
Precursor-Directed Biosynthesis of Aminofulvenes: New Chalanilines from Endophytic Fungus Chalara sp.


Abstract
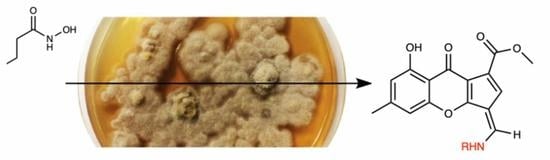
:
1. Introduction
2. Results
2.1. Structure Elucidation
2.2. Bioactivity
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Alberti, F.; Foster, G.D.; Bailey, A.M. Natural products from filamentous fungi and production by heterologous expression. Appl. Microbiol. Biotechnol. 2017, 101, 493–500. [Google Scholar] [CrossRef] [Green Version]
- Keller, N.P. Fungal secondary metabolism: Regulation, function and drug discovery. Nat. Rev. Microbiol. 2019, 17, 167–180. [Google Scholar] [CrossRef]
- Okada, B.K.; Seyedsayamdost, M.R. Antibiotic dialogues: Induction of silent biosynthetic gene clusters by exogenous small molecules. FEMS Microbiol. Rev. 2017, 41, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Lou, H.X. Strategies to diversify natural products for drug discovery. Med. Res. Rev. 2018, 38, 1255–1294. [Google Scholar] [CrossRef]
- Greco, C.; Keller, N.P.; Rokas, A. Unearthing fungal chemodiversity and prospects for drug discovery. Curr. Opin. Microbiol. 2019, 51, 22–29. [Google Scholar] [CrossRef]
- Aly, A.H.; Debbab, A.; Proksch, P. Fungal endophytes: Unique plant inhabitants with great promises. Appl. Microbiol. Biotechnol. 2011, 90, 1829–1845. [Google Scholar] [CrossRef]
- Hyde, K.D.; Xu, J.; Rapior, S.; Jeewon, R.; Lumyong, S.; Niego, A.G.T.; Abeywickrama, P.D.; Aluthmuhandiram, J.V.S.; Brahamanage, R.S.; Brooks, S.; et al. The amazing potential of fungi: 50 ways we can exploit fungi industrially. Fungal Divers. 2019, 97, 1–136. [Google Scholar] [CrossRef] [Green Version]
- Vylkova, S. Environmental pH modulation by pathogenic fungi as a strategy to conquer the host. PLoS Pathog. 2017, 13, e1006149. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Dai, C.-C. Endophytes: A potential resource for biosynthesis, biotransformation, and biodegradation. Ann. Microbiol. 2011, 61, 207–215. [Google Scholar] [CrossRef]
- Biotechnological Use of Fungal Enzymes. In Fungi; Wiley: Hoboken, NJ, USA, 2017; pp. 201–225.
