
First-Principles Study of Bi-Doping Effects in Hg0.75Cd0.25Te

(This article belongs to the Section Materials Chemistry)
Abstract
:1. Introduction
2. Results and Discussion
2.1. Atomic Relaxations
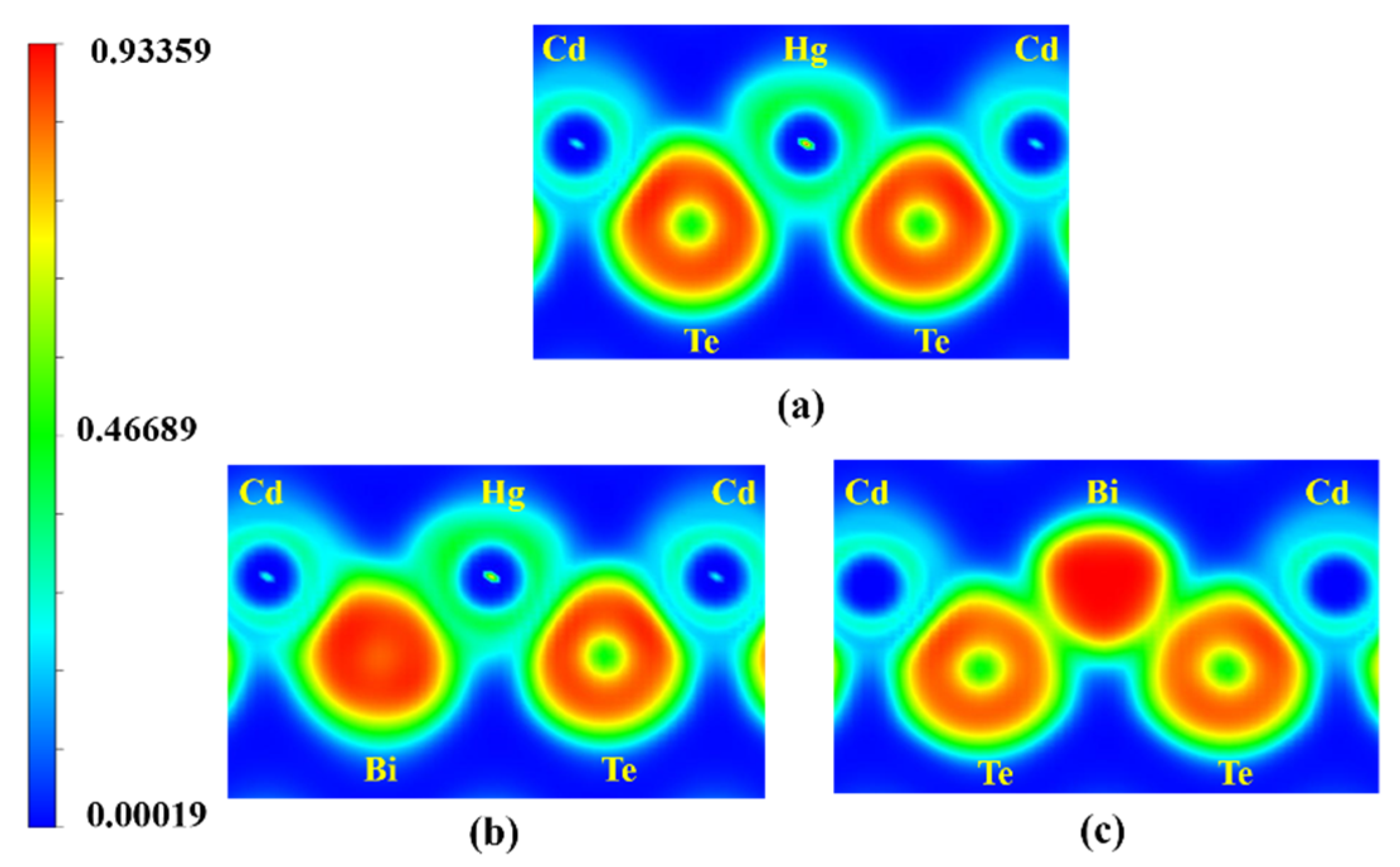
2.2. Bonding Mechanism
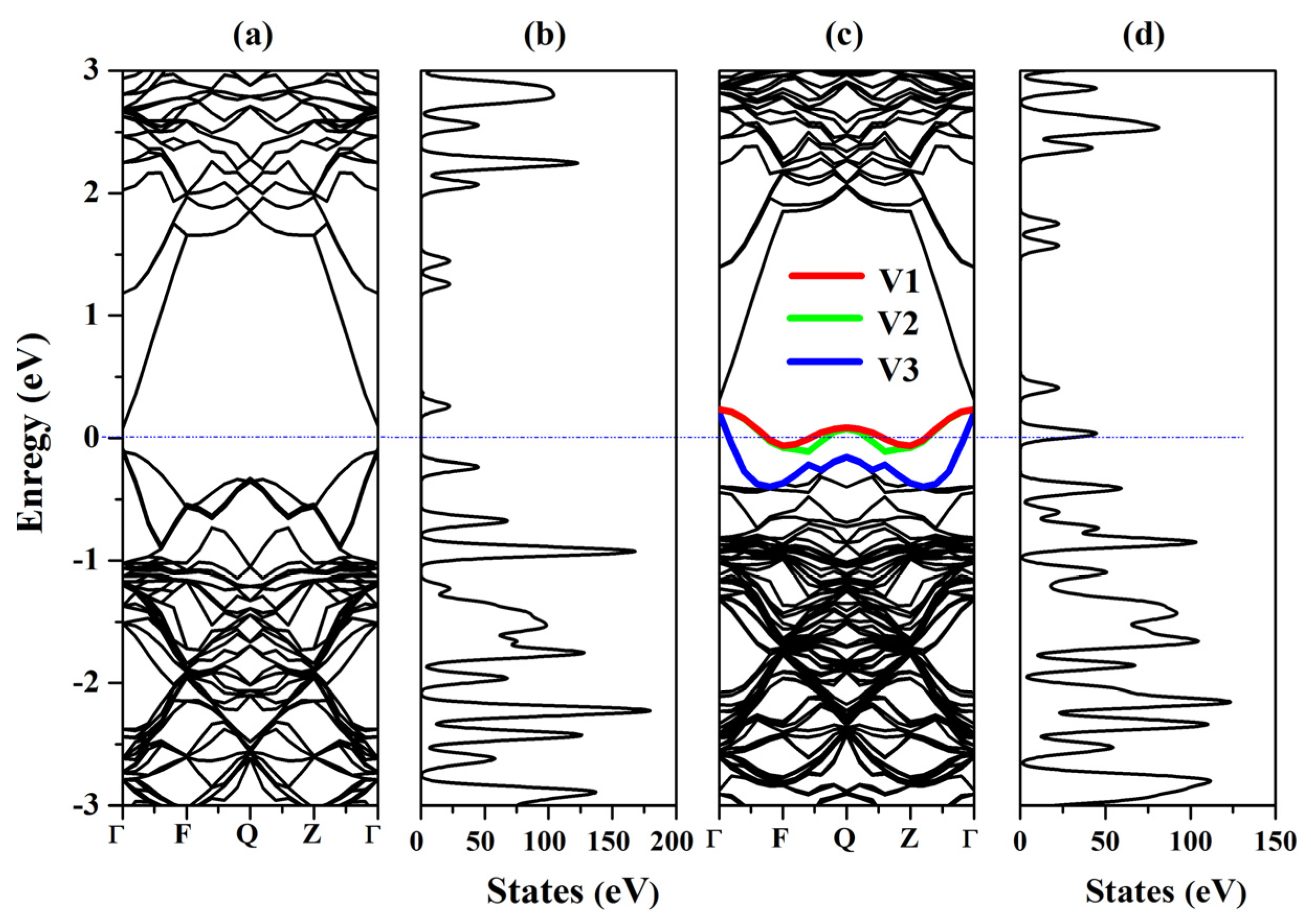
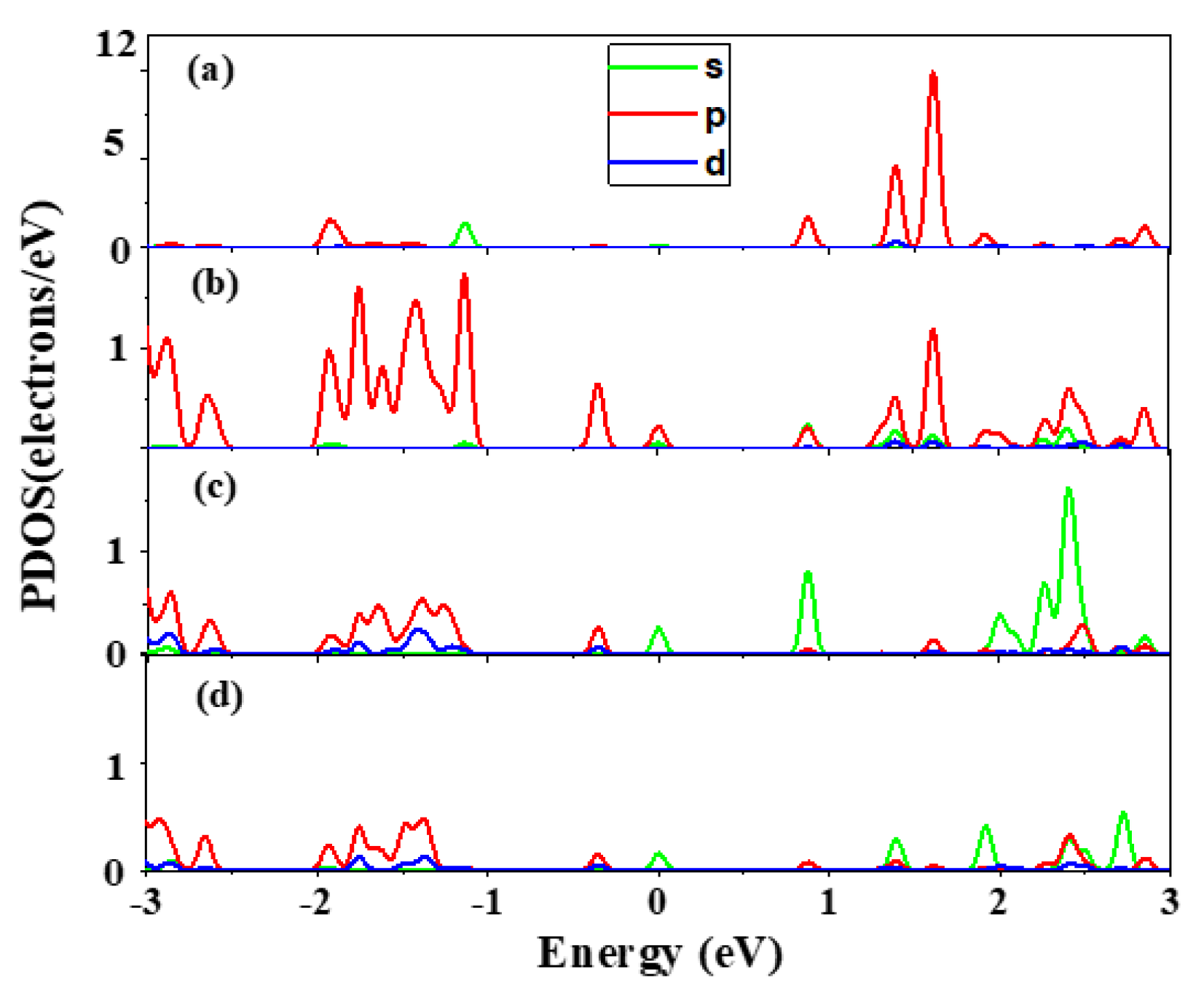
2.3. Electronic Properties
3. Materials and Methods
4. Conclusions
- The covalent radii difference between dopant cation Bi and host atoms (Te and Hg) causes the relaxations of both bond lengths and bond angles;
- The results of charge density and ELFs indicate that the Bi impurity maintains relatively strong bonding characteristics with the host atom in Hg0.75Cd0.25Te;
- The impurity Bi shows a complicated amphoteric behavior in Hg0.75Cd0.25Te, which in situ substitutes the cation Hg behavior as n-type and in situ substitutes the anion Te behavior as p-type Hg0.75Cd0.25Te.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Rogalski, A. Toward third generation HgCdTe infrared detectors. J. Alloys Compd. 2004, 371, 53–57. [Google Scholar] [CrossRef]
