Biochimie 2019, 158, 130–138. [Google Scholar] [CrossRef]
- Zeifman, Y.S.; Boyko, K.M.; Nikolaeva, A.Y.; Timofeev, V.I.; Rakitina, T.V.; Popov, V.O.; Bezsudnova, E.Y. Functional characterization of PLP fold type IV transaminase with a mixed type of activity from Haliangium ochraceum. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 575–585. [Google Scholar] [CrossRef]
- Radkov, A.D.; Moe, L.A. Bacterial synthesis of D-amino acids. Appl. Microbiol. Biotechnol. 2014, 98, 5363–5374. [Google Scholar] [CrossRef]
- Grishin, D.V.; Zhdanov, D.D.; Pokrovskaya, M.V.; Sokolov, N.N. D-amino acids in nature, agriculture and biomedicine. All Life 2020, 13, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Yonaha, K.; Misono, H.; Yamamoto, T.; Soda, K. D-amino acid aminotransferase of Bacillus sphaericus. Enzymologic and spectrometric properties. J. Biol. Chem. 1975, 250, 6983–6989. [Google Scholar] [CrossRef]
- Tanizawa, K.; Masus, Y.; Asano, S.; Tanaka, H.; Sodas, K. Thermostable D-amino acid aminotransferase from a thermophilic Bacillus Species. Purification, characterization, and active site sequence determination. J. Biol. Chem. 1989, 264, 2445–2449. [Google Scholar] [CrossRef]
- Van Ophem, P.W.; Peisach, D.; Erickson, S.D.; Soda, K.; Ringe, D.; Manning, J.M. Effects of the E177K mutation in D-amino acid transaminase. Studies on an essential coenzyme anchoring group that contributes to stereochemical fidelity. Biochemistry 1999, 38, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-G.; Hong, S.-P.; Song, J.J.; Kim, S.-J.; Kwak, M.-S.; Sung, M.-H. Functional and structural characterization of thermostable D-amino acid aminotransferases from Geobacillus spp. Appl. Environ. Microbiol. 2006, 72, 1588–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, J.; Shimizu, Y.; Mutaguchi, Y.; Doi, K.; Ohshima, T. Characterization of D-amino acid aminotransferase from Lactobacillus salivarius. J. Mol. Catal. B Enzym. 2013, 94, 15–22. [Google Scholar] [CrossRef]
- Sugio, S.; Petsko, G.A.; Manning, J.M.; Soda, K.; Ringe, D. Crystal structure of a D-amino acid aminotransferase: How the protein controls stereoselectivity. Biochemistry 1995, 34, 9661–9669. [Google Scholar] [CrossRef] [PubMed]
- Park, E.-S.; Dong, J.-Y.; Shin, J.-S. Biocatalytic asymmetric synthesis of unnatural amino acids through the cascade transfer of amino groups from primary amines onto keto acids. ChemCatChem 2013, 5, 3538–3542. [Google Scholar] [CrossRef]
- Zhou, H.; Meng, L.; Yin, X.; Liu, Y.; Xu, G.; Wu, J.; Wu, M.; Yang, L. Artificial biocatalytic cascade with three enzymes in one pot for asymmetric synthesis of chiral unnatural amino acids. Eur. J. Org. Chem. 2019, 2019, 6470–6477. [Google Scholar] [CrossRef]
- Parmeggiani, F.; Rué Casamajo, A.; Walton, C.J.W.; Galman, J.L.; Turner, N.J.; Chica, R.A. One-pot biocatalytic synthesis of substituted D-tryptophans from indoles enabled by an engineered aminotransferase. ACS Catal. 2019, 9, 3482–3486. [Google Scholar] [CrossRef]
- Silva, M.V.D.M.; Costa, I.C.R.; de Souza, R.O.M.A.; Bornscheuer, U.T. Biocatalytic cascade reaction for the asymmetric synthesis of L- and D-homoalanine. ChemCatChem 2019, 11, 407–411. [Google Scholar] [CrossRef]
