
Design, Synthesis, Biological Evaluation, and Computational Studies of Novel Tri-Aryl Imidazole-Benzene Sulfonamide Hybrids as Promising Selective Carbonic Anhydrase IX and XII Inhibitors

 , , and
, , and
Abstract
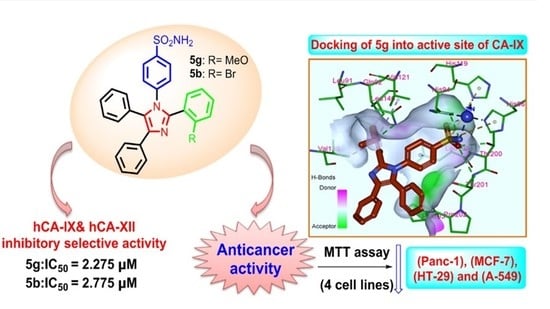
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biology
2.2.1. Structure Activity Relationship Study
2.2.2. In Vitro Anticancer Activity
2.3. Docking Study
3. Materials and Methods
3.1. Chemistry
General Procedure for Synthesis of 4-(4,5-Diphenyl-2-(substituted benzene)-1H-imidazol-1-yl)benzene Sulfonamides 5a–n
3.2. Biology
3.2.1. Carbonic Anhydrase Assay
3.2.2. Cell Viability Assay
3.2.3. Antiproliferative Activity
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Appendix A. Experimental
Appendix A.1. Chemistry
Appendix A.2. In Vitro Carbonic Anhydrase Inhibitory Assay
Appendix A.3. Cytotoxic Activity Using MTT Assay and Evaluation of IC50
Appendix A.3.1. MTT Assay
Appendix A.3.2. Assay for Antiproliferative Effect
Appendix A.4. Statistical Analysis
References
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef]
- Nishimori, I.; Minakuchi, T.; Onishi, S.; Vullo, D.; Cecchi, A.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors: Cloning, characterization, and inhibition studies of the cytosolic isozyme III with sulphonamides. Bioorg. Med. Chem. 2007, 15, 7229–7236. [Google Scholar] [CrossRef]
- Supuran, C.T.; Scozzafava, A.; Casini, A. Carbonic anhydrase inhibitors. Med. Res. Rev. 2003, 23, 146–189. [Google Scholar] [CrossRef]
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef]
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Multiple binding modes of inhibitors to carbonic anhydrases: How to design specific drugs targeting 15 different isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef] [Green Version]
- Supuran, C.T.; Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Carta, F.; Monti, S.M.; De Simone, G. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: Three for the price of one. Med. Res. Rev. 2018, 38, 1799–1836. [Google Scholar] [CrossRef]
- Waheed, A.; Sly, W.S. Carbonic anhydrase XII functions in health and disease. Gene 2017, 623, 33–40. [Google Scholar] [CrossRef]
- Proescholdt, M.A.; Merrill, M.J.; Stoerr, E.-M.; Lohmeier, A.; Pohl, F.; Brawanski, A. Function of carbonic anhydrase IX in glioblastoma multiforme. Neuro-Oncology 2012, 14, 1357–1366. [Google Scholar] [CrossRef]
- Dorai, T.; Sawczuk, I.S.; Pastorek, J.; Wiernik, P.H.; Dutcher, J.P. The role of carbonic anhydrase IX overexpression in kidney cancer. Eur. J. Cancer 2005, 41, 2935–2947. [Google Scholar] [CrossRef]
- Robertson, N.; Potter, C.; Harris, A.L. Role of carbonic anhydrase IX in human tumor cell growth, survival, and invasion. Cancer Res. 2004, 64, 6160–6165. [Google Scholar] [CrossRef] [Green Version]
- Wingo, T.; Tu, C.; Laipis, P.J.; Silverman, D.N. The catalytic properties of human carbonic anhydrase IX. Biochem. Biophys. Res. Commun. 2001, 288, 666–669. [Google Scholar] [CrossRef]
- Barnett, D.H.; Sheng, S.; Charn, T.H.; Waheed, A.; Sly, W.S.; Lin, C.-Y. Estrogen receptor regulation of carbonic anhydrase XII through a distal enhancer in breast cancer. Cancer Res. 2008, 68, 3505–3515. [Google Scholar] [CrossRef] [Green Version]
- Melissa, A.P.; Brian, M.; Robert, M. Probing the Surface of Human Carbonic Anhydrase for Clues towards the Design of Isoform Specific Inhibitors. BioMed Res. Int. 2015, 2015, 453543. [Google Scholar]
- Bozdag, M.; Ferraroni, M.; Nuti, E.; Vullo, D.; Rossello, A.; Carta, F.; Scozzafava, A.; Supuran, C.T. Combining the tail and the ring approaches for obtaining potent and isoform-selective carbonic anhydrase inhibitors: Solution and X-ray crystallographic studies. Bioorg. Med. Chem. 2014, 22, 334–340. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/ct2/show/NCT02215850 (accessed on 13 August 2014).