- Du, L.; You, J.; Nicholas, K.M.; Cichewicz, R.H. Chemoreactive Natural Products that Afford Resistance against Disparate Antibiotics and Toxins. Angew. Chem. Int. Ed. Engl. 2016, 55, 4220–4225. [Google Scholar] [CrossRef] [Green Version]
- Adpressa, D.A.; Stalheim, K.J.; Proteau, P.J.; Loesgen, S. Unexpected Biotransformation of the HDAC Inhibitor Vorinostat Yields Aniline-Containing Fungal Metabolites. ACS Chem. Biol. 2017, 12, 1842–1847. [Google Scholar] [CrossRef]
- Khoshbakht, M.; Thanaussavadate, B.; Zhu, C.; Cao, Y.; Zakharov, L.N.; Loesgen, S.; Blakemore, P.R. Total Synthesis of Chalaniline B: An Antibiotic Aminoxanthone from Vorinostat-Treated Fungus Chalara sp. 6661. J. Org. Chem. 2021. [Google Scholar] [CrossRef]
- Dunn, T.B.; Ellis, J.M.; Kofink, C.C.; Manning, J.R.; Overman, L.E. Asymmetric Construction of Rings A−D of Daphnicyclidin-Type Alkaloids. Org. Lett. 2009, 11, 5658–5661. [Google Scholar] [CrossRef] [Green Version]
- Hong, B.-C.; Shr, Y.-J.; Wu, J.-L.; Gupta, A.K.; Lin, K.-J. Novel [6 + 2] Cycloaddition of Fulvenes with Alkenes: A Facile Synthesis of the Anislactone and Hirsutane Framework. Org. Lett. 2002, 4, 2249–2252. [Google Scholar] [CrossRef]
- Naksomboon, K.; Poater, J.; Bickelhaupt, F.M.; Fernández-Ibáñez, M.Á. para-Selective C–H Olefination of Aniline Derivatives via Pd/S,O-Ligand Catalysis. J. Am. Chem. Soc. 2019, 141, 6719–6725. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, J.; Xie, Z.; Li, J.; Li, J.; Hu, L. Design, synthesis and biological evaluation of mogrol derivatives as a novel class of AMPKα2β1γ1 activators. Bioorg. Med. Chem. Lett. 2020, 30, 126790. [Google Scholar] [CrossRef]
