
Synthesis and Biological Assessment of 4,1-Benzothiazepines with Neuroprotective Activity on the Ca2+ Overload for the Treatment of Neurodegenerative Diseases and Stroke

 , and
, and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
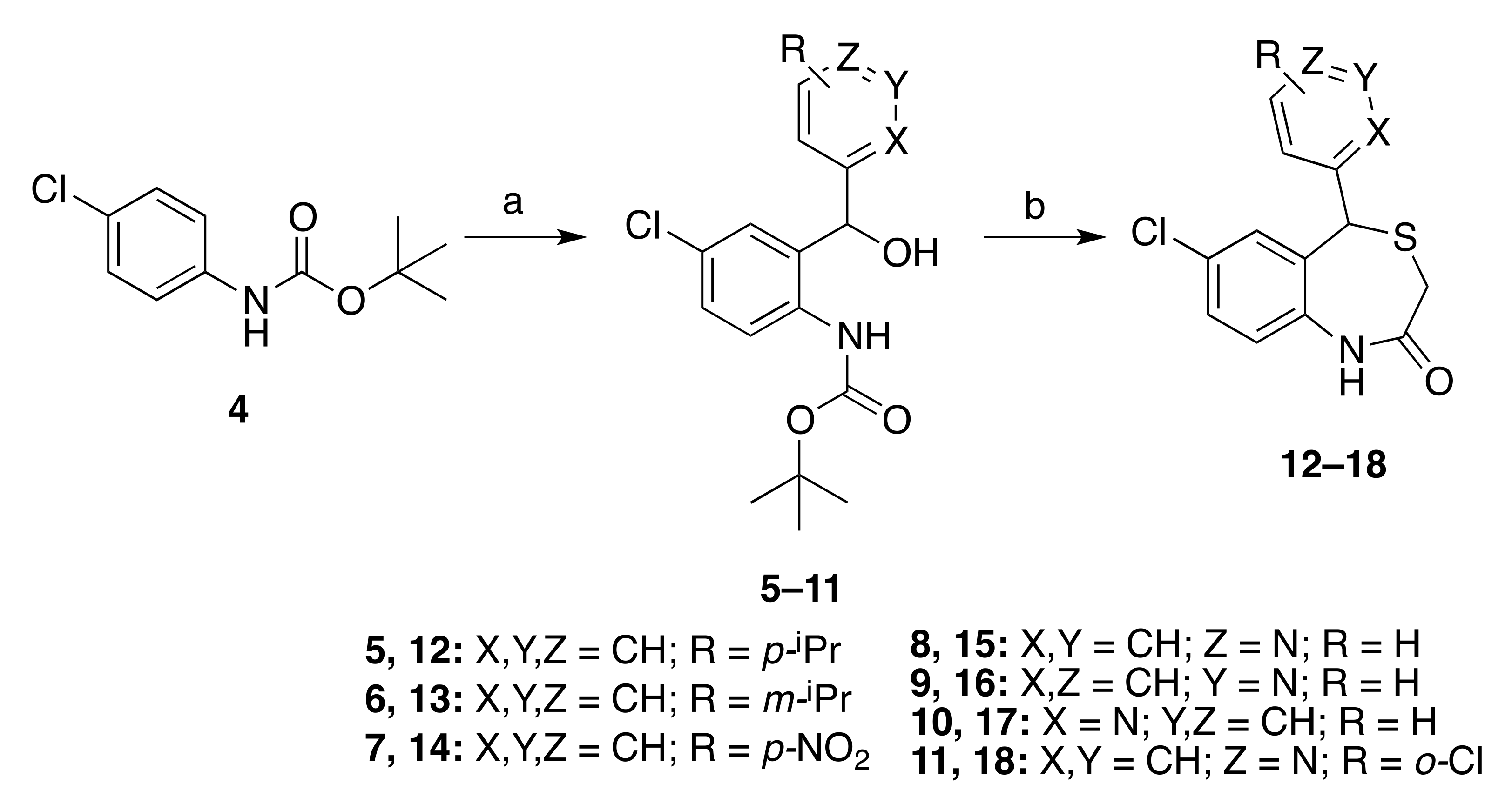
2.1. Chemistry
2.2. Pharmacology
2.2.1. Assessment of per se Toxicity of the New Derivatives in Cell Cultures
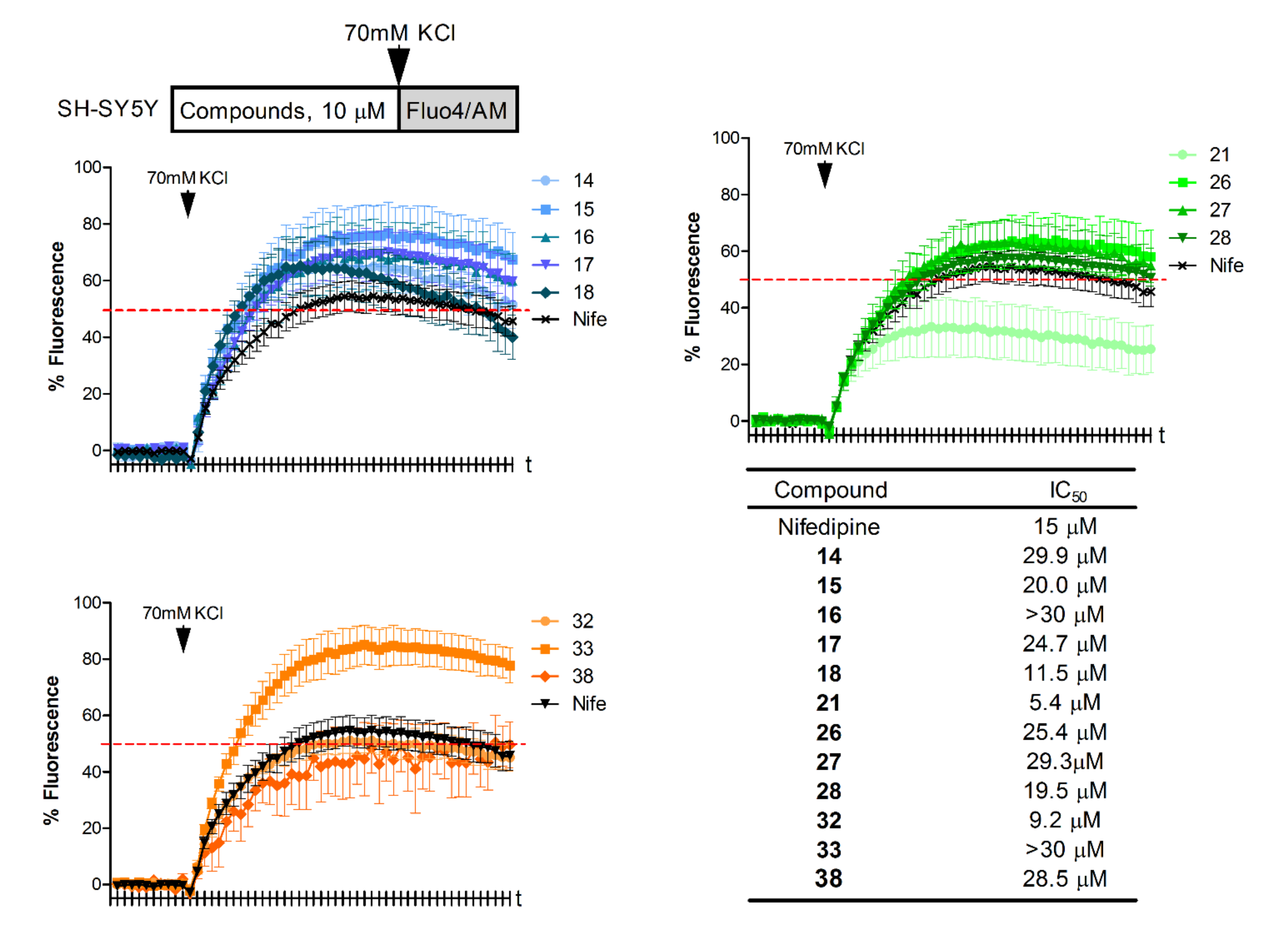
2.2.2. Effect of Compounds on the Membrane Depolarization-Induced and NMDA-Induced Ca2+ Influx
2.2.3. Effect of Compounds against Toxic Stimuli Related to Neurodegeneration
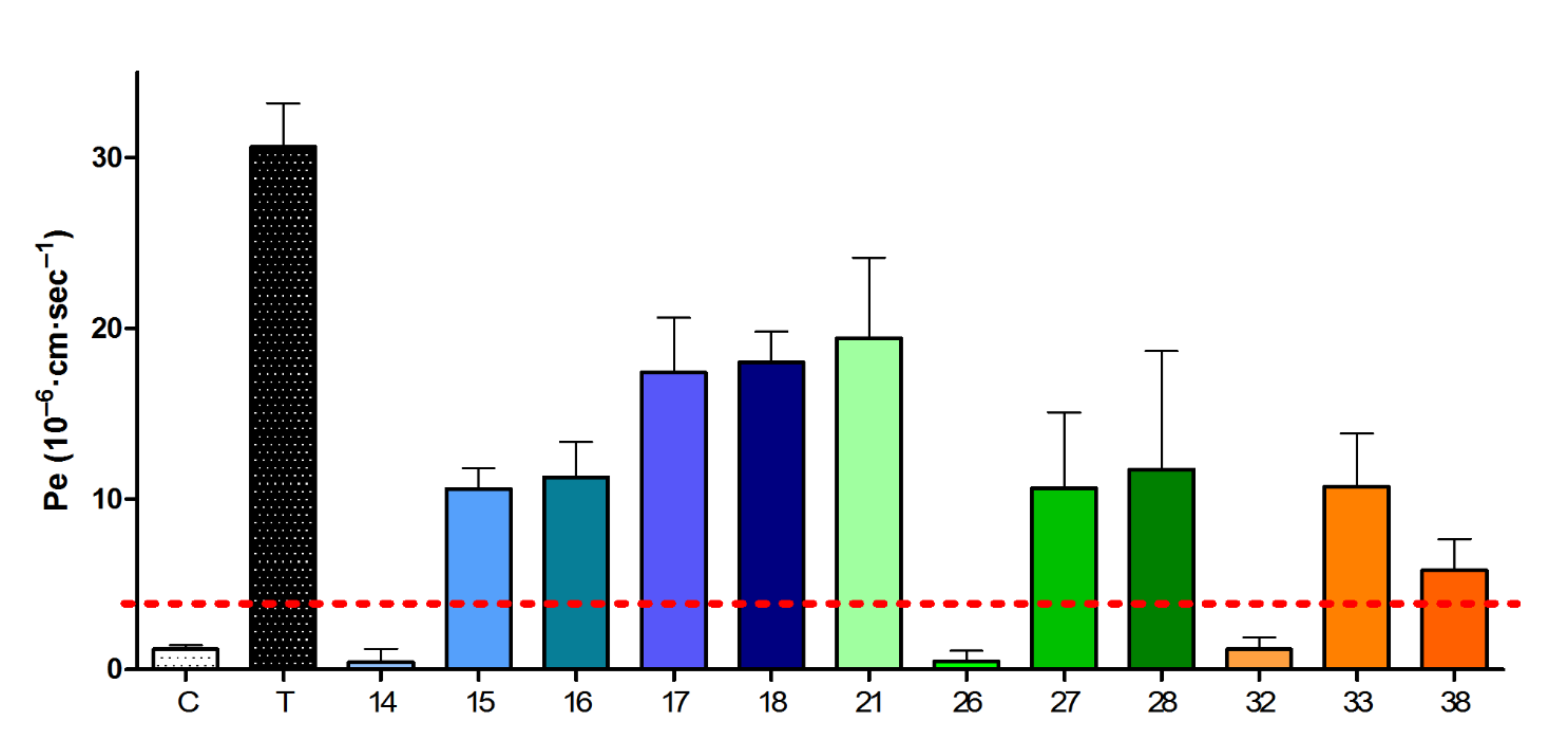
2.2.4. Blood–Brain Barrier Crossing Ability
3. Materials and Methods
3.1. General
3.2. General Procedure for the Synthesis of Intermediates 5–11, 20, 26–28, 31, and 35
3.2.1. 4-Chloro-2-[hydroxy(4′-isopropylphenyl)methyl]-N-tert-butoxycarbonylaniline (5)
3.2.2. 4-Chloro-2-[hydroxy(3′-isopropylphenyl)methyl]-N-tert-butoxycarbonylaniline (6)
3.2.3. 4-Chloro-2-[hydroxy(4′-nitrophenyl)methyl]-N-tert-butoxycarbonylaniline (7)
3.2.4. 4-Chloro-2-[hydroxy(pyridin-4-yl)methyl]-N-tert-butoxycarbonylaniline (8)
3.2.5. 4-Chloro-2-[hydroxy(pyridin-3-yl)methyl]-N-tert-butoxycarbonylaniline (9)
3.2.6. 4-Chloro-2-[hydroxy(pyridin-2-yl)methyl]-N-tert-butoxycarbonylaniline (10)
3.2.7. 4-Chloro-2-[(3-chloropyridin-4-yl)hydroxymethyl]-N-tert-butoxycarbonylaniline (11)
3.2.8. 4-Dimethylamino-2-[hydroxy(2′-Isopropylphenyl)methyl]-N-tert-butoxycarbonylaniline (20)
3.2.9. 2-[(2′-Chlorophenyl)hydroxymethyl]-4-dimethylamino-N-tert-butylcarbonylaniline (23)
3.2.10. 4-Dimethylamino-2-[hydroxy(2′-Methoxyphenyl)methyl]-N-tert-butylcarbonylaniline (24)
3.2.11. 4-Dimethylamino-2-[(4′-fluorophenyl)hydroxymethyl]-N-tert-butylcarbonylaniline (25)
3.2.12. 2-[Hydroxy(2′-methoxyphenyl)methyl]-N-tert-butylcarbonylaniline (31)
3.2.13. N-[3-[hydroxy(phenyl)methyl]pyrydin-4-yl]-2,2-dimethylpropanamide (35)
3.3. Synthesis of 2-[(4′-Fluorophenyl)hydroxymethyl]-N-tert-butylcarbonylaniline (30)