- Walton, C.J.W.; Parmeggiani, F.; Barber, J.E.B.; McCann, J.L.; Turner, N.J.; Chica, R.A. Engineered aminotransferase for the production of D-phenylalanine derivatives using biocatalytic cascades. ChemCatChem 2018, 10, 470–474. [Google Scholar] [CrossRef]
- Daligault, H.; Lapidus, A.; Zeytun, A.; Nolan, M.; Lucas, S.; Del Rio, T.G.; Tice, H.; Cheng, J.-F.; Tapia, R.; Han, C.; et al. Complete genome sequence of Haliscomenobacter hydrossis type strain (OT). Stand. Genom. Sci. 2011, 4, 352–360. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Kuramitsu, S.; Aki, K.; Watanabe, Y.; Takagi, T.; Nishigai, M.; Ikai, A.; Kagamiyama, H. Branched-chain amino acid aminotransferase of Escherichia coli: Overproduction and properties. J. Biochem. 1988, 104, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Wybenga, G.G.; Crismaru, C.G.; Janssen, D.B.; Dijkstra, B.W. Structural determinants of the β-selectivity of a bacterial aminotransferase. J. Biol. Chem. 2012, 287, 28495–28502. [Google Scholar] [CrossRef] [Green Version]
- Ro, H.S.; Hong, S.P.; Seo, H.J.; Yoshimura, T.; Esaki, N.; Soda, K.; Kim, H.S.; Sung, M.H. Site-directed mutagenesis of the amino acid residues in beta-strand III [Val30-Val36] of D-amino acid aminotransferase of Bacillus sp. YM-1. FEBS Lett. 1996, 398, 141–145. [Google Scholar] [CrossRef] [Green Version]
- Voss, M.; Xiang, C.; Esque, J.; Nobili, A.; Menke, M.J.; André, I.; Höhne, M.; Bornscheuer, U.T. Creation of (R)-amine transaminase activity within an α-amino acid transaminase scaffold. ACS Chem. Biol. 2020, 15, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Boyko, K.; Gorbacheva, M.; Rakitina, T.; Korzhenevskiy, D.; Vanyushkina, A.; Kamashev, D.; Lipkin, A.; Popov, V. Expression, purification, crystallization and preliminary X-ray crystallographic analysis of the histone-like HU protein from Spiroplasma melliferum KC3. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2015, 71, 24–27. [Google Scholar] [CrossRef] [Green Version]
- Pace, C.N.; Vajdos, F.; Fee, L.; Grimsley, G.; Gray, T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995, 4, 2411–2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarova, O.; Kamberov, E.; Margolis, B. Generation of deletion and point mutations with one primer in a single cloning step. Biotechniques 2000, 29, 970–972. [Google Scholar] [CrossRef] [Green Version]
- Mikhailova, A.G.; Rakitina, T.V.; Timofeev, V.I.; Karlinsky, D.M.; Korzhenevskiy, D.A.; Agapova, Y.K.; Vlaskina, A.V.; Ovchinnikova, M.V.; Gorlenko, V.A.; Rumsh, L.D. Activity modulation of the oligopeptidase B from Serratia proteamaculans by site-directed mutagenesis of amino acid residues surrounding catalytic triad histidine. Biochimie 2017, 139, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Boyko, K.M.; Stekhanova, T.N.; Nikolaeva, A.Y.; Mardanov, A.V.; Rakitin, A.L.; Ravin, N.V.; Bezsudnova, E.Y.; Popov, V.O. First structure of archaeal branched-chain amino acid aminotransferase from Thermoproteus uzoniensis specific for L-amino acids and R-amines. Extremophiles 2016, 20, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Schätzle, S.; Höhne, M.; Redestad, E.; Robins, K.; Bornscheuer, U.T. Rapid and sensitive kinetic assay for characterization of omega-transaminases. Anal. Chem. 2009, 81, 8244–8248. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.-S.; Lee, S.-G.; Hong, S.-P.; Kwak, M.-S.; Esaki, N.; Soda, K.; Sung, M.-H. Production of aromatic D-amino acids from α-keto acids and ammonia by coupling of four enzyme reactions. J. Mol. Catal. B Enzym. 1999, 6, 241–247. [Google Scholar] [CrossRef]
- Beilsten-Edmands, J.; Winter, G.; Gildea, R.; Parkhurst, J.; Waterman, D.; Evans, G. Scaling diffraction data in the DIALS software package: Algorithms and new approaches for multi-crystal scaling. Acta Crystallogr. Sect. D Struct. Biol. 2020, 76, 385–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, P.R. An introduction to data reduction: Space-group determination, scaling and intensity statistics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 282–292. [Google Scholar] [CrossRef] [Green Version]
- Vagin, A.A.; Isupov, M.N. Spherically averaged phased translation function and its application to the search for molecules and fragments in electron-density maps. Acta Crystallogr. Sect. D Biol. Crystallogr. 2001, 57, 1451–1456. [Google Scholar] [CrossRef] [Green Version]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP 4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Krissinel, E.; Henrick, K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2256–2268. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the additive CHARMM all-atom protein force field targeting Improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denning, E.J.; Priyakumar, U.D.; Nilsson, L.; Mackerell, A.D. Impact of 2′-hydroxyl sampling on the conformational properties of RNA: Update of the CHARMM all-atom additive force field for RNA. J. Comput. Chem. 2011, 32, 1929–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2009, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TA, (Numbering Corresponds to) | Specificity-Determining Sequence Motifs | |
|---|---|---|
| Motif 1 | Motif 2 | |
| DAAT, (DAAT from Bacillus sp. YM-1) | 26FxxxxYxV[IVA][KR]35 | 86HxY88…98[RK]xH100 |
| BCAT, (BCAT from Escherichia coli) | 31YxxxxF[ED]Gx[KR]40 | 95YxR97…107[LMVI]G[VL]109 |
| R-TA, (R-TA from Aspergillus terrus) | 55HxxxxYD[VT]x[STAHP]64 | 115[FY]V[EQAWNS]117… 128[RKFGP]x[STANER]130 |
| TA from C. pusillum | 51RxxxxFETIA60 | 115GxK117…123GIEGEGR129 |
| Halhy | 28RxxxxFDYFL57 | 88GxR90…96GYSPDGYTPVN106 |
| Amino Donor | Amino Acceptor | Initial Activity, U/mg | Assay |
|---|---|---|---|
| D-alanine | α-ketoglutarate | 50 ± 3 | LDH |
| D-leucine | 0.70 ± 0.08 | HPLC | |
| L-alanine | ND | LDH | |
| L-leucine | ND | HPLC | |
| (S)-PEA | ND | Acetophenone assay | |
| (R)-PEA | ND | Acetophenone assay | |
| D-glutamate | pyruvate | 74 ± 2 | HGDH |
| (S)-PEA | ND | Acetophenone assay | |
| (R)-PEA | ND | Acetophenone assay |
| α-Keto Acid | Initial Activity, U/mg |
|---|---|