- Yang, R.; Du, W.; Yuan, H.; Qin, T.; He, R.; Ma, Y.; Du, H. Synthesis and biological evaluation of 2-phenyl-4-aminoquinolines as potential antifungal agents. Mol. Divers. 2020, 24, 1065–1075. [Google Scholar] [CrossRef]
- Srivastava, P.; Tripathi, P.N.; Sharma, P.; Shrivastava, S.K. Design, synthesis, and evaluation of novel N-(4-phenoxybenzyl)aniline derivatives targeting acetylcholinesterase, β-amyloid aggregation and oxidative stress to treat Alzheimer’s disease. Bioorg. Med. Chem. 2019, 27, 3650–3662. [Google Scholar] [CrossRef]
- Soli, E.D.; Braun, M.P. Synthesis of [phenyl-U-14C]aryl and [8-14C]carboxy labeled tracers of vorinostat. J. Labelled Comp. Radiopharm. 2006, 49, 437–443. [Google Scholar] [CrossRef]
- Salmi-Smail, C.; Fabre, A.; Dequiedt, F.; Restouin, A.; Castellano, R.; Garbit, S.; Roche, P.; Morelli, X.; Brunel, J.M.; Collette, Y. Modified cap group suberoylanilide hydroxamic acid histone deacetylase inhibitor derivatives reveal improved selective antileukemic activity. J. Med. Chem. 2010, 53, 3038–3047. [Google Scholar] [CrossRef] [PubMed]
- Rebelo, B.A.; Santos, R.B.; Ascenso, O.S.; Nogueira, A.C.; Lousa, D.; Abranches, R.; Ventura, M.R. Synthesis and biological effects of small molecule enhancers for improved recombinant protein production in plant cell cultures. Bioorg. Chem. 2020, 94, 103452. [Google Scholar] [CrossRef]
- Remiszewski, S.W.; Sambucetti, L.C.; Atadja, P.; Bair, K.W.; Cornell, W.D.; Green, M.A.; Howell, K.L.; Jung, M.; Kwon, P.; Trogani, N.; et al. Inhibitors of human histone deacetylase: Synthesis and enzyme and cellular activity of straight chain hydroxamates. J. Med. Chem. 2002, 45, 753–757. [Google Scholar] [CrossRef]
