
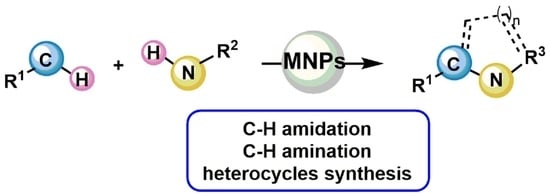
Metal Nanoparticles as Sustainable Tools for C–N Bond Formation via C–H Activation



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
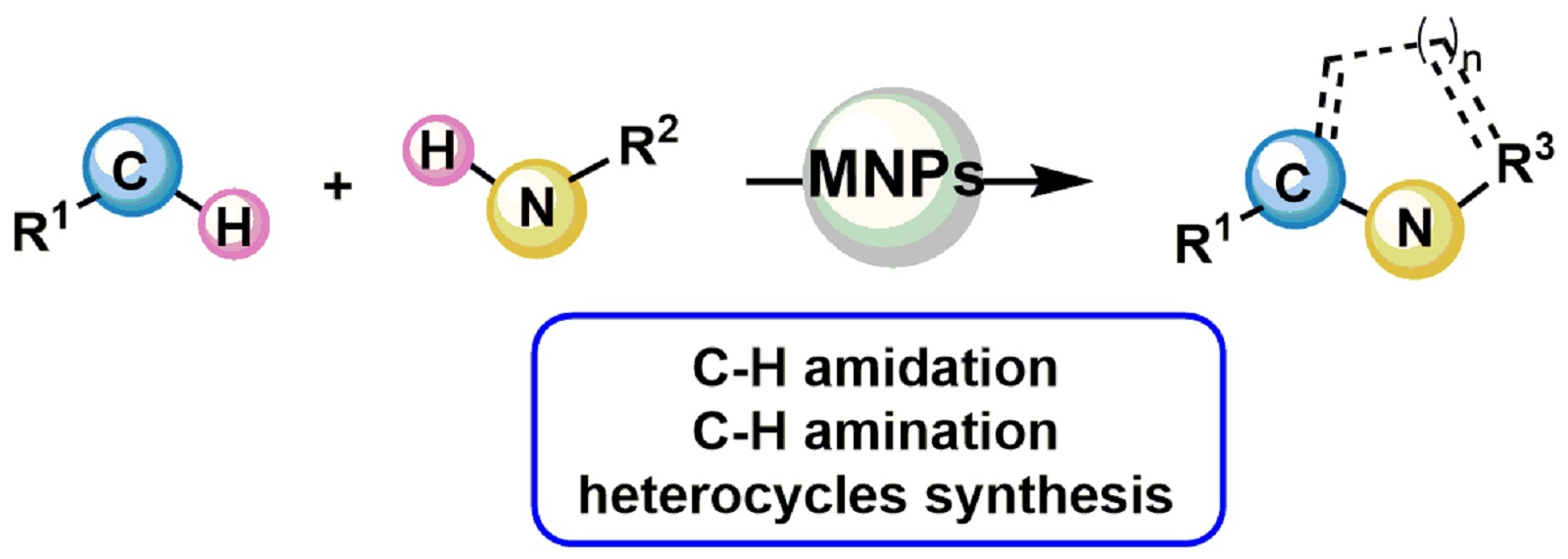
2. C–H Amidation
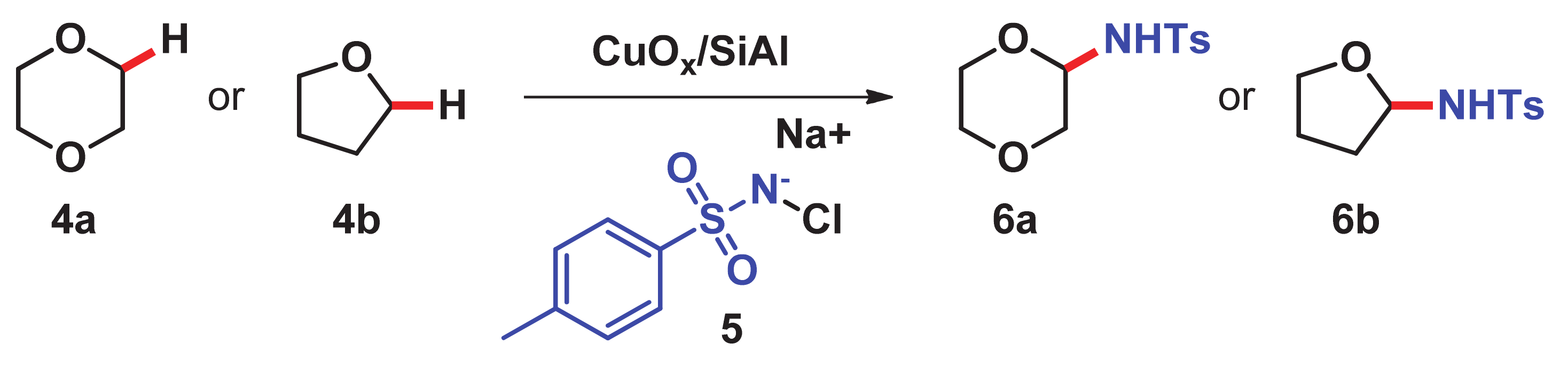
3. C–H Amination
4. C–N Bond Formation in the Synthesis of Heterocycles
5. Conclusions and Future Outlooks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ballini, R. (Ed.) Green Synthetic Processes and Procedures; Royal Society of Chemistry: Croydon, UK, 2019; Volume 61. [Google Scholar]
- Sheldon, R.A.; Arends, I.; Hanefeld, U. Green Chemistry and Catalysis; John Wiley & Sons: Weinheim, Germany, 2007. [Google Scholar]
- Dalton, T.; Faber, T.; Glorius, F. C–H activation: Toward sustainability and applications. ACS Cent. Sci. 2021, 7, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Santoro, S.; Ferlin, F.; Vaccaro, L. Sustainable Approaches to C–H Functionalizations Through Flow Techniques-Flow Chemistry; RSC Green Chemistry: Cambridge, UK, 2020; pp. 199–216. [Google Scholar]
- Santoro, S.; Ferlin, F.; Ackermann, L.; Vaccaro, L. C–H functionalization reactions under flow conditions. Chem. Soc. Rev. 2019, 48, 2767–2782. [Google Scholar] [CrossRef]
- Santoro, S.; Marrocchi, A.; Lanari, D.; Ackermann, L.; Vaccaro, L. Towards sustainable C–H functionalization reactions: The emerging role of bio-based reaction media. Chem. Eur. J. 2018, 24, 13383–13390. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, K.I.; Goldman, A.S. Large-scale selective functionalization of alkanes. Acc. Chem. Res. 2017, 50, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, R.H.; Lei, A. Introduction: CH activation. Chem. Rev. 2017, 117, 8481–8482. [Google Scholar] [CrossRef] [Green Version]
- Davies, H.M.L.; Morton, D. Recent advances in C–H functionalization. J. Org. Chem. 2016, 81, 343–350. [Google Scholar] [CrossRef] [Green Version]
- Girard, S.A.; Knauber, T.; Li, C.-J. The cross-dehydrogenative coupling of Csp3–H bonds: A versatile strategy for C–C bond formations. Angew. Chem. Int. Ed. 2014, 53, 74–100. [Google Scholar] [CrossRef]
- Rouquet, G.; Chatani, N. Catalytic functionalization of C(sp2)–H and C(sp3)–H bonds by using bidentate directing groups. Angew. Chem. Int. Ed. 2013, 52, 11726–11743. [Google Scholar] [CrossRef]
- Wencel-Delord, J.; Glorius, F. C–H bond activation enables the rapid construction and latestage diversification of functional molecules. Nat. Chem. 2013, 5, 369–375. [Google Scholar] [CrossRef]