| pyruvate | 74 ± 2 |
| α-ketoglutarate | 50 ± 3 (with 5 mM D-alanine) |
| 2-oxobutyrate | 42 ± 2 |
| 2-oxovalerate | 15.0 ± 0.4 |
| 2-oxohexanoate | 0.320 ± 0.005 |
| 3-metyl-2-oxovalerate | 0.085 ± 0.004 |
| 4-metyl-2-oxovalerate | 0.220 ± 0.007 |
| phenylpyruvate | 1.88 ± 0.02 |
| Substrate | Cosubstrate | kcat, c−1 | Km, mM | kcat/Km, c−1 M −1 |
|---|---|---|---|---|
| Wild-type Halhy | ||||
| α-ketoglutarate | D-alanine 40 mM | 146 ± 4 | 2.3 ± 0.2 | 63,000 ± 7000 |
| D-alanine | α-ketoglutarate 5 mM | 146 ± 4 | 23 ± 1 | 6300 ± 400 |
| pyruvate | D-glutamate 4 mM | 215 ± 6 | 2.1 ± 0.1 | 103,000 ± 8000 |
| D-glutamate | pyruvate 2.5 mM | 215 ± 6 | 10.3 ± 0.7 | 21,000 ± 2000 |
| D-leucine | α-ketoglutarate 5 mM | 10.5 ± 0.4 | 83 ± 8 | 130 ± 20 |
| 4-methyl-2-oxovalerate | D-alanine 40 mM | 4.2 ± 0.1 | 70 ± 6 | 60 ± 7 |
| R179G variant | ||||
| α-ketoglutarate | D-alanine 40 mM | 8.6 ± 0.6 | 8.1 ± 0.8 | 1100 ± 200 |
| D-alanine | α-ketoglutarate 5 mM | 8.6 ± 0.6 | 321 ± 32 | 27 ± 5 |
| Diffraction Source | ID30A-3, ESRF |
|---|---|
| Wavelength (Å) | 0.97 |
| Temperature (K) | 100 |
| Detector | Eiger X 4M |
| Crystal-to-detector distance (mm) | 131.78 |
| Rotation range per image (°) | 0.2 |
| Total rotation range (°) | 160 |
| Space group | C2 |
| a, b, c (Å) | 86.88, 71.75, 52.99 |
| α, β, γ (°) | 90.0, 100.96, 90.0 |
| Resolution range (Å) | 52.03–2.00 (2.04-2.00) |
| Number of unique reflections | 21,396 (1419) |
| Completeness (%) | 97.4 (92.5) |
| Average redundancy | 3.0 (3.2) |
| 〈I/σ(I)〉 | 5.2 (1.9) |
| Rmeas (%) (Diederichs and Karplus 1997) | 13.4 (57.6) |
| CC1/2 (Diederichs and Karplus 1997) | 98.7 (87.1) |
| Number of reflections used for the refinement | 20,186 |
| Rfact (%) | 18.0 |
| Rfree (%) | 24.4 |
| Bonds (Å) | 0.02 |
| Angles (°) | 1.95 |
| Average B-factors (Å2) | |
| Protein | 27.9 |
| Water | 30.5 |
| PLP | 25.5 |
| Ramachandran plot | |
| Most favoured (%) | 92.5 |
| Allowed (%) | 5.7 |
| PDB entry code | 7P7X |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakunova, A.K.; Nikolaeva, A.Y.; Rakitina, T.V.; Isaikina, T.Y.; Khrenova, M.G.; Boyko, K.M.; Popov, V.O.; Bezsudnova, E.Y. The Uncommon Active Site of D-Amino Acid Transaminase from Haliscomenobacter hydrossis: Biochemical and Structural Insights into the New Enzyme. Molecules 2021, 26, 5053. https://doi.org/10.3390/molecules26165053
Bakunova AK, Nikolaeva AY, Rakitina TV, Isaikina TY, Khrenova MG, Boyko KM, Popov VO, Bezsudnova EY. The Uncommon Active Site of D-Amino Acid Transaminase from Haliscomenobacter hydrossis: Biochemical and Structural Insights into the New Enzyme. Molecules. 2021; 26(16):5053. https://doi.org/10.3390/molecules26165053
Chicago/Turabian StyleBakunova, Alina K., Alena Yu. Nikolaeva, Tatiana V. Rakitina, Tatiana Y. Isaikina, Maria G. Khrenova, Konstantin M. Boyko, Vladimir O. Popov, and Ekaterina Yu. Bezsudnova. 2021. "The Uncommon Active Site of D-Amino Acid Transaminase from Haliscomenobacter hydrossis: Biochemical and Structural Insights into the New Enzyme" Molecules 26, no. 16: 5053. https://doi.org/10.3390/molecules26165053