- Thangavel, S.; Rajamanikandan, R.; Friedrich, H.B.; Ilanchelian, M.; Omondi, B. Binding interaction, conformational change, and molecular docking study of N-(pyridin-2-ylmethylene)aniline derivatives and carbazole Ru(II) complexes with human serum albumins. Polyhedron 2016, 107, 124–135. [Google Scholar] [CrossRef]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]
- CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, 7th ed.; National Committee for Clinical Laboratory Standards: Wayne, PA, USA, 2018. [Google Scholar]
- Kennedy, J. Mutasynthesis, chemobiosynthesis, and back to semi-synthesis: Combining synthetic chemistry and biosynthetic engineering for diversifying natural products. Nat. Prod. Rep. 2008, 25, 25–34. [Google Scholar] [CrossRef]
- Hughes, C.C. Chemical labeling strategies for small molecule natural product detection and isolation. Nat. Prod. Rep. 2021. [Google Scholar] [CrossRef]
- Haney, W.A.; Moussaoui, B.; Strother, J.A. Prolonged exposure to stressors suppresses exploratory behavior in zebrafish larvae. J. Exp. Biol. 2020, 223, jeb224964. [Google Scholar] [CrossRef]
- Lösgen, S.; Magull, J.; Schulz, B.; Draeger, S.; Zeeck, A. Isofusidienols: Novel chromone-3-oxepines produced by the endophytic fungus Chalara sp. Eur. JOC 2008, 4, 698–703. [Google Scholar]
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing M100, 30th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2020. [Google Scholar]
- Kmail, A.; Lyoussi, B.; Zaid, H.; Saad, B. In vitro assessments of cytotoxic and cytostatic effects of Asparagus aphyllus, Crataegus aronia, and Ephedra alata in monocultures and co-cultures of Hepg2 and THP-1-derived macrophages. Pharmacogn. Commun. 2015, 5, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]




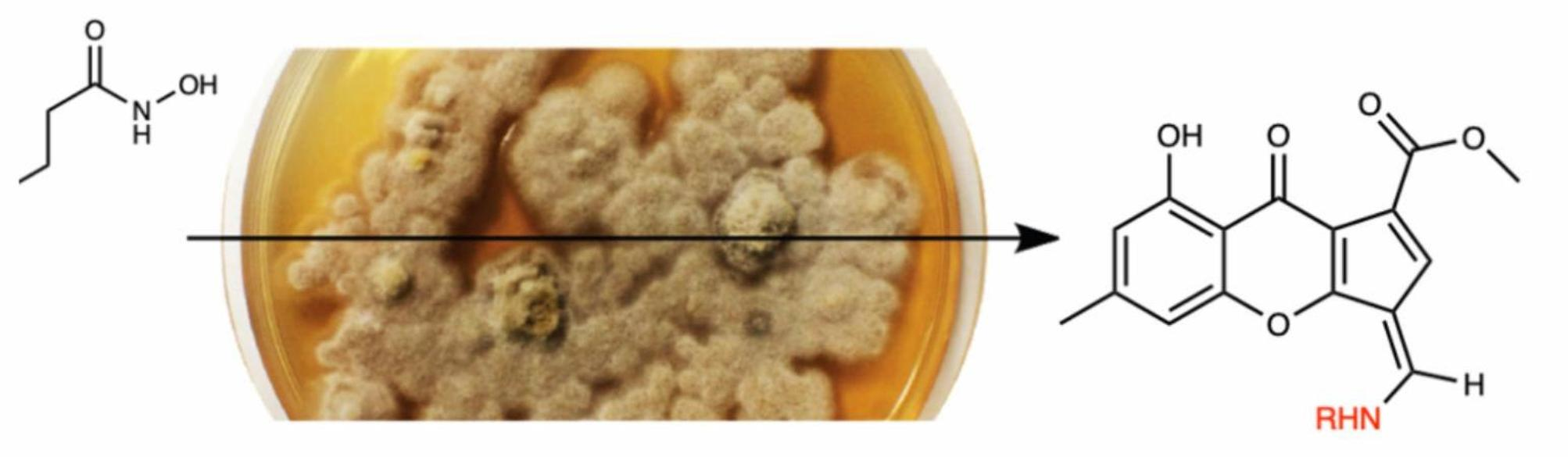{kind=link}
{kind=link}
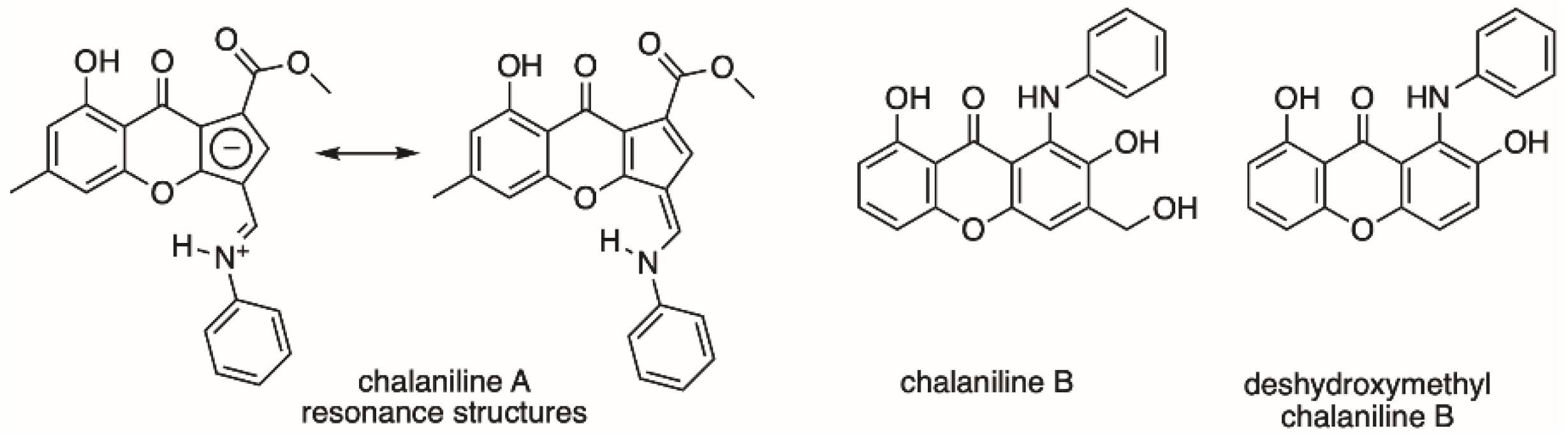
{kind=link}