3.4. General Procedure for the Synthesis of 4,1-Benzothiazepines 12–18, 21, 26–28
3.4.1. 7-Chloro-5-(4′-isopropylphenyl)-3,5-dihydro-4,1-benzothiazepin-2-(1H)-one (12)
3.4.2. 7-Chloro-5-(3′-isopropylphenyl)-3,5-dihydro-4,1-benzothiazepin-2-(1H)-one (13)
3.4.3. 7-Chloro-5-(4′-nitrophenyl)-3,5-dihydro-4,1-benzothiazepin-2-(1H)-one (14)
3.4.4. 7-Chloro-5-(pyridin-4-yl)-3,5-dihydro-4,1-benzothiazepin-2-(1H)-one (15)
3.4.5. 7-Chloro-5-(pyridin-3-yl)-3,5-dihydro-4,1-benzothiazepin-2-(1H)-one (16)
3.4.6. 7-Chloro-5-(pyridin-2-yl)-3,5-dihydro-4,1-benzothiazepin-2-(1H)-one (17)
3.4.7. 7-Chloro-5-(3′-chloropyridin-4-yl)-3,5-dihydro-4,1-benzothiazepin-2-(1H)-one (18)
3.4.8. 7-Dimethylamino-5-(2′-isopropylphenyl)-3,5-dihydro-4,1-benzothiazepin-2-(1H)-one (21)
3.4.9. 5-(2′-Chlorophenyl)-7-dimethylamino-3,5-dihydro-4,1-benzothiazepin-2-(1H)-one (26)
3.4.10. 7-Dimethylamino-5-(2′-methoxyphenyl)-3,5-dihydro-4,1-benzothiazepin-2-(1H)-one (27)
3.4.11. 7-Dimethylamino-5-(4′-fluorophenyl)-3,5-dihydro-4,1-benzothiazepin-2-(1H)-one (28)
3.4.12. 5-(4′-Fluorophenyl)-3,5-dihydro-4,1-benzothiazepin-2-(1H)-one (32)
3.4.13. 5-(2′-Methoxyphenyl)-3,5-dihydro-4,1-benzothiazepin-2-(1H)-one (33)
3.5. Synthesis of (4-Aminopyridin-3-yl)(phenyl)methanol (36)
3.6. Synthesis of Methyl 2-[(4-Aminopyridin-3-yl)(phenyl)methylthio]acetate (37)
3.7. Synthesis of 5-Phenyl-3,5-dihydropyrido[4,3-e][1,4]thiazepin-2-(1H)-one (38)
3.8. Experimental Use of Animals
3.9. Cell Cultures
3.10. Per se Toxicity Assay
3.11. Blockade of Cytosolic Ca2+ Fluctuations Assay
3.12. Neuroprotection Assays
3.13. Blood–Brain Barrier Permeation Assay (PAMPA)
3.14. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium Signalling: Dynamics, Homeostasis and Remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, R.G.M. NMDA Receptors and Memory Encoding. Neuropharmacology 2013, 74, 32–40. [Google Scholar] [CrossRef]
- Chang, P.K.; Verbich, D.; McKinney, R.A. AMPA Receptors as Drug Targets in Neurological Disease—Advantages, Caveats, and Future Outlook. Eur. J. Neurosci. 2012, 35, 1908–1916. [Google Scholar] [CrossRef]