- Rehm, R.; Walther, M.; Fleissner, J.; Schmitz, J.; Ziegler, J.; Cabanski, W.; Breiter, R. Bispectral thermal imaging with quantum well infrared photodetectors and InAs/GaSb type-II superlattices. In Infrared Technology and Applications XXXII, Proceedings of the Defense and Security Symposium, Orlando, FL, USA, 17 May 2006; SPIE DIGIT LIBRARY: Bellingham, WA, USA, 2006; Volume 6206, p. 62060Y. [Google Scholar]
- Rogalski, A. Material considerations for third generation infrared photon detectors. Infrared Phys. Technol. 2007, 50, 240–252. [Google Scholar] [CrossRef]
- Rogalski, A. HgCdTe infrared detector material: History, status and outlook. Rep. Prog. Phys. 2005, 68, 2267–2336. [Google Scholar] [CrossRef] [Green Version]
- Ghandhi, S.K.; Taskar, N.R.; Parat, K.K.; Bhat, I.B. Indium doping of n-type HgCdTe layers grown by organometallic vapor phase epitaxy. Appl. Phys. Lett. 1990, 57, 252–254. [Google Scholar] [CrossRef]
- Sun, L.Z.; Chen, X.S.; Sun, Y.L.; Zhou, X.H.; Quan, Z.J.; Duan, H.; Lu, W. Relaxations and bonding mechanism in Hg1−xCdxTe with mercury vacancy defect: First-principles study. Phys. Rev. B 2006, 73, 195206. [Google Scholar] [CrossRef]
- Berding, M.A.; Sher, A.; Chen, A.-B. Vacancy formation and extraction energies in semiconductor compounds and alloys. J. Appl. Phys. 1990, 68, 5064–5076. [Google Scholar] [CrossRef]
- Berding, M.A.; Van Schilfgaarde, M.; Sher, A. First-principles calculation of native defect densities in Hg0.8Cd0.2Te. Phys. Rev. B 1994, 50, 1519–1534. [Google Scholar] [CrossRef]