- Available online: https://clinicaltrials.gov/ct2/show/NCT03450018 (accessed on 1 March 2018).
- Congiu, C.; Onnis, V.; Balboni, G.; Supuran, C.T. Synthesis and carbonic anhydrase I, II, IX and XII inhibition studies of 4-N,N-disubstituted sulfanilamides incorporating 4,4,4-trifluoro-3-oxo-but-1-enyl, phenacyl thiourea and imidazol-2 (3H)-one/thione moieties. Bioorg. Med. Chem. Lett. 2014, 24, 1776–1779. [Google Scholar] [CrossRef]
- Georgey, H.H.; Manhi, F.M.; Mahmoud, W.R.; Mohamed, N.A.; Berrino, E.; Supuran, C.T. 1,2,4-Trisubstituted imidazolinones with dual carbonic anhydrase and p38 mitogen-activated protein kinase inhibitory activity. Bioorg. Chem. 2019, 82, 109–116. [Google Scholar] [CrossRef]
- Khalifah, R.G. The carbon dioxide hydration activity of carbonic anhydrase I. Stop flow kinetic studies on the native human isoenzymes B and C. J. Biol. Chem. 1971, 246, 2561–2573. [Google Scholar] [CrossRef]
- Elbastawesy, M.A.I.; Aly, A.A.; Ramadan, M.Y.; Elshaier, A.M.M.; Youssif, G.M.B.; Brown, A.B.; Abuo-Rahma, G.E. A Novel Pyrazoloquinolin-2-ones: Design, Synthesis, Docking Studies, and Biological Evaluation as Antiproliferative EGFR- TK Inhibitors. Bioorg. Chem. 2019, 90, 103045. [Google Scholar] [CrossRef]
- Al-Wahaibi, L.H.; Gouda, A.M.; Abou-Ghadir, O.F.; Salem, O.I.; Ali, A.T.; Farghaly, H.S.; Abdelrahman, M.H.; Trembleau, L.; Abdu-Allah, H.H.; Youssif, B.G. Design and synthesis of novel 2,3-dihydropyrazino[1,2-a]indole-1,4-dione derivatives as antiproliferative EGFR and BRAFV600E dual inhibitors. Bioorg. Chem. 2020, 104, 104260. [Google Scholar] [CrossRef]
- Youssif, B.G.; Mohamed, A.M.; Osman, E.E.A.; Abou-Ghadir, O.F.; Elnaggar, D.H.; Abdelrahman, M.H.; Treamblu, L.; Gomaa, H.A. 5-Chlorobenzofuran-2-carboxamides: From allosteric CB1 modulators to potential apoptotic antitumor agents. Eur. J. Med. Chem. 2019, 177, 1–11. [Google Scholar] [CrossRef]
- Abdelbaset, M.S.; Abdel-Aziz, M.; Abuo-Rahma, G.E.D.A.; Abdelrahman, M.H.; Ramadan, M.; Youssif, B.G. Novel quinoline derivatives carrying nitrones/oximes nitric oxide donors: Design, synthesis, antiproliferative and caspase-3 activation activities. Arch. Pharm. 2019, 352, 1800270. [Google Scholar] [CrossRef] [Green Version]
- Salmas, R.E.; Senturk, M.; Yurtsever, M.; Durdagi, S. Discovering novel carbonic anhydrase type IX (CA IX) inhibitors from seven million compounds using virtual screening and in vitro analysis. J. Enzym. Inhib. Med. Chem. 2016, 31, 425–433. [Google Scholar]