- Shen, J.; Yakel, J.L. Nicotinic Acetylcholine Receptor-Mediated Calcium Signaling in the Nervous System. Acta. Pharmacol. Sin. 2009, 30, 673–680. [Google Scholar] [CrossRef] [Green Version]
- Zamponi, G.W.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and their Future Therapeutic Potential. Pharmacol. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef] [Green Version]
- Carafoli, E. Calcium--a Universal Carrier of Biological Signals. Delivered on 3 July 2003 at the Special FEBS Meeting in Brussels. FEBS J. 2005, 272, 1073–1089. [Google Scholar] [CrossRef]
- Carafoli, E. The Interplay of Mitochondria with Calcium: An Historical Appraisal. Cell Calcium 2012, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Guerini, D.; Coletto, L.; Carafoli, E. Exporting Calcium from Cells. Cell Calcium 2005, 38, 281–289. [Google Scholar] [CrossRef]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A Forty-Kilodalton Protein of the Inner Membrane is the Mitochondrial Calcium Uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Fernández-Morales, J.; Arranz-Tagarro, J.; Calvo-Gallardo, E.; Maroto, M.; Padín, J.; García, A.G. Stabilizers of Neuronal and Mitochondrial Calcium Cycling as a Strategy for Developing a Medicine for Alzheimer’s Disease. ACS Chem. Neurosci. 2012, 3, 873–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agostini, M.; Fasolato, C. When, Where and how? Focus on Neuronal Calcium Dysfunctions in Alzheimer’s Disease. Cell Calcium 2016, 60, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Di Scala, C.; Yahi, N.; Boutemeur, S.; Flores, A.; Rodriguez, L.; Chahinian, H.; Fantini, J. Common Molecular Mechanism of Amyloid Pore Formation by Alzheimer’s Β-Amyloid Peptide and A-Synuclein. Sci. Rep. 2016, 6, 28781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbel-Ornath, M.; Hudry, E.; Boivin, J.R.; Hashimoto, T.; Takeda, S.; Kuchibhotla, K.V.; Hou, S.; Lattarulo, C.R.; Belcher, A.M.; Shakerdge, N.; et al. Soluble Oligomeric Amyloid-Β Induces Calcium Dyshomeostasis that Precedes Synapse Loss in the Living Mouse Brain. Mol. Neurodegener. 