- Yeung, C.S.; Dong, V.M. Catalytic dehydrogenative cross-coupling: Forming carbon–carbon bonds by oxidizing two carbon–hydrogen bonds. Chem. Rev. 2011, 111, 1215–1292. [Google Scholar] [CrossRef]
- Dyker, G. (Ed.) Handbook of C–H Transformations: Applications in Organic Synthesis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; Volume 2. [Google Scholar]
- Crabtree, R.H.A.E. Shilov’s influence on early work in organometallic CH activation and functionalization. J. Organomet. Chem. 2015, 793, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Li, J.J. C–H Bond Activation in Organic Synthesis; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar]
- Shilov, A.E.; Shul’pin, G.B. Activation of C–H bonds by metal complexes. Chem. Rev. 1997, 97, 2879–2932. [Google Scholar] [CrossRef]
- Guo, X.-X. Copper-Catalyzed C–H Activation. In Copper Catalysis in Organic Synthesis; Anilkumar, G., Saranya, S., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2020; pp. 329–348. [Google Scholar]
- Zhang, M.; Wang, Q.; Peng, Y.; Chen, Z.; Wan, C.; Chen, J.; Zhao, Y.; Zhang, R.; Zhang, A.Q. Transition metal-catalyzed sp3 C–H activation and intramolecular C–N coupling to construct nitrogen heterocyclic scaffolds. Chem. Commun. 2019, 55, 13048–13065. [Google Scholar] [CrossRef]
- Borpatra, P.J.; Deka, B.; Deb, M.L.; Baruah, P.K. Recent advances in intramolecular C–O/C–N/C–S bond formation via C–H functionalization. Org. Chem. Front. 2019, 6, 3445–3489. [Google Scholar] [CrossRef]
- Gandeepan, P.; Kaplaneris, N.; Santoro, S.; Vaccaro, L.; Ackermann, L. Biomass-Derived Solvents for Sustainable Transition Metal-Catalyzed C–H Activation. ACS Sustain. Chem. Eng. 2019, 7, 8023–8040. [Google Scholar] [CrossRef]
- Gandeepan, P.; Muller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. 3d Transition metals for C–H activation. Chem. Rev. 2019, 119, 2192–2452. [Google Scholar] [CrossRef]
- Chu, J.C.K.; Rovis, T. Complementary strategies for directed C(sp3)–H functionalization: A comparison of transition-metal-catalyzed activation, hydrogen atom transfer, and carbene/nitrene transfer. Angew. Chem. Int. Ed. 2018, 57, 62–101. [Google Scholar] [CrossRef]
- Gandeepan, P.; Ackermann, L. Transient directing groups for transformative C–H activation by synergistic metal catalysis. Chem 2018, 4, 199–222. [Google Scholar] [CrossRef] [Green Version]
- Arockiam, P.B.; Bruneau, C.; Dixneuf, P.H. Ruthenium(II)-catalyzed C–H bond activation and functionalization. Chem. Rev. 2012, 112, 5879–5918. [Google Scholar] [CrossRef]
- Hashiguchi, B.G.; Bischof, S.M.; Konnick, M.M.; Periana, R.A. Designing catalysts for functionalization of unactivated C–H bonds based on the CH activation reaction. Acc. Chem. Res. 2012, 45, 885–898. [Google Scholar] [CrossRef]