- Vydyanath, H.R.; Ellsworth, J.A.; Devaney, C.M. Electrical activity, mode of incorporation and distribution coefficient of group V elements in Hg1−xCdxTe grown from tellurium rich liquid phase epitxial growth solutions. J. Electron. Mater. 1987, 16, 13–25. [Google Scholar] [CrossRef]
- Vydyanath, H.R.; Abbott, R.C.; Nelson, D.A. Mode of incorporation of phosphorus in Hg0.8Cd0.2Te. J. Appl. Phys. 1983, 54, 1323. [Google Scholar] [CrossRef]
- Sivananthan, S.; Wijewarnasuriya, P.S.; Aqariden, F.; Vydyanath, H.R.; Zandian, M.; Edwall, D.D.; Arias, J.M. Mode of arsenic incorporation in HgCdTe grown by MBE. J. Electron. Mater. 1997, 26, 621–624. [Google Scholar] [CrossRef]
- Capper, P. The behaviour of selected impurities in CdxHg1−xTe. J. Cryst. Growth 1982, 57, 280–299. [Google Scholar] [CrossRef]
- Vydyanath, H.R. Amphoteric behaviour of group V dopants in (Hg, Cd)Te. Semicond. Sci. Technol. 1990, 5, S213–S216. [Google Scholar] [CrossRef]
- Li, D.C.; Yang, M.; Zhao, S.Z.; Cai, Y.Q.; Feng, Y.P. First principles study of Bismuth alloying effects in GaAs saturable ab-sorber. Opt. Express 2012, 20, 11574–11580. [Google Scholar] [CrossRef] [PubMed]