2017, 12, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The Antibody Aducanumab Reduces Aβ Plaques in Alzheimer’s Disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Gamage, K.K.; Kumar, S. Aducanumab Therapy Ameliorates Calcium Overload in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2017, 37, 4430–4432. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.; Lingenfelter, K.S.; Bender, A.M.; Lindsley, C.W. Classics in Chemical Neuroscience: Memantine. ACS Chem. Neurosci. 2017, 8, 1823–1829. [Google Scholar] [CrossRef]
- Urushitani, M.; Nakamizo, T.; Inoue, R.; Sawada, H.; Kihara, T.; Honda, K.; Akaike, A.; Shimohama, S. N-Methyl-D-Aspartate Receptor-Mediated Mitochondrial Ca2+ Overload in Acute Excitotoxic Motor Neuron Death: A Mechanism Distinct from Chronic Neurotoxicity After Ca2+ Influx. J. Neurosci. Res. 2001, 63, 377–387. [Google Scholar] [CrossRef]
- Van Den Bosch, L.; Vandenberghe, W.; Klaassen, H.; Van Houtte, E.; Robberecht, W. Ca(2+)-Permeable AMPA Receptors and Selective Vulnerability of Motor Neurons. J. Neurol. Sci. 2000, 180, 29–34. [Google Scholar] [CrossRef]
- Chang, Q.; Martin, L.J. Voltage-Gated Calcium Channels are Abnormal in Cultured Spinal Motoneurons in the G93A-SOD1 Transgenic Mouse Model of ALS. Neurobiol. Dis. 2016, 93, 78–95. [Google Scholar] [CrossRef] [Green Version]
- Calzaferri, F.; Ruiz-Ruiz, C.; de Diego, A.M.G.; de Pascual, R.; Méndez-López, I.; Cano-Abad, M.F.; Maneu, V.; de Los Ríos, C.; Gandía, L.; García, A.G. The Purinergic P2X7 Receptor as a Potential Drug Target to Combat Neuroinflammation in Neurodegenerative Diseases. Med. Res. Rev. 2020, 40, 2427–2465. [Google Scholar] [CrossRef]
- Liu, J.; Prell, T.; Stubendorff, B.; Keiner, S.; Ringer, T.; Gunkel, A.; Tadic, V.; Goldhammer, N.; Malci, A.; Witte, O.W.; et al. Down-Regulation of Purinergic P2X7 Receptor Expression and Intracellular Calcium Dysregulation in Peripheral Blood Mononuclear Cells of Patients with Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2016, 630, 77–83. [Google Scholar] [CrossRef]