- Alterio, V.; Hilvo, M.; Di Fiore, A.; Supuran, C.T.; Pan, P.; Parkkila, S.; Scaloni, A.; Pastorek, J.; Pastorekova, S.; Pedone, C.; et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc. Natl. Acad. Sci. USA 2009, 106, 16233–16238. [Google Scholar] [CrossRef] [Green Version]
- Leitans, J.; Kazaks, A.; Balode, A.; Ivanova, J.; Zalubovskis, R.; Supuran, C.T.; Tars, K. Efficient Expression and Crystallization System of Cancer-Associated Carbonic Anhydrase Isoform IX. J. Med. Chem. 2015, 58, 9004–9009. [Google Scholar] [CrossRef]
- El-Sherief, H.A.M.; Youssif, B.G.M.; Bukhari, S.N.A.; Abdelazeem, A.H.; Abdel-Aziz, M.; Abdel-Rahman, H.M. Synthesis, anticancer activity and molecular modeling studies of 1,2,4-triazole derivatives as EGFR inhibitors. Eur. J. Med. Chem. 2018, 156, 774–789. [Google Scholar] [CrossRef]
- Abdelatef, S.A.; El-Saadi, M.T.; Amin, N.H.; Abdelazeem, A.H.; Omar, H.A.; Abdellatif, K.R.A. Design, synthesis and anticancer evaluation of novel spirobenzo[h]chromene and spirochromane derivatives with dual EGFR and B-RAF inhibitory activities. Eur. J. Med. Chem. 2018, 150, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Dassault Systems. Dassault Systems BIOVIA, Discovery Studio Visualizer, v16.1.0.15350; Dassault Systems: San Diego, CA, USA, 2016. [Google Scholar]
- Parkkila, S.; Innocenti, A.; Kallio, H.; Hilvo, M.; Scozzafava, A.; Supuran, C.T. The protein tyrosine kinase inhibitors imatinib and nilotinib strongly inhibit several mammalian α-carbonic anhydrase isoforms. Bioorg. Med. Chem. Lett. 2009, 19, 4102–4106. [Google Scholar] [CrossRef]
- Závada, J.; Závadová, Z.; Pastorek, J.; Biesová, Z.; Jezek, J.; Velek, J. Human tumour-associated cell adhesion protein MN/CA IX: Identification of M75 epitope and of the region mediating cell adhesion. Br. J. Cancer 2000, 82, 1808–1813. [Google Scholar] [CrossRef]
- Zavadova, Z.; Zavada, J. Carbonic anhydrase IX (CA IX) mediates tumor cell interactions with microenvironment. Oncol. Rep. 2005, 13, 977–982. [Google Scholar] [CrossRef]
- Brzozowski, Z.; Slawinski, J.; Saczewski, F.; Innocenti, A.; Supuran, C.T. Carbonic anhydrase inhibitors: Synthesis and inhibition of the human cytosolic isozymes I and II and transmembrane isozymes IX, XII (cancer-associated) and XIV with 4-substituted 3-pyridinesulfonamides. Eur. J. Med. Chem. 2010, 45, 2396–2404. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, Z.; Slawinski, J.; Innocenti, A.; Supuran, C.T. Carbonic anhydrase inhibitors. Regioselective synthesis of novel 1-substituted 1,4-dihydro-4-oxo-3-pyridinesulfonamides and their inhibition of the human cytosolic isozymes I and II and transmembrane cancer-associated isozymes IX and XII. Eur. J. Med. Chem. 2010, 45, 3656–3661. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | hCA I | hCA II | hCA IX | hCA XII | Selectivity Ratio |