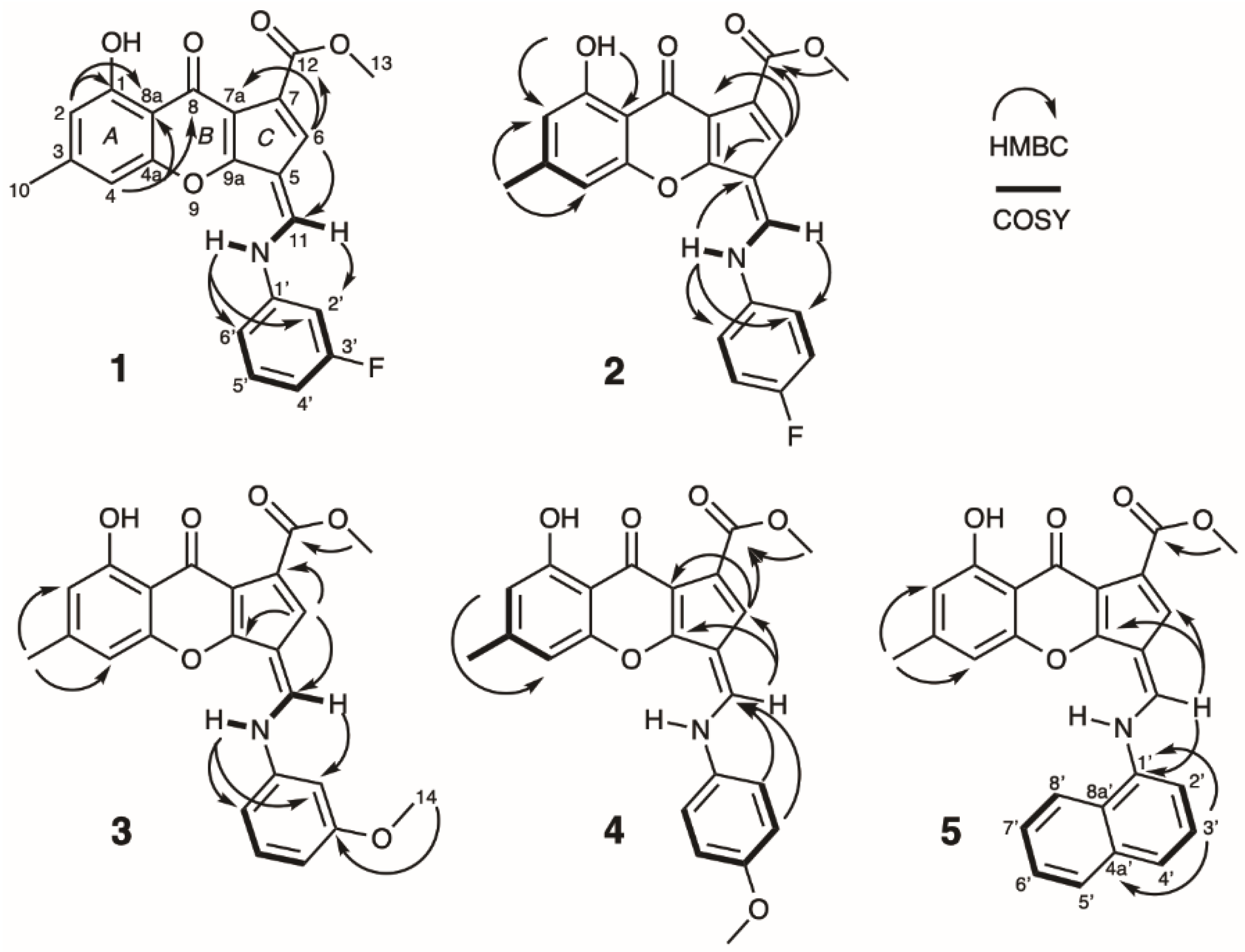{kind=link}
| 1 | 2 | 3 | 4 | 5 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Pos. | δC Type | δH | δC Type | δH | δC Type | δH | δC Type | δH | δC Type | δH |
| 1 | 161.3, C | 161.8, C | 161.3, C | 161.9, C | 161.7, C | |||||
| 2 | 111.2, CH | 6.57, s | 111.5, CH | 6.56, s | 111.0, CH | 6.56, s | 109.3, CH | 6.37, s | 109.8, CH | 6.41, s |
| 3 | 145.0, C | 145.3, C | 144.9, C | 142.5, C | 143.3, C | |||||
| 4 | 106.9, CH | 6.88, s | 107.3, CH | 6.87, s | 106.9, CH | 6.89, s | 106.0, CH | 6.70, s | 106.3, CH | 6.71, s |
| 4a | 155.0, C | 155.5, C | 155.0, C | 154.9, C | 155.1, C | |||||
| 5 | 105.3 C | 105.0, C | 104.8, C | 107.8, C | 107.1, C | |||||
| 6 | 120.6, CH | 7.69, s | 121.0, CH | 7.69, s | 120.7, CH | 7.73, s | 122.0, CH | 7.31, s | 122.7, CH | |
| 7 | 119.6, CH | 119.2, C | 118.9, C | 124.7, C | 122.3, C | |||||
| 7a | 111.1, C | 111.4, C | 111.1, C | 110.7, C | 110.9, C | |||||
| 8 | 176.2, C | 176.6, C | 176.2, C | 174.7, C | 175.7, C | |||||
| 8a | 107.7, C | 108.1, C | 107.6, C | 107.3, C | 107.4, C | |||||
| 9 | ||||||||||
| 9a | 160.6, C | 160.7, C | 160.3, C | 154.2, C | 155.0, C | |||||
| 10 | 21.7, CH3 | 2.38, s | 21.7, CH3 | 2.38, s | 21.7, CH3 | 2.38, s | 21.5, CH3 | 2.32, s | 21.6, CH3 | 2.32, s |
| 11 | 145.1, CH | 8.74, d (14.5) a | 146.1, C | 8.68, d, (14.5) a | 145.2, CH | 8.73, d (14.7) a | 147.0, CH | 8.50, s | 152.1, CH | 8.61, s |
| 12 | 164.2, C | 164.7, C | 164.2, C | 164.9, C | 165.0, C | |||||
| 13 | 51.2, CH3 | 3.77, s | 51.6, CH3 | 3.77, s | 51.1, CH3 | 3.76, s | 50.0, CH3 | 3.65, s | 50.3, CH3 | 3.76, s |
| 14 | 55.5, CH3 | 3.84, s | 55.1, CH3 | 3.75, s | ||||||
| 1′ | 140.7, C (10.3) b | 135.5, C (2.6) b | 140.1, C | 133.1, C | 133.7, C * | |||||