- Damiano, M.; Starkov, A.A.; Petri, S.; Kipiani, K.; Kiaei, M.; Mattiazzi, M.; Flint Beal, M.; Manfredi, G. Neural Mitochondrial Ca2+ Capacity Impairment Precedes the Onset of Motor Symptoms in G93A Cu/Zn-Superoxide Dismutase Mutant Mice. J. Neurochem. 2006, 96, 1349–1361. [Google Scholar] [CrossRef]
- Carriedo, S.G.; Sensi, S.L.; Yin, H.Z.; Weiss, J.H. AMPA Exposures Induce Mitochondrial Ca(2+) Overload and ROS Generation in Spinal Motor Neurons in Vitro. J. Neurosci. 2000, 20, 240–250. [Google Scholar] [CrossRef] [Green Version]
- Volles, M.J.; Lee, S.J.; Rochet, J.C.; Shtilerman, M.D.; Ding, T.T.; Kessler, J.C.; Lansbury, P.T. Vesicle Permeabilization by Protofibrillar Alpha-Synuclein: Implications for the Pathogenesis and Treatment of Parkinson’s Disease. Biochemistry 2001, 40, 7812–7819. [Google Scholar] [CrossRef] [PubMed]
- Danzer, K.M.; Haasen, D.; Karow, A.R.; Moussaud, S.; Habeck, M.; Giese, A.; Kretzschmar, H.; Hengerer, B.; Kostka, M. Different Species of Alpha-Synuclein Oligomers Induce Calcium Influx and Seeding. J. Neurosci. 2007, 27, 9220–9232. [Google Scholar] [CrossRef]
- Surmeier, D.J. Calcium, Ageing, and Neuronal Vulnerability in Parkinson’s Disease. Lancet Neurol. 2007, 6, 933–938. [Google Scholar] [CrossRef]
- Tang, T.; Tu, H.; Chan, E.Y.W.; Maximov, A.; Wang, Z.; Wellington, C.L.; Hayden, M.R.; Bezprozvanny, I. Huntingtin and Huntingtin-Associated Protein 1 Influence Neuronal Calcium Signaling Mediated by Inositol-(1,4,5) Triphosphate Receptor Type 1. Neuron 2003, 39, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Zeron, M.M.; Hansson, O.; Chen, N.; Wellington, C.L.; Leavitt, B.R.; Brundin, P.; Hayden, M.R.; Raymond, L.A. Increased Sensitivity to N-Methyl-D-Aspartate Receptor-Mediated Excitotoxicity in a Mouse Model of Huntington’s Disease. Neuron 2002, 33, 849–860. [Google Scholar] [CrossRef] [Green Version]