- Kim, K.; Jung, Y.; Lee, S.; Kim, M.; Shin, D.; Byun, H.; Cho, S.J.; Song, H.; Kim, H. Directed C− H activation and tandem cross-coupling reactions using palladium nanocatalysts with controlled oxidation. Angew. Chem. Int. Ed. 2017, 56, 6952–6956. [Google Scholar] [CrossRef]
- Ferlin, F.; Luque Navarro, P.M.; Gu, Y.; Lanari, D.; Vaccaro, L. Waste minimized synthesis of pharmaceutically active compounds via heterogeneous manganese catalysed C–H oxidation in flow. Green Chem. 2020, 22, 397–403. [Google Scholar] [CrossRef]
- Ferlin, F.; van der Hulst, M.K.; Santoro, S.; Lanari, D.; Vaccaro, L. Continuous flow/waste minimized synthesis of benzoxazoles catalysed by heterogeneous manganese systems. Green Chem. 2019, 21, 5298–5305. [Google Scholar] [CrossRef]
- Ferlin, F.; Yetra, S.R.; Warratz, S.; Vaccaro, L.; Ackermann, L. Reusable Pd@PEG catalyst for aerobic dehydrogenative C–H/C–H arylations of 1,2,3-triazoles. Chem. Eur. J. 2019, 25, 11427–11431. [Google Scholar] [CrossRef]
- Sciosci, D.; Valentini, F.; Ferlin, F.; Chen, S.; Gu, Y.; Piermatti, O.; Vaccaro, L. A heterogeneous and recoverable palladium catalyst to access the regioselective C–H alkenylation of quinoline N-oxides. Green Chem. 2020, 22, 6560–6566. [Google Scholar] [CrossRef]
- Asensio, J.M.; Bouzouita, D.; van Leeuwen, P.W.N.M.; Chaudret, B. σ-H–H, σ-C–H, and σ-Si–H bond activation catalyzed by metal nanoparticles. Chem. Rev. 2020, 120, 1042–1084. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, A.; Asiri, A.M.; Garcia, H. Formation of C–C and C–heteroatom bonds by C–H activation by metal organic frameworks as catalysts or supports. ACS Catal. 2019, 9, 1081–1102. [Google Scholar] [CrossRef]
- Liu, M.; Wu, J.; Hou, H. Metal–organic framework (MOF)-based materials as heterogeneous catalysts for C–H bond activation. Chem. Eur. J. 2019, 25, 2935–2948. [Google Scholar] [CrossRef]
- Pla, D.; Gómez, M. Metal and metal oxide nanoparticles: A lever for C–H functionalization. ACS Catal. 2016, 6, 3537–3552. [Google Scholar] [CrossRef]
- Reay, A.J.; Fairlamb, I.J.S. Catalytic C–H bond functionalisation chemistry: The case for quasi-heterogeneous catalysis. Chem. Commun. 2015, 51, 16289–16307. [Google Scholar] [CrossRef] [PubMed]
- Valentini, F.; Brufani, G.; Latterini, L.; Vaccaro, L. Metal nanoparticles catalyzed C–C bond formation via C–H activation. In Advanced Heterogeneous Catalysts Volume 1: Applications at the Nano-Scale; American Chemical Society: Washington, DC, USA, 2020; pp. 513–543. [Google Scholar] [CrossRef]