- Li, D.C.; Yang, M.; Zhao, S.Z.; Cai, Y.Q.; Lu, Y.H.; Bai, Z.Q.; Feng, Y.P. First-principles study of the effect of Bi-Ga heteroan-tisites in GaAs:Bi alloy. Comput. Mater. Sci. 2012, 63, 178–181. [Google Scholar] [CrossRef]
- Li, D.; Yang, M.; Cai, Y.; Zhao, S.; Feng, Y. First principles study of the ternary complex model of EL2 defect in GaAs saturable absorber. Opt. Express 2012, 20, 6258–6266. [Google Scholar] [CrossRef]
- Savin, A.; Jepsen, O.; Flad, J.; Andersen, O.K.; Preuss, H.; Von Schnering, H.G. Electron Localization in Solid-State Structures of the Elements: The Diamond Structure. Angew. Chem. Int. Ed. 1992, 31, 187–188. [Google Scholar] [CrossRef]
- Wang, Q.X.; Yang, J.R.; Sun, T.; Wei, Y.F.; Fang, W.Z.; He, L. Relationship between lattice parameters and compositions of molecular beam epitaxial Hg1−xCdxTe films. Acta Phys. Sin. 2005, 54, 3726–3733. [Google Scholar]
- Lento, J.; Mozos, J.L.; Nieminen, R.M. Charged point defects in semiconductors and the supercell approximation. J. Phys. Condens. Matter 2002, 14, 2637–2645. [Google Scholar]
- Simak, S.I.; Häußermann, U.; Abrikosov, I.A.; Eriksson, O.; Wills, J.M.; Lidin, S.; Johansson, B. Stability of the Anomalous Large-Void CoSn Structure. Phys. Rev. Lett. 1997, 79, 1333–1336. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.Z.; Chen, X.S.; Sun, Y.L.; Zhou, X.H.; Quan, Z.J.; Duan, H.; Lu, W. Electronic properties of the Au impurity in Hg0.75Cd0.25Te. First-Principles Study. Phys. B 2009, 404, 131–137. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Defect | Bond | Before (Å) | After (Å) | Change (Å) | Change Ratio (Å) |
|---|---|---|---|---|---|
| BiTe | Bi-Hg1 | 2.802 | 2.783 (0) | −0.019 | −0.68% |
| Bi-Cd1 | 2.794 (0) | 2.797 (0) | +0.003 | +0.11% | |
| Hg1-Cd1 | 4.572 | 4.559 | −0.013 | −0.29% | |
| BiHg | Te1-Bi | 2.802 | 2.991 | +0.189 | +6.31% |
| Cd2-Bi | 4.572 | 4.632 | +0.06 | +0.13% | |
| Hg2-Bi | 4.572 | 4.640 | +0.068 | +0.15% | |
| Te1-Hg2 | 2.802 | 2.833 | +0.031 | +1.1% | |
| Te1-Cd2 | 2.794 | 2.810 | +0.015 | +0.05% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Su, X.; Li, D.; Cao, L. First-Principles Study of Bi-Doping Effects in Hg0.75Cd0.25Te. Molecules 2021, 26, 4847. https://doi.org/10.3390/molecules26164847
Sun X, Su X, Li D, Cao L. First-Principles Study of Bi-Doping Effects in Hg0.75Cd0.25Te. Molecules. 2021; 26(16):4847. https://doi.org/10.3390/molecules26164847
Chicago/Turabian StyleSun, Xueli, Xuejun Su, Dechun Li, and Lihua Cao. 2021. "First-Principles Study of Bi-Doping Effects in Hg0.75Cd0.25Te" Molecules 26, no. 16: 4847. https://doi.org/10.3390/molecules26164847