- Kaltenbach, L.S.; Romero, E.; Becklin, R.R.; Chettier, R.; Bell, R.; Phansalkar, A.; Strand, A.; Torcassi, C.; Savage, J.; Hurlburt, A.; et al. Huntingtin Interacting Proteins are Genetic Modifiers of Neurodegeneration. PLoS Genet. 2007, 3, e82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sattler, R.; Charlton, M.P.; Hafner, M.; Tymianski, M. Distinct Influx Pathways, Not Calcium Load, Determine Neuronal Vulnerability to Calcium Neurotoxicity. J. Neurochem. 1998, 71, 2349–2364. [Google Scholar] [CrossRef]
- Koh, P. Melatonin Regulates the Calcium-Buffering Proteins, Parvalbumin and Hippocalcin, in Ischemic Brain Injury. J. Pineal. Res. 2012, 53, 358–365. [Google Scholar] [CrossRef]
- Cox, D.A.; Conforti, L.; Sperelakis, N.; Matlib, M.A. Selectivity of Inhibition of Na(+)-Ca2+ Exchange of Heart Mitochondria by Benzothiazepine CGP-37157. J. Cardiovasc. Pharmacol. 1993, 21, 595–599. [Google Scholar] [CrossRef]
- González-Lafuente, L.; Egea, J.; León, R.; Martínez-Sanz, F.J.; Monjas, L.; Perez, C.; Merino, C.; García-De Diego, A.M.; Rodríguez-Franco, M.I.; Garcia, A.G.; et al. Benzothiazepine CGP37157 and its Isosteric 2′-Methyl Analogue Provide Neuroprotection and Block Cell Calcium Entry. ACS Chem. Neurosci. 2012, 3, 519–529. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Sanz, F.J.; Lajarín-Cuesta, R.; Moreno-Ortega, A.J.; González-Lafuente, L.; Fernández-Morales, J.C.; López-Arribas, R.; Cano-Abad, M.F.; de los Ríos, C. Benzothiazepine CGP37157 Analogues Exert Cytoprotection in various in Vitro Models of Neurodegeneration. ACS Chem. Neurosci. 2015, 6, 1626–1636. [Google Scholar] [CrossRef]
- Moreno-Ortega, A.J.; Martínez-Sanz, F.J.; Lajarín-Cuesta, R.; de Los Rios, C.; Cano-Abad, M.F. Benzothiazepine CGP37157 and its 2’-Isopropyl Analogue Modulate Ca2+ Entry through CALHM1. Neuropharmacology 2015, 95, 503–510. [Google Scholar] [CrossRef]