- Fairlamb, I.J.S.; Scott, N.W.J. Pd Nanoparticles in C–H Activation and Cross-coupling Catalysis. In Nanoparticles in Catalysis; Springer: Cham, Switzerland, 2020; pp. 171–205. [Google Scholar] [CrossRef]
- Serp, P.; Philippot, K. (Eds.) Nanomaterials in Catalysis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013. [Google Scholar]
- Astruc, D. Nanoparticles and Catalysis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008. [Google Scholar]
- Heiz, U.; Landman, U. (Eds.) Nanocatalysis; Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- Cao, G.; Wang, Y. Nanostructures and Nanomaterials: Synthesis, Properties and Applications, 2nd ed.; World Scientific Series in Nanoscience and Nanotechnology; World Scientific Pub Co Inc. Imperial College Press: London, UK, 2011; Volume 2. [Google Scholar]
- Calvino-Casilda, V.; Lopez-Peinado, A.J.; Martin-Aranda, R.M.; Mayoral, E.P. (Eds.) Nanocatalysis: Applications and Technologies; CRC Press: Boca Raton, FL, USA, 2019. [Google Scholar]
- Chopra, R.; Kumar, M.; Bhalla, V. Development of a supramolecular ensemble of an AIEE active hexaphenylbenzene derivative and Ag@Cu2O core–shell NPs: An efficient photocatalytic system for C–H activation. Chem. Commun. 2016, 52, 10179–10182. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Kaur, M.; Walia, P.K.; Kumar, M.; Bhalla, V. Encapsulating Au-Fe3O4Nanodots into AIE-active SupramolecularAssemblies:Ambient Visible-light Harvesting “Dip-Strip” Photocatalyst for C-C/C-N Bond Formation Reactions. Chem. Asian J. 2019, 14, 809–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Chung, J.; Byun, S.M.; Kim, B.M.; Lee, C. Direct catalytic C–H arylation of imidazo[1,2-α]pyridine with aryl bromides using magnetically recyclable Pd–Fe3O4 nanoparticles. Tetrahedron 2013, 69, 5660–5664. [Google Scholar] [CrossRef]
- Kaur, S.; Kumar, M.; Bhalla, V. Aggregates of perylene bisimide stabilized superparamagnetic Fe3O4 nanoparticles: An efficient catalyst for the preparation of propargylamines and quinolines via C–H activation. Chem. Commun. 2015, 51, 16327–16330. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; Pham, L.T.; Phan, N.T.S.; Truong, T. Efficient and robust superparamagnetic copper ferrite nanoparticle-catalyzed sequential methylation and C–H activation: Aldehyde free propargylamine synthesis. Catal. Sci. Technol. 2014, 4, 4281–4288. [Google Scholar] [CrossRef]
- Rafiee, F.; Rezaie, F. Karder, Bio-crosslinking of chitosan with oxidized starch, its functionalization with amino acid and magnetization: As a green magnetic support for silver immobilization and its catalytic activity investigation. Int. J. Biol. Macromol. 2020, 146, 1124–1132. [Google Scholar] [CrossRef]