|---|---|---|---|---|---|
| hCA XII/ hCA IX | |||||
| 5a | >100 | >100 | 2.9 | 5.5 | 1.90 |
| 5b | >100 | >100 | 0.4 | 4.6 | 11.50 |
| 5c | >100 | >100 | 8.2 | 3.7 | 0.45 |
| 5d | >100 | >100 | 0.7 | 4.9 | 7.00 |
| 5e | >100 | >100 | 0.8 | 5.2 | 6.50 |
| 5f | >100 | >100 | 8.4 | 7.1 | 0.90 |
| 5g | >100 | >100 | 0.3 | 3.7 | 12.00 |
| 5h | >100 | >100 | 7.4 | 3.9 | 0.50 |
| 5i | >100 | >100 | 9.2 | 6.6 | 0.70 |
| 5j | >100 | >100 | 1.1 | 6.2 | 5.60 |
| 5k | >100 | >100 | 9.3 | 6.8 | 0.70 |
| 5l | >100 | >100 | 8.5 | 6.8 | 0.80 |
| 5m | >100 | >100 | 8.8 | 6.2 | 0.70 |
| 5n | >100 | >100 | 1.3 | 6.4 | 5.00 |
| AAZ | 0.25 | 0.012 | 0.026 | 0.006 | 0.20 |
| Comp. | Cell Viability % | Antiproliferative Activity IC50 ± SEM (µM) | ||||
|---|---|---|---|---|---|---|
| A-549 | MCF-7 | Panc-1 | HT-29 | Average | ||
| 5b | 91 | 2.8 ± 0.5 | 2.7 ± 0.4 | 2.5 ± 0.2 | 3.1 ± 0.1 | 2.8 |
| 5d | 87 | 3.1 ± 0.6 | 3.9 ± 0.4 | 3.8 ± 0.3 | 3.1 ± 0.4 | 3.5 |
| 5e | 86 | 4.4 ± 0.7 | 3.6 ± 0.8 | 3.7 ± 0.5 | 3.9 ± 0.6 | 3.9 |
| 5g | 90 | 2.4 ± 0.5 | 2.1 ± 0.2 | 2.2 ± 0.4 | 2.4 ± 0.4 | 2.3 |
| 5j | 88 | 4.6 ± 0.6 | 3.6 ± 0.2 | 4.6 ± 0.9 | 4.8 ± 0.4 | 4.4 |
| 5n | 90 | 4.7 ± 0.6 | 4.6 ± 0.6 | 4.8 ± 0.3 | 4.8 ± 0.2 | 4.7 |
| Doxorubicin | - | 1.2 ± 0.8 | 0.9± 0.6 | 1.4 ±0.6 | 1.0 ± 0.8 | 1.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Wahaibi, L.H.; Youssif, B.G.M.; Taher, E.S.; Abdelazeem, A.H.; Abdelhamid, A.A.; Marzouk, A.A. Design, Synthesis, Biological Evaluation, and Computational Studies of Novel Tri-Aryl Imidazole-Benzene Sulfonamide Hybrids as Promising Selective Carbonic Anhydrase IX and XII Inhibitors. Molecules 2021, 26, 4718. https://doi.org/10.3390/molecules26164718
Al-Wahaibi LH, Youssif BGM, Taher ES, Abdelazeem AH, Abdelhamid AA, Marzouk AA. Design, Synthesis, Biological Evaluation, and Computational Studies of Novel Tri-Aryl Imidazole-Benzene Sulfonamide Hybrids as Promising Selective Carbonic Anhydrase IX and XII Inhibitors. Molecules. 2021; 26(16):4718. https://doi.org/10.3390/molecules26164718
Chicago/Turabian StyleAl-Wahaibi, Lamya H., Bahaa G. M. Youssif, Ehab S. Taher, Ahmed H. Abdelazeem, Antar A. Abdelhamid, and Adel A. Marzouk. 2021. "Design, Synthesis, Biological Evaluation, and Computational Studies of Novel Tri-Aryl Imidazole-Benzene Sulfonamide Hybrids as Promising Selective Carbonic Anhydrase IX and XII Inhibitors" Molecules 26, no. 16: 4718. https://doi.org/10.3390/molecules26164718