- Martínez-Sanz, F.J.; Lajarín-Cuesta, R.; González-Lafuente, L.; Moreno-Ortega, A.J.; Punzón, E.; Cano-Abad, M.F.; de los Ríos, C. Neuroprotective Profile of Pyridothiazepines with Blocking Activity of the Mitochondrial Na(+)/Ca(2+) Exchanger. Eur. J. Med. Chem. 2016, 109, 114–123. [Google Scholar] [CrossRef]
- López-Gil, A.; Nanclares, C.; Méndez-López, I.; Martínez-Ramírez, C.; de Los Rios, C.; Padín-Nogueira, J.F.; Montero, M.; Gandía, L.; García, A.G. The Quantal Catecholamine Release from Mouse Chromaffin Cells Challenged with Repeated ACh Pulses is Regulated by the Mitochondrial Na+ /Ca2+ Exchanger. J. Physiol. 2017, 595, 2129–2146. [Google Scholar] [CrossRef] [Green Version]
- Pei, Y.; Lilly, M.J.; Owen, D.J.; D’Souza, L.J.; Tang, X.; Yu, J.; Nazarbaghi, R.; Hunter, A.; Anderson, C.M.; Glasco, S.; et al. Efficient Syntheses of Benzothiazepines as Antagonists for the Mitochondrial Sodium-Calcium Exchanger: Potential Therapeutics for Type II Diabetes. J. Org. Chem. 2003, 68, 92–103. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Bading, H. Synaptic Versus Extrasynaptic NMDA Receptor Signalling: Implications for Neurodegenerative Disorders. Nature reviews. Neuroscience 2010, 11, 682–696. [Google Scholar] [CrossRef] [Green Version]
- Kulikov, A.V.; Rzhaninova, A.A.; Goldshtein, D.V.; Boldyrev, A.A. Expression of NMDA Receptors in Multipotent Stromal Cells of Human Adipose Tissue Under Conditions of Retinoic Acid-Induced Differentiation. Bull. Exp. Biol. Med. 2007, 144, 626–629. [Google Scholar] [CrossRef]
- Mattson, M.P.; Cheng, B.; Davis, D.; Bryant, K.; Lieberburg, I.; Rydel, R.E. Β-Amyloid Peptides Destabilize Calcium Homeostasis and Render Human Cortical Neurons Vulnerable to Excitotoxicity. J. Neurosci. 1992, 12, 376–389. [Google Scholar] [CrossRef] [Green Version]
- Lewerenz, J.; Maher, P. Chronic Glutamate Toxicity in Neurodegenerative Diseases-what is the Evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; Carter, G.T. High Throughput Artificial Membrane Permeability Assay for Blood-Brain Barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef]