- Veisi, H.; Mohammadi, L.; Hemmati, S.; Tamoradi, T.; Mohammadi, P. In situ immobilized silver nanoparticles on rubia tinctorum extract-coated ultrasmall iron oxide nanoparticles: An efficient nanocatalyst with magnetic recyclability for synthesis of propargylamines by A3 coupling reaction. ACS Omega 2019, 4, 13991–14003. [Google Scholar] [CrossRef] [Green Version]
- Aghahosseini, H.; Rezaei, S.J.T.; Tadayyon, M.; Ramazani, A.; Amani, V.; Ahmadi, R.; Abdolahnjadian, D. Highly efficient aqueous synthesis of propargylamines through C–H activation catalyzed by magnetic organosilica-supported gold nanoparticles as an artificial metalloenzyme. Eur. J. Inorg. Chem. 2018, 2589–2598. [Google Scholar] [CrossRef]
- Gulati, U.; Rajesh, U.C.; Rawat, D.S. CuO/Fe2O3 NPs: Robust and magnetically recoverable nanocatalyst for decarboxylative A3 and KA2 coupling reactions under neat conditions. Tetrahedron Lett. 2016, 57, 4468–4472. [Google Scholar] [CrossRef]
- Panwar, V.; Kumar, P.; Bansal, A.; Ray, S.S.; Jain, S.L. PEGylated magnetic nanoparticles (PEG@Fe3O4) as cost effective alternative for oxidative cyanation of tertiary amines via C-H activation. Appl. Catal. A: Gen. 2015, 498, 25–31. [Google Scholar] [CrossRef]
- Gruttadauria, M.; Giacalone, F.; Noto, R. “Release and catch” catalytic systems. Green Chem. 2013, 15, 2608–2618. [Google Scholar] [CrossRef]
- Fairlamb, I.J.S.; Lee, A.F. Fundamental Pd(0)/Pd(II) redox steps in cross-coupling reactions: Homogeneous, hybrid homogeneous–heterogeneous to heterogeneous mechanistic pathways for C–C couplings. In Catalytic C–H/C–X Bond. Functionalization: Transition Metal. Mediation; Ribas, X., Ed.; RSC Catalysis Series; RSC Publishing: Cambridge, UK, 2013; pp. 72–107. [Google Scholar]
- Song, S.-Z.; Meng, Y.-N.; Li, Q.; Wei, W.-T. Recent progress in the construction of C−N bonds via metal-free radical C(sp3)−H functionalization. Adv. Synth. Catal. 2020, 362, 2120–2134. [Google Scholar] [CrossRef]
- Timsina, Y.N.; Gupton, B.F.; Ellis, K.C. Palladium-catalyzed C–H amination of C(sp2) and C(sp3)–H bonds: Mechanism and scope for N-based molecule synthesis. ACS Catal. 2018, 8, 5732–5776. [Google Scholar] [CrossRef]
- Henry, M.C.; Mostafa, M.A.; Sutherland, A. Recent advances in transition-metal-catalyzed, directed aryl C–H/N–H cross-coupling reactions. Synthesis 2017, 49, 4586–4598. [Google Scholar] [CrossRef] [Green Version]
- Dauban, P.; Darses, B. C—N Bond Formation by Arene C—H Activation Using a Palladium Catalyst. In Catalytic Transformations via C-H Activation; Yu, J.-Q., Ed.; Georg Thieme Verlag KG: New York, NY, USA, 2015; Volume 2, pp. 221–248. [Google Scholar]