| 2′ | 105.28, CH (26.2) b | 7.61, d (10.7) c | 120.5, CH, (8.3) b | 7.68, m | 104.0, CH | 7.23, t (2.0) a | 121.4, CH | 7.10, d (8.7) a | 112.7, CH * | 7.10, brd |
| 3′ | 162.8, C (244.9) b | 116.5, CH, (23.0) b | 7.36, t (8.7) a | 160.4, C | 114.2, CH | 6.91, d (8.8) a | 125.5, CH * | 7.49, m * | ||
| 4′ | 112.6, CH (21.4) b | 7.12, t (8.0) c | 160.6, C (244.8) b | 111.9, CH | 6.87, dd (8.0, 2.0) a | 156.1, C | 123.3, CH | 7.65, d (7.3) a | ||
| 4a′ | 134.2, C | |||||||||
| 5′ | 131.5, CH (9.4) b | 7.53, q (8.0) a | 130.7, CH | 7.40, t (8.1) a | 127.8, CH | 7.88, d (7.0) a | ||||
| 6′ | 114.8, CH (2.8) b | 7.44, d (8.0) a | 110.7, CH | 7.21, dd (8.1, 2.0) a | 123.6, CH * | 7.49, m * | ||||
| 7′ | 126.7, CH * | 7.49, m * | ||||||||
| 8′ | 124.0, CH | 8.39, d (7.0) a | ||||||||
| 8a′ | 129.0, C | |||||||||
| OH | 13.75, brd | 13.84, brd | 13.83, brd | 14.97, brd | 14.91, brd | |||||
| NH | 11.84, d (14.5) a | 11.89, d (14.5) a | 11.83, d (14.7) a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khoshbakht, M.; Srey, J.; Adpressa, D.A.; Jagels, A.; Loesgen, S. Precursor-Directed Biosynthesis of Aminofulvenes: New Chalanilines from Endophytic Fungus Chalara sp. Molecules 2021, 26, 4418. https://doi.org/10.3390/molecules26154418
Khoshbakht M, Srey J, Adpressa DA, Jagels A, Loesgen S. Precursor-Directed Biosynthesis of Aminofulvenes: New Chalanilines from Endophytic Fungus Chalara sp. Molecules. 2021; 26(15):4418. https://doi.org/10.3390/molecules26154418
Chicago/Turabian StyleKhoshbakht, Mahsa, Jason Srey, Donovon A. Adpressa, Annika Jagels, and Sandra Loesgen. 2021. "Precursor-Directed Biosynthesis of Aminofulvenes: New Chalanilines from Endophytic Fungus Chalara sp." Molecules 26, no. 15: 4418. https://doi.org/10.3390/molecules26154418