- Lajarín-Cuesta, R.; Nanclares, C.; Arranz-Tagarro, J.; González-Lafuente, L.; Arribas, R.L.; Araujo de Brito, M.; Gandía, L.; de los Ríos, C. Gramine Derivatives Targeting Ca2+ Channels and Ser/Thr Phosphatases: A New Dual Strategy for the Treatment of Neurodegenerative Diseases. J. Med. Chem. 2016, 59, 6265–6280. [Google Scholar] [CrossRef] [Green Version]
- Espadinha, M.; Dourado, J.; Lajarin-Cuesta, R.; Herrera-Arozamena, C.; Gonçalves, L.M.D.; Rodríguez-Franco, M.I.; de los Rios, C.; Santos, M.M.M. Optimization of Bicyclic Lactam Derivatives as NMDA Receptor Antagonists. ChemMedChem 2017, 12, 537–545. [Google Scholar] [CrossRef] [Green Version]
- Denizot, F.; Lang, R. Rapid Colorimetric Assay for Cell Growth and Survival: Modifications to the Tetrazolium Dye Procedure Giving Improved Sensitivity and Reliability. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viejo, L.; Rubio-Alarcón, M.; Arribas, R.L.; Moreno-Castro, M.; Pérez-Marín, R.; Braun-Cornejo, M.; Estrada-Valencia, M.; de los Ríos, C. Synthesis and Biological Assessment of 4,1-Benzothiazepines with Neuroprotective Activity on the Ca2+ Overload for the Treatment of Neurodegenerative Diseases and Stroke. Molecules 2021, 26, 4473. https://doi.org/10.3390/molecules26154473
Viejo L, Rubio-Alarcón M, Arribas RL, Moreno-Castro M, Pérez-Marín R, Braun-Cornejo M, Estrada-Valencia M, de los Ríos C. Synthesis and Biological Assessment of 4,1-Benzothiazepines with Neuroprotective Activity on the Ca2+ Overload for the Treatment of Neurodegenerative Diseases and Stroke. Molecules. 2021; 26(15):4473. https://doi.org/10.3390/molecules26154473
Chicago/Turabian StyleViejo, Lucía, Marcos Rubio-Alarcón, Raquel L. Arribas, Manuel Moreno-Castro, Raquel Pérez-Marín, María Braun-Cornejo, Martín Estrada-Valencia, and Cristóbal de los Ríos. 2021. "Synthesis and Biological Assessment of 4,1-Benzothiazepines with Neuroprotective Activity on the Ca2+ Overload for the Treatment of Neurodegenerative Diseases and Stroke" Molecules 26, no. 15: 4473. https://doi.org/10.3390/molecules26154473