- Harry, N.A.; Jagadeesh, R.V. Copper-Catalyzed Aminations. In Copper Catalysis in Organic Synthesis; Anilkumar, G., Saranya, S., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2020; pp. 239–259. [Google Scholar]
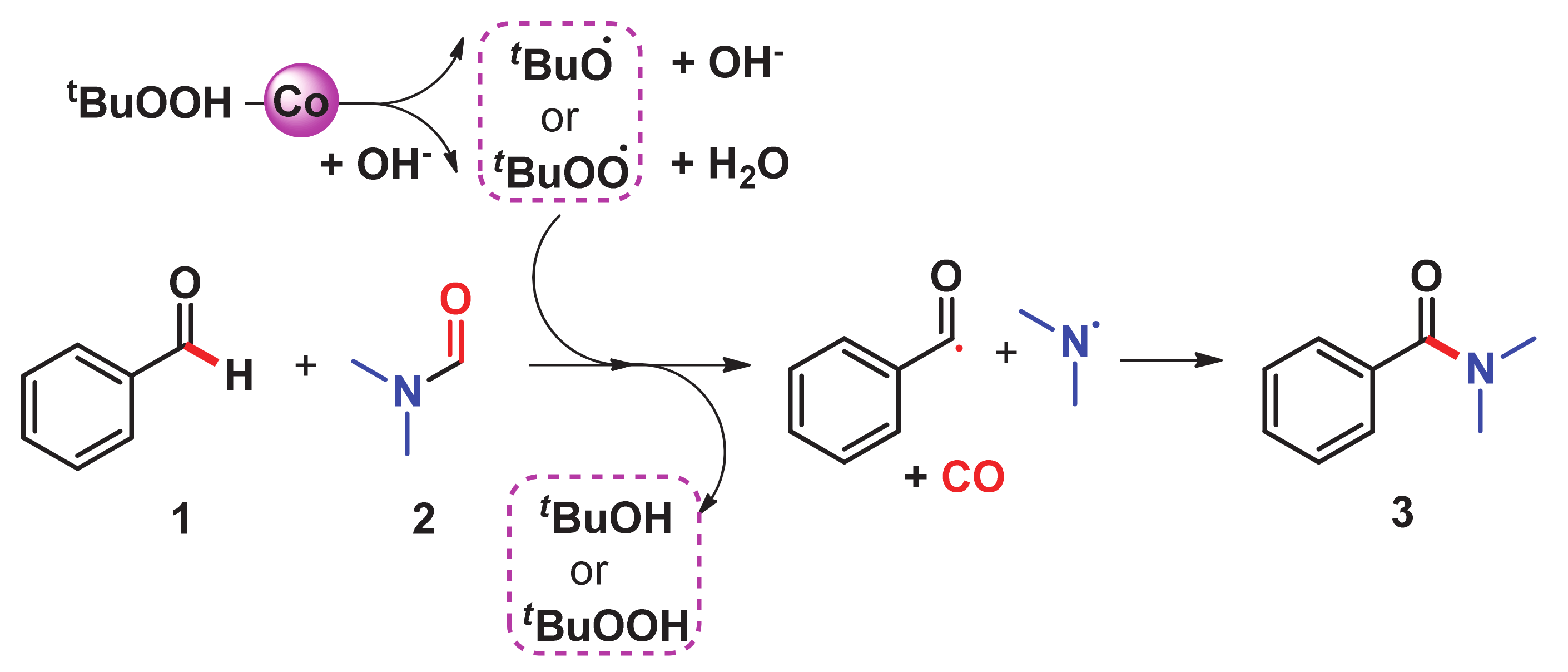
- Bai, C.; Yao, X.; Li, Y. Easy access to amides through aldehydic C–H bond functionalization catalyzed by heterogeneous Co-based catalysts. ACS Catal. 2015, 5, 884–891. [Google Scholar] [CrossRef]
- Gava, R.; Biffis, A.; Tubaro, C.; Zaccheria, F.; Ravasio, N. Heterogeneous copper-based catalysts for the amidation of activated C–H bonds. Catal. Commun. 2013, 40, 63–65. [Google Scholar] [CrossRef]
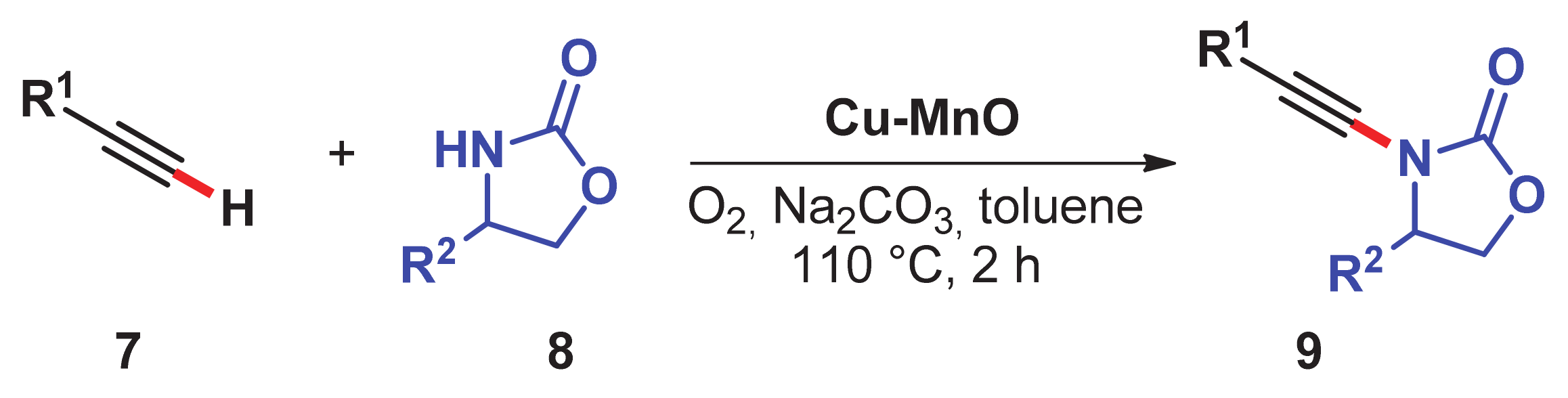
- Singh, H.; Sahoo, T.; Sen, C.; Galani, S.M.; Ghosh, S.C. Aerobic oxidative alkynylation of H-phosphonates and amides: An efficient route for the synthesis of alkynylphosphonates and ynamides using a recyclable Cu–MnO catalyst. Catal. Sci. Technol. 2019, 9, 1691–1698. [Google Scholar] [CrossRef]
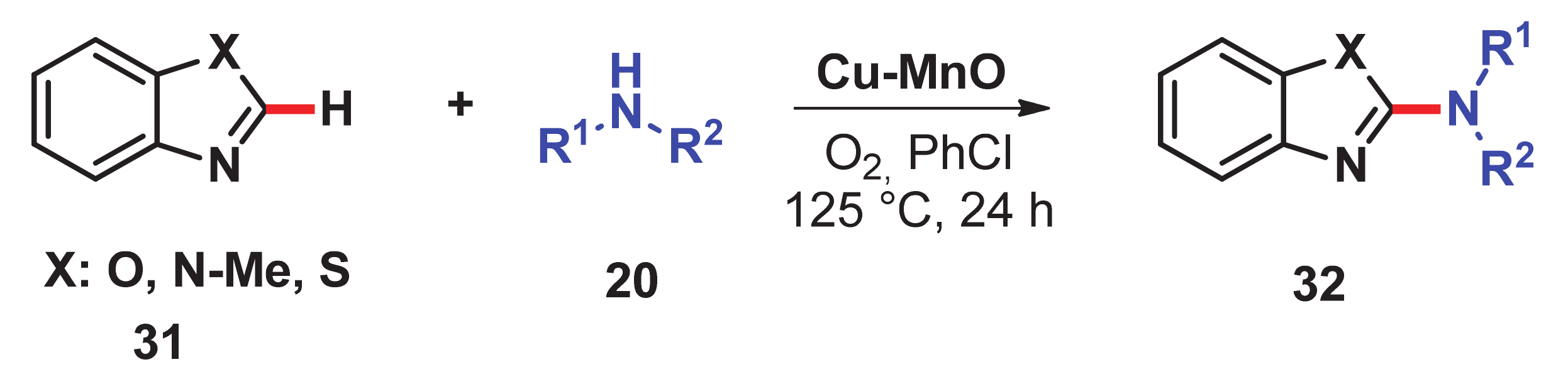
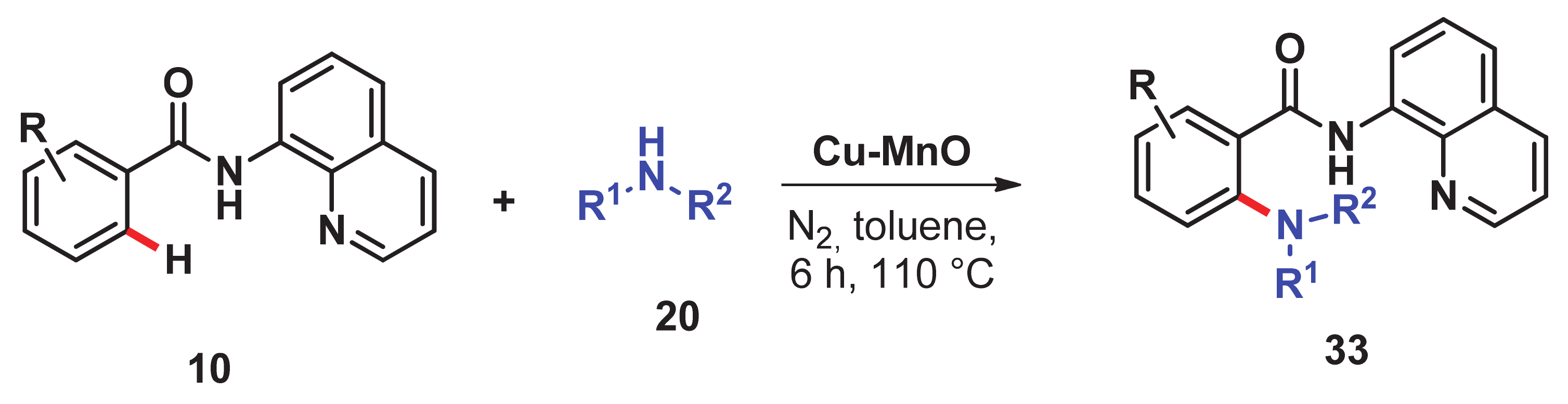
- Singh, H.; Sen, C.; Suresh, E.; Panda, A.B.; Ghosh, S.C. C–H amidation and amination of arenes and heteroarenes with amide and amine using Cu-MnO as a reusable catalyst under mild conditions. J. Org. Chem. 2021, 86, 3261–3275. [Google Scholar] [CrossRef]
- Meng, X.; Wang, Y.; Wang, Y.; Chen, B.; Jing, Z.; Chen, G.; Zhao, P. OMS-2-supported Cu hydroxide-catalyzed benzoxazoles synthesis from catechols and amines via domino oxidation process at room temperature. J. Org. Chem. 2017, 82, 6922–6931. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Yu, C.; Chen, G.; Zhao, P. Heterogeneous biomimetic aerobic synthesis of 3-iodoimidazo[1,2-a]pyridines via CuOx/OMS-2-catalyzed tandem cyclization/iodination and their late-stage functionalization. Catal. Sci. Technol. 2015, 5, 372–379. [Google Scholar] [CrossRef]
- Biswas, S.; Mullick, K.; Chen, S.-Y.; Kriz, D.A.; Shakil, M.D.; Kuo, C.-H.; Angeles-Boza, A.M.; Rossi, A.R.; Suib, S.L. Mesoporous copper/manganese oxide catalyzed coupling of alkynes: Evidence for synergistic cooperative catalysis. ACS Catal. 2016, 6, 5069–5080. [Google Scholar] [CrossRef]
- Oishi, T.; Yamaguchi, K.; Mizuno, N. Conceptual design of heterogeneous oxidation catalyst: Copper hydroxide on manganese oxide-based octahedral molecular sieve for aerobic oxidative alkyne homocoupling. ACS Catal. 2011, 1, 1351–1354. [Google Scholar] [CrossRef]
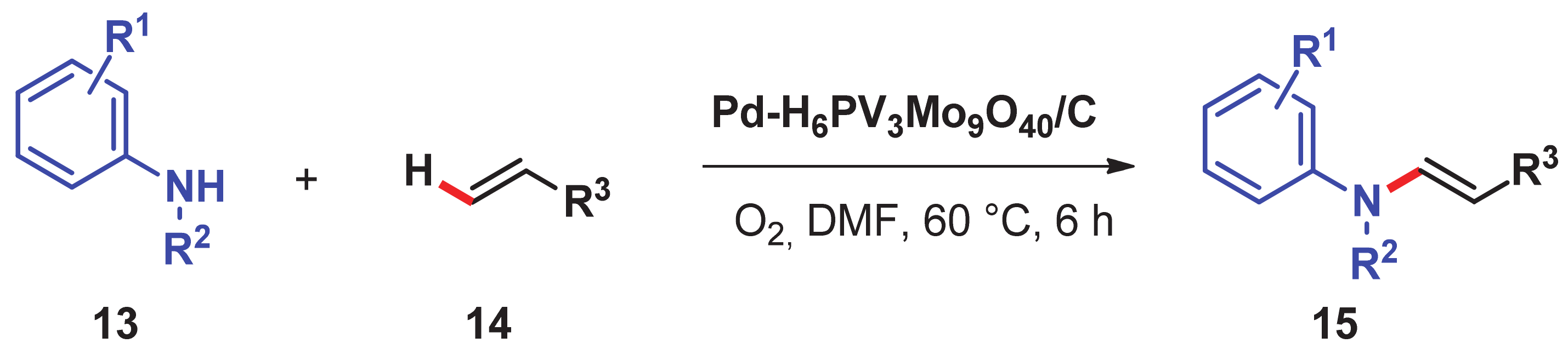
- Chng, L.L.; Zhang, J.; Yang, J.; Amoura, M.; Ying, J.Y. C–C bond formation via C–H activation and C–N bond formation via oxidative amination catalyzed by palladium- polyoxometalate nanomaterials using dioxygen as the terminal oxidant. Adv. Synth. Catal. 2011, 353, 2988–2998. [Google Scholar] [CrossRef]
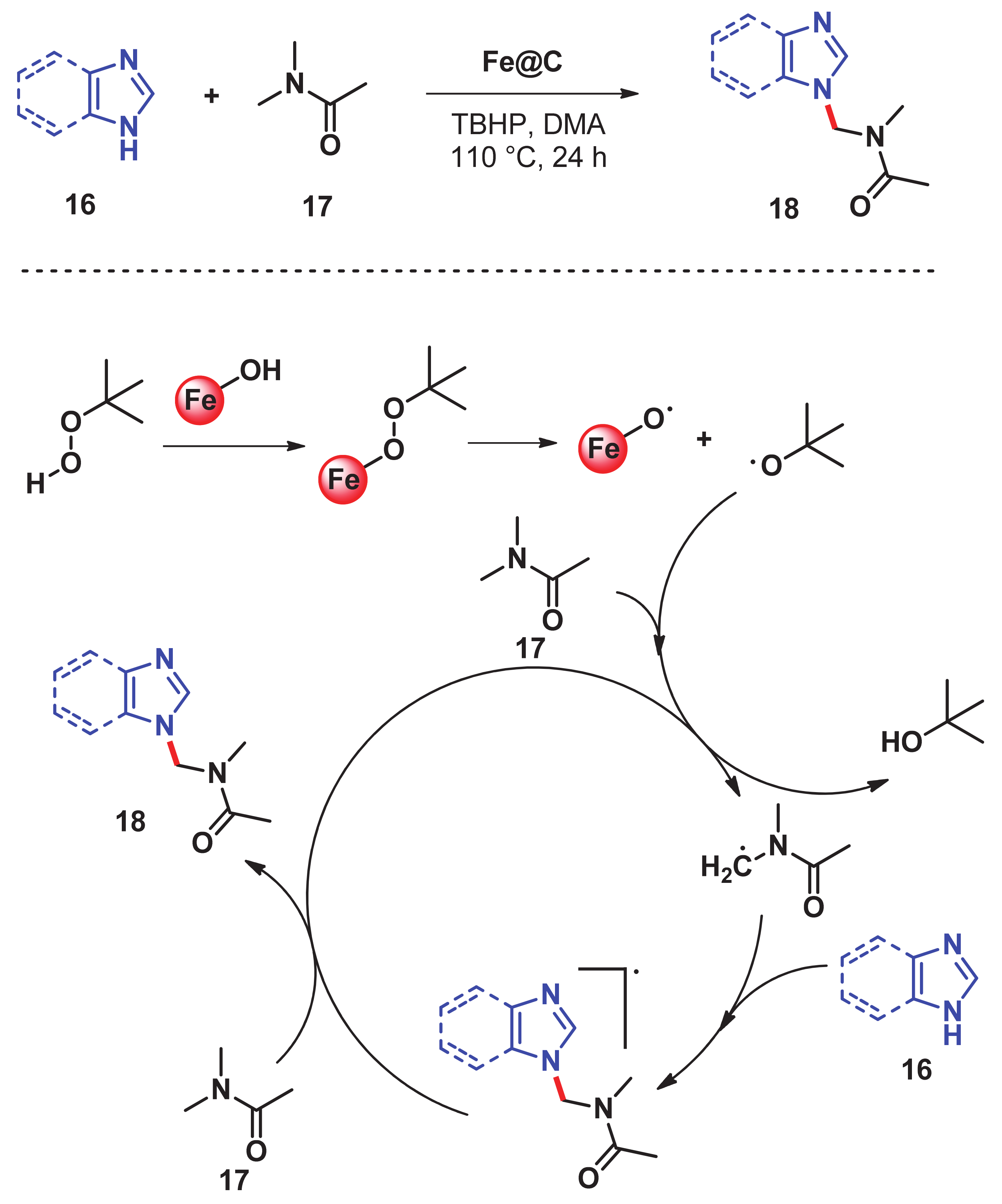
- He, J.; Dhakshinamoorthy, A.; Primo Arnau, A.M.; García Gómez, H. Iron nanoparticles embedded in graphitic carbon matrix as heterogeneous catalysts for the oxidative CN coupling of aromatic NH compounds and amides. Chem. Cat. Chem. 2017, 9, 3003–3012. [Google Scholar] [CrossRef]
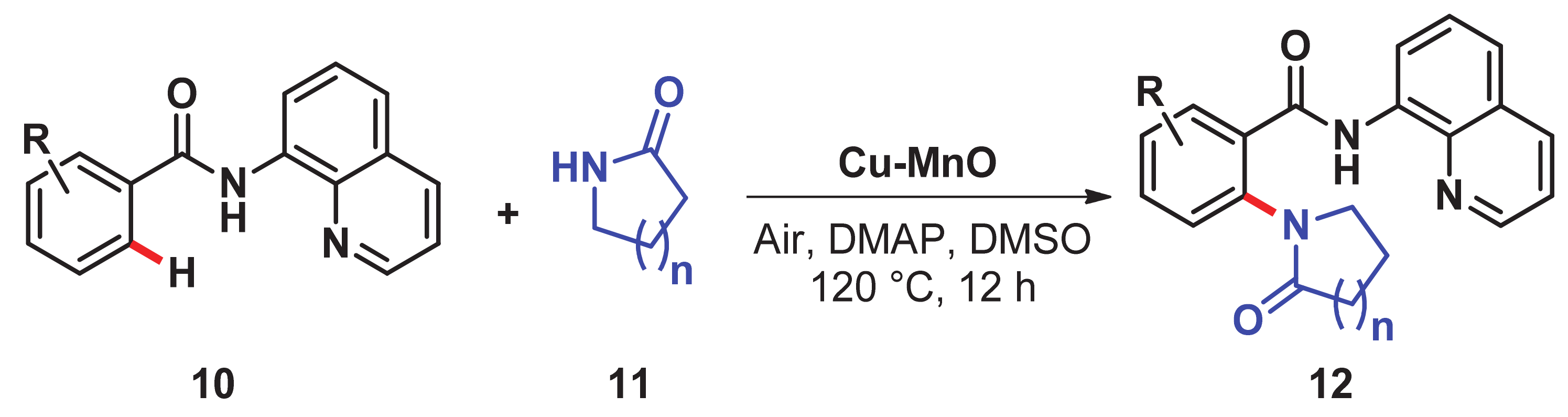
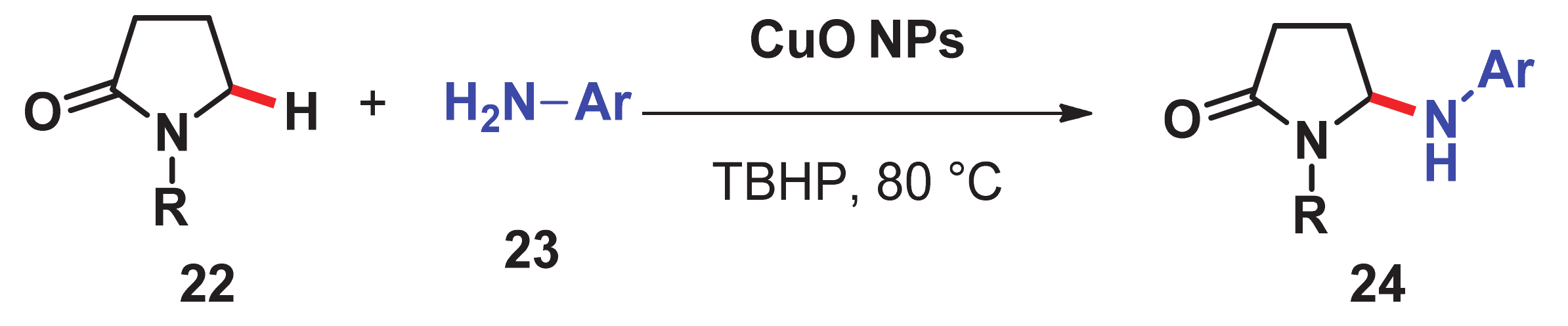
- Priyadarshini, S.; Joseph, P.A.; Kantam, M.L. Copper catalyzed oxidative cross-coupling of aromatic amines with 2-pyrrolidinone: A facile synthesis of N-aryl-γ-amino-γ-lactams. Tetrahedron 2014, 70, 6068–6074. [Google Scholar] [CrossRef]
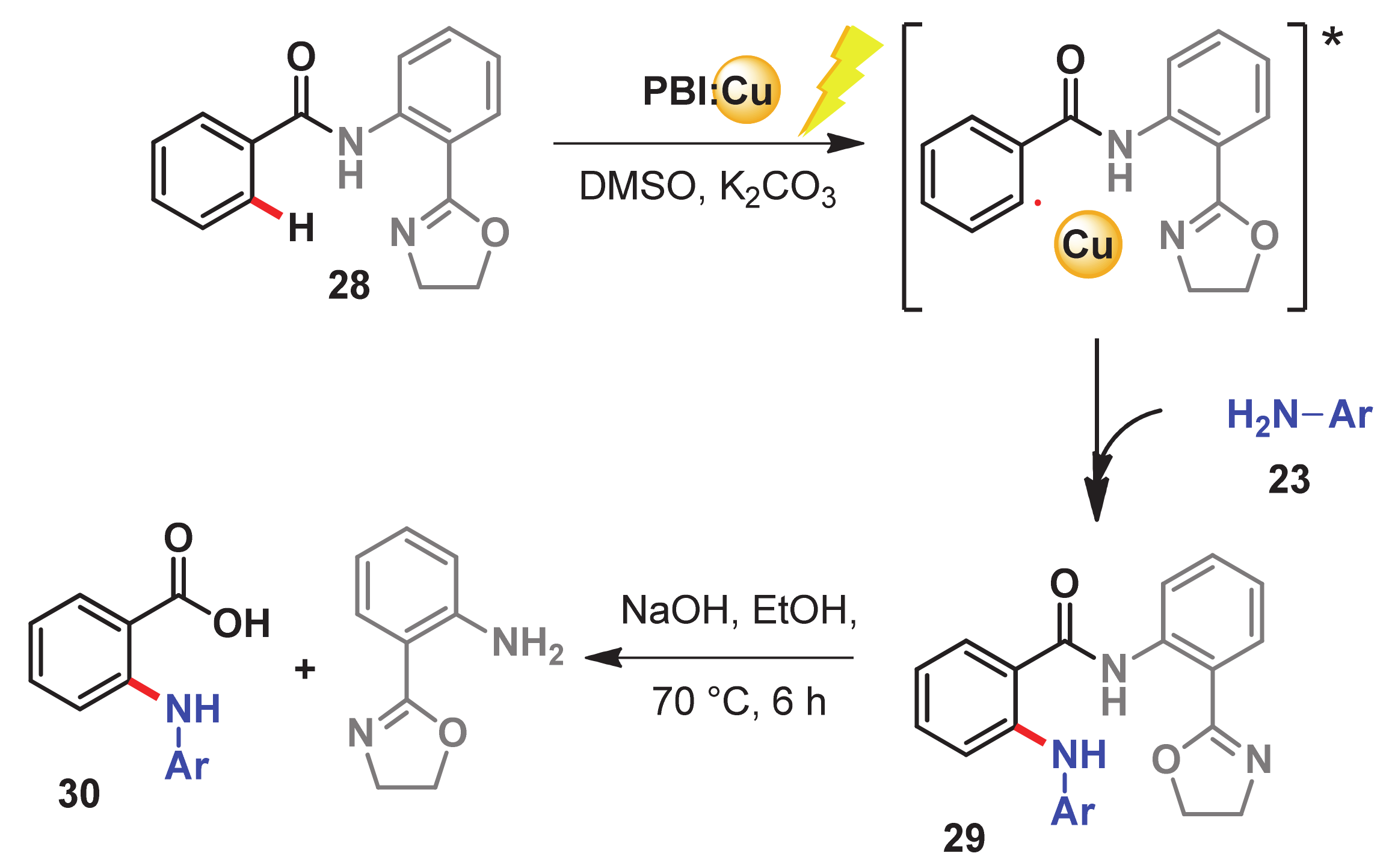
- Kaur, S.; Kumar, M.; Bhalla, V. Supramolecular ensemble of PBI derivative and copper nanoparticles: A light harvesting antenna for photocatalytic C (sp2)–H functionalization. Green Chem. 2016, 18, 5870–5883. [Google Scholar] [CrossRef]
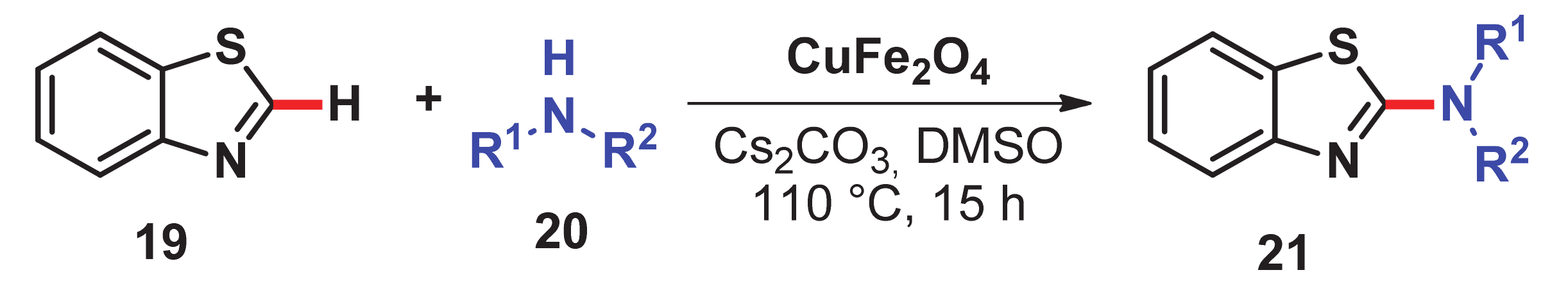
- Satish, G.; Reddy, K.H.V.; Anil, B.S.P.; Shankar, J.; Kumar, R.U.; Nageswar, Y.V.D. Direct C–H amination of benzothiazoles by magnetically recyclable CuFe2O4 nanoparticles under ligand-free conditions. Tetrahedron Lett. 2014, 55, 5533–5538. [Google Scholar] [CrossRef]
- Singh, H.; Pal, P.; Sen, C.; Panda, A.B.; Ghosh, S.C. Heterogeneous Cu-MnO-Catalyzed Direct C–H Amination of Azoles Using O2 as the Sole Oxidant. Asian J. Org. Chem. 2017, 6, 702–706. [Google Scholar] [CrossRef]
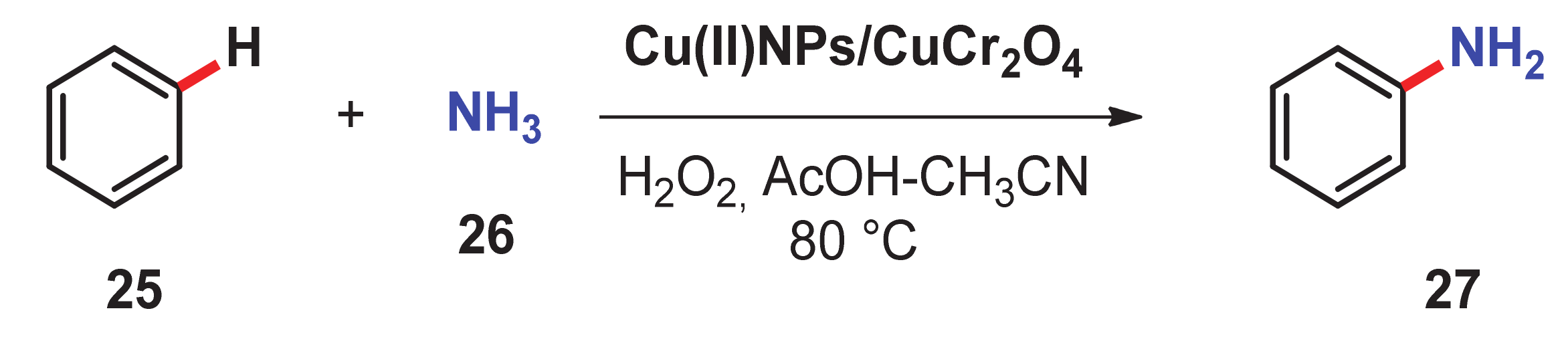
- Acharyya, S.S.; Ghosh, S.; Bal, R. Direct catalytic oxyamination of benzene to aniline over Cu(II) nanoclusters supported on CuCr2O4 spinel nanoparticles via simultaneous activation of C–H and N–H bonds. Chem. Commun. 2014, 50, 13311–13314. [Google Scholar] [CrossRef]
- Pal, P.; Giri, A.K.; Singh, H.; Ghosh, S.C.; Panda, A.B. Heterogeneously porous γ-MnO2-catalyzed direct oxidative amination of benzoxazole through C-H activation in the presence of O2. Chem. Asian J. 2014, 9, 2392–2396. [Google Scholar] [CrossRef]
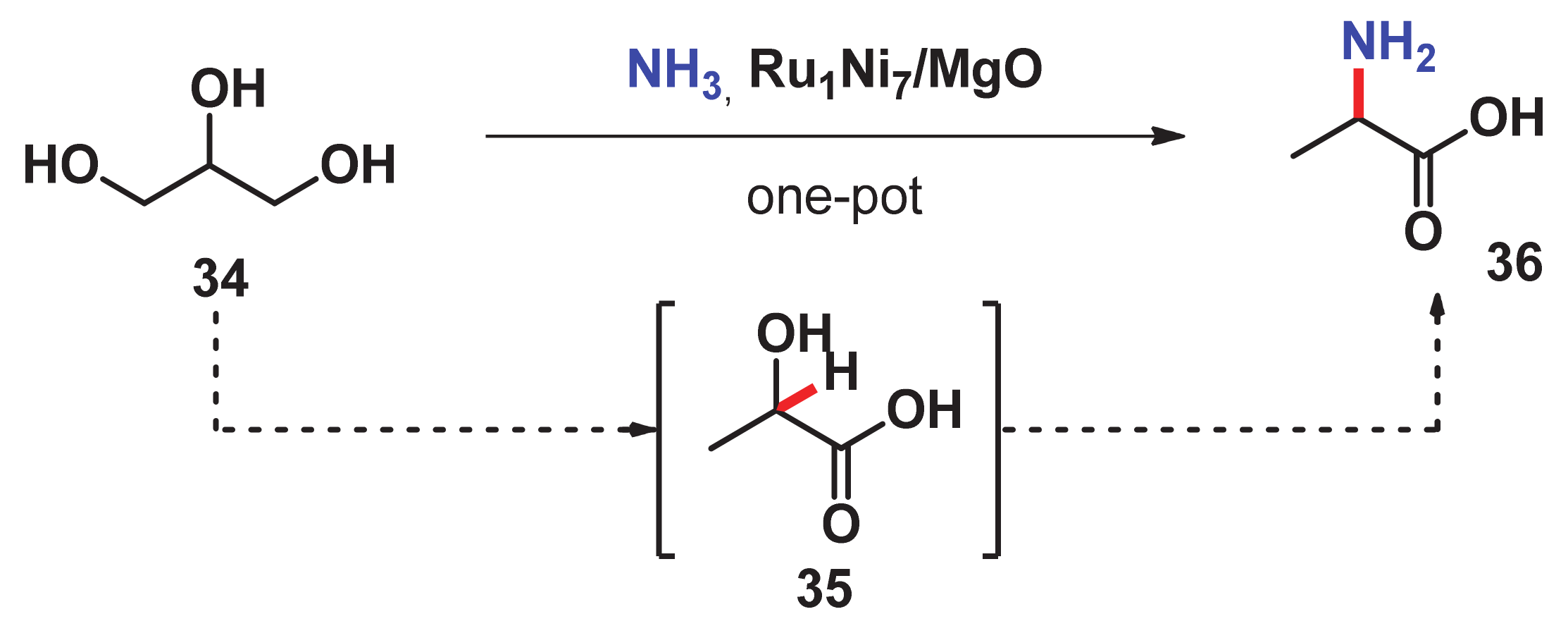
- Wang, Y.; Furukawa, S.; Song, S.; He, Q.; Asakura, H.; Yan, N. Catalytic production of alanine from waste glycerol. Angew. Chem. Int. Ed. 2020, 59, 2289–2293. [Google Scholar] [CrossRef] [PubMed]
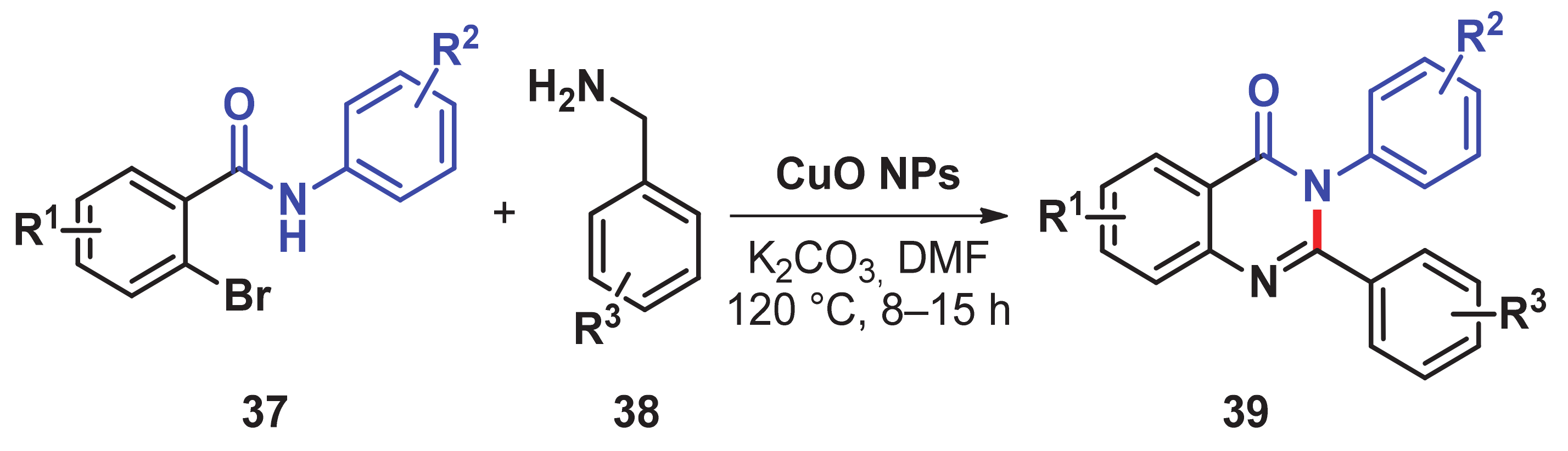
- Modi, A.; Ali, W.; Mohanta, P.R.; Khatun, N.; Patel, B.K. CuO nanoparticle catalyzed synthesis of 2,3-disubstituted quinazolinones via sequential N-Arylation and oxidative C–H amidation. ACS Sustain. Chem. Eng. 2015, 3, 2582–2590. [Google Scholar] [CrossRef]
- Chng, L.L.; Yang, J.; Wei, Y.; Ying, J.Y. Palladium nanomaterials in catalytic intramolecular C–H amination reactions. Chem. Commun. 2014, 50, 9049–9052. [Google Scholar] [CrossRef] [PubMed]
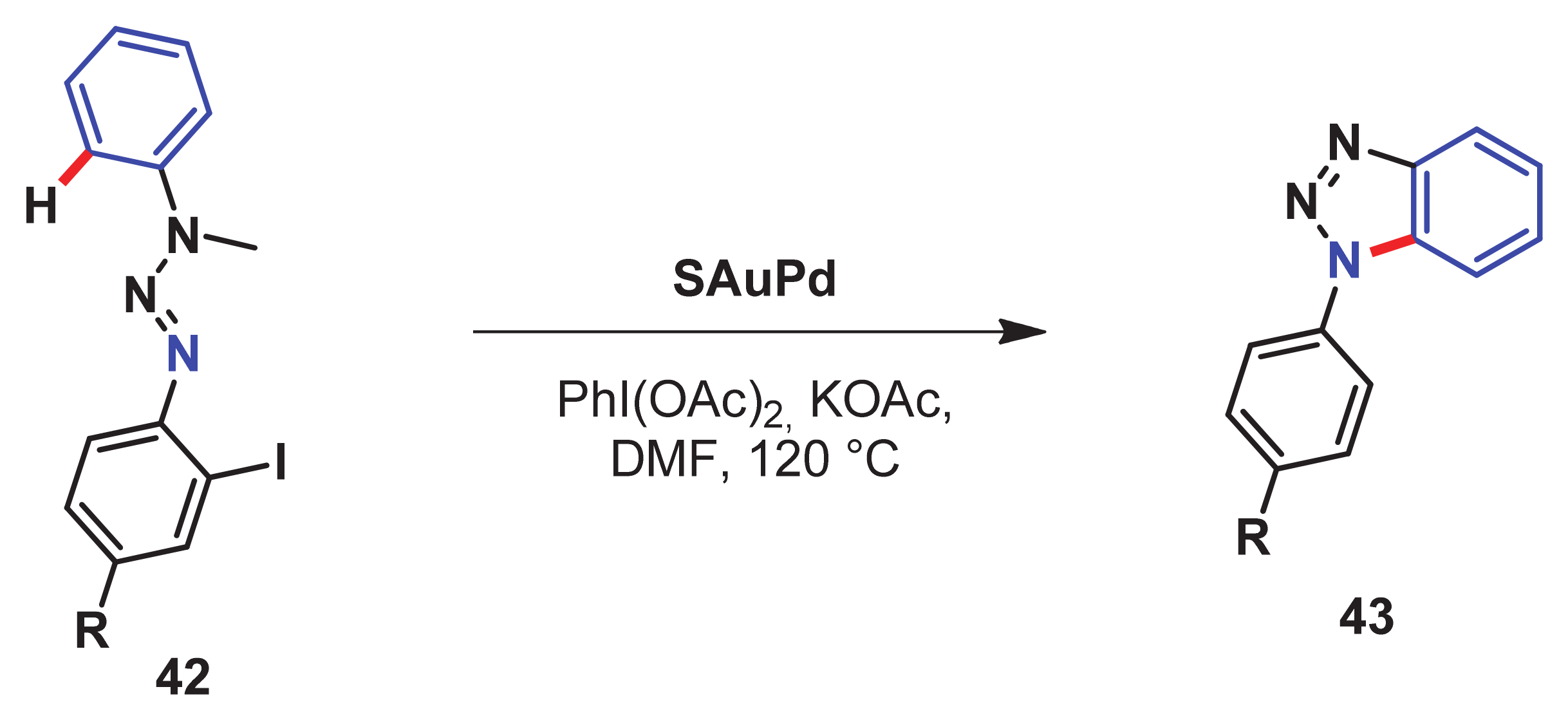
- Takagi, K.; Al-Amin, M.; Hoshiya, N.; Wouters, J.; Sugimoto, H.; Shiro, Y.; Fukuda, H.; Shuto, S.; Arisawa, M. Palladium-nanoparticle-catalyzed 1, 7-palladium migration involving C–H activation, followed by intramolecular amination: Regioselective synthesis of N 1-arylbenzotriazoles and an evaluation of their inhibitory activity toward indoleamine 2, 3-dioxygenase. J. Org. Chem. 2014, 79, 6366–6371. [Google Scholar] [CrossRef]
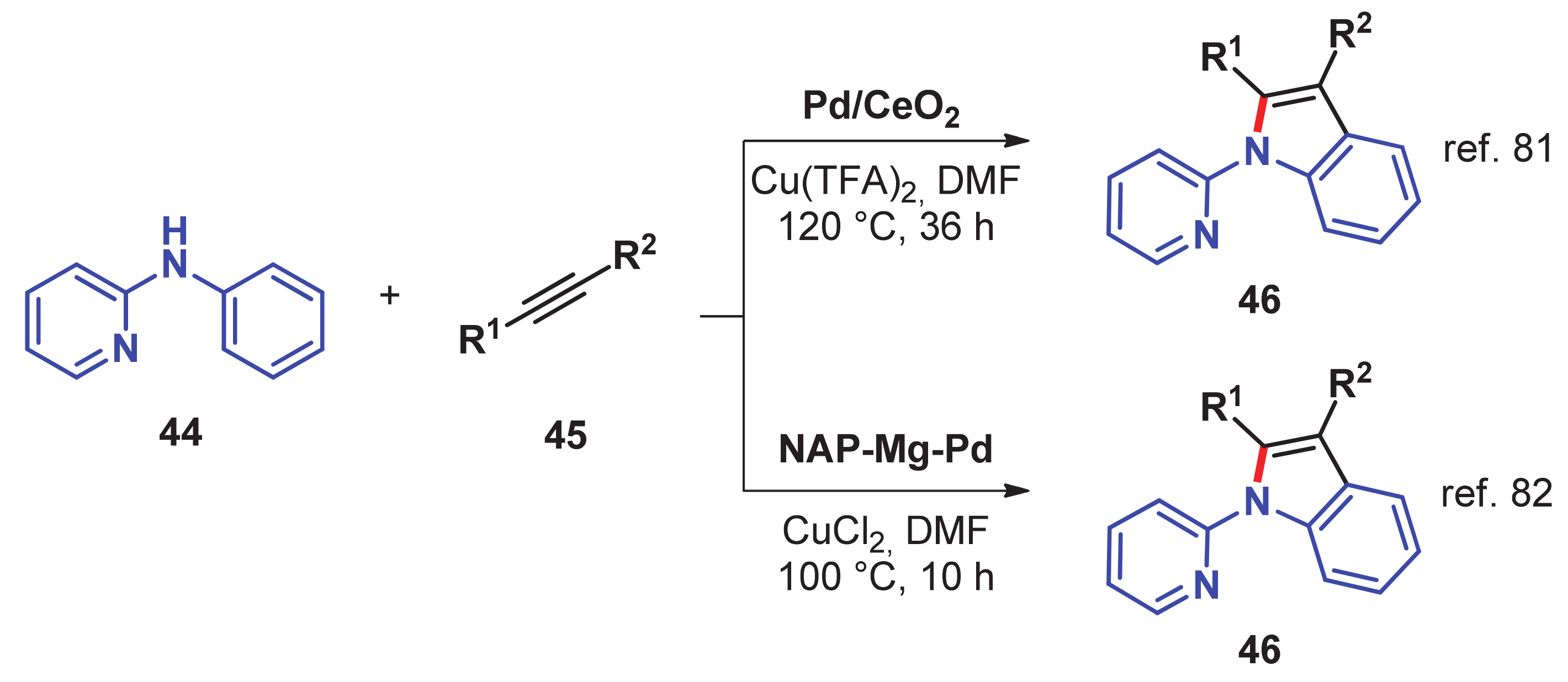
- Chen, J.; He, L.; Natte, K.; Neumann, H.; Beller, M.; Wu, X.-F. Palladium@Cerium(IV) oxide-catalyzed oxidative synthesis of n-(2-pyridyl)indoles via C–H activation reaction. Adv. Synth. Catal. 2014, 356, 2955–2959. [Google Scholar] [CrossRef]
- Reddy, P.V.; Annapurna, M.; Srinivas, P.; Likhar, P.R.; Kantam, M.L. Nanocrystalline magnesium oxide-stabilized palladium (0): An efficient and reusable catalyst for synthesis of N-(2-pyridyl) indoles. New J. Chem. 2015, 39, 3399–3404. [Google Scholar] [CrossRef]
- Saha, R.; Sekar, G. Stable Pd-nanoparticles catalyzed domino CH activation/CN bond formation strategy: An access to phenanthridinones. J. Catal. 2018, 366, 176–188. [Google Scholar] [CrossRef]
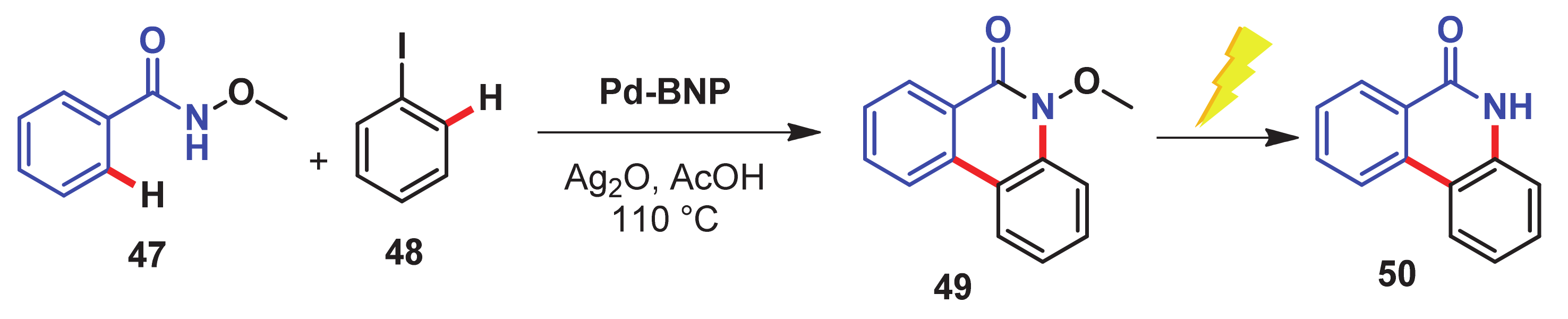
- Jayarajan, R.; Kumar, R.; Gupta, J.; Dev, G.; Kadu, P.; Chatterjee, D.; Bahadur, D.; Maiti, D.; Maji, S.K. Fabrication of an amyloid fibril-palladium nanocomposite: A sustainable catalyst for C–H activation and the electrooxidation of ethanol. J. Mater. Chem. A 2019, 7, 4486–4493. [Google Scholar] [CrossRef]



















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valentini, F.; Piermatti, O.; Vaccaro, L. Metal Nanoparticles as Sustainable Tools for C–N Bond Formation via C–H Activation. Molecules 2021, 26, 4106. https://doi.org/10.3390/molecules26134106
Valentini F, Piermatti O, Vaccaro L. Metal Nanoparticles as Sustainable Tools for C–N Bond Formation via C–H Activation. Molecules. 2021; 26(13):4106. https://doi.org/10.3390/molecules26134106
Chicago/Turabian StyleValentini, Federica, Oriana Piermatti, and Luigi Vaccaro. 2021. "Metal Nanoparticles as Sustainable Tools for C–N Bond Formation via C–H Activation" Molecules 26, no. 13: 4106. https://doi.org/10.3390/